QC-7129B鲎试剂灵敏度复核操作规程
- 格式:doc
- 大小:30.00 KB
- 文档页数:3


USP这一章节的部分内容已与欧洲药典和日本药典协调一致,不一致的部分以符号*标出。
本章阐述了关于检查和定量供试品内毒素的方法。
本法利用鲎(Limuluspolyhemus或 Tachypleus tridentatus)血细胞提取物制备的用于内毒素检测的鲎试剂(LAL)检定内毒素。
*1该检查包括两种方法:一为凝胶法,系利用鲎试剂与内毒素产生凝集反应的原理;另一种为光度测定法,该法利用鲎试剂与内毒素反应过程中的光学变化来实现内毒素的测定,这种方法又可分为浊度法(基于形成凝胶的过程中,溶菌液的浊度变化)和显色法(得到的肽-呈色基团复合物断裂后,检测反应混合物的色度)。
检测时,可用其中任一种方法进行试验。
当测定结果可疑或有争议时,除非各论中另有规定,以凝胶法测定结果为准。
直接比较供试品溶液与标准内毒素溶液,判断凝胶法的反应终点。
内毒素含量以USP内毒素单位(USP-EU)表示。
[注-1USP EU相当于1个内毒素单位。
]LAL试剂专用于浊度检查法或显色法,因此,使用这两种方法进行检定时,必须符合它们各自的要求。
两种检查法都要求建立标准曲线,以检定供试品的内毒素含量。
主要的实验步骤有:在预定的时间内将内毒素和对照品分别与LAL试剂保温培养;读取相应波长处的吸光度等。
使用终点浊度法时,应在孵育时间结束时马上读数;对终点显色法,则要在孵育终止时添加酶反应-终止制剂,反应停止后,方可读数。
动态浊度法和动态显色法分析整个反应时间内吸光度的变化,并通过这些读数计算比值。
仪器所有玻璃器皿及由其他耐热材料制成的器皿需用已验证的工艺在热烘箱内进行去热原处理。
*2去热原时,常用的最小时间和温度设置分别为30分钟和250℃。
若使用塑料器械,如微孔板和微量进样器配套的吸头等,它们必须标明无内毒素并确对试验无干扰。
[注-本章内,“管”也包括任何其他反应容器,如微孔板的孔等。
]内毒素储备标准溶液和内毒素标准溶液的制备USP内毒素RS的效价规定为10000 USP内毒素单位(EU)/西林瓶。

鲎试验检查细菌内毒素应注意的问题鲎试验检查细菌内毒素应注意的问题发布时间02年09月05日 09时18分方法:通过对干扰鲎试验准确性干扰因素的分析(包括鲎试剂本身、操作条件、配制原料等),提出了鲎试验中应注意的问题。
结果:只要克服了检查中的干扰因素,鲎法具有比家兔法更简便、快速、灵敏度高、重现性好等优点。
鲎试验是利用鲎的变形细胞与微量内毒素产生凝集反应的现象,作为判断供试品中细菌内毒素限量是否符合规定的一种方法[1]。
它具有比家兔法更简便、快速、灵敏度高、重现性好等优点。
中国药典1995年版已收载细菌内毒素检查法(简称鲎试验),但只用于几种输液的检查。
根据现有的实验证明,凡不干扰细菌内毒素检查或虽有干扰,但可以克服的药品注射剂,将能够全部以鲎法代替家兔热原检查法。
但是该法受到许多因素的影响,故在做鲎试验检查时,应注意下列问题。
1鲎试剂本身1.1鲎试剂灵敏度 为了保证检查细菌内毒素结果的准确性,用于试验的鲎试剂应首先核对灵敏度。
因为在我们的实际工作中,发现鲎试剂均符合药典规定标准前提下,由于生产厂家不同,部分鲎试剂的实测灵敏度与标示值有差异而导致检测同一检品时,用相同标示值的鲎试剂,出现不同结果。
因而先核对灵敏度可避免因鲎试剂本身造成的判断失误。
建议法定部门和生产单位应注意鲎试剂的质量检查。
1.2供试品稀释 在取样品时,一定要熟悉中国药典的内容,不能直接取大输液原液0.1ml进行检查,这是排除干扰因素的简单有效的方法。
因药典已明确规定试验中鲎试剂的灵敏度不超过0.5EU/ml,根据供试品的稀释公式: 供试品稀释倍数= L/λυL :供试品的内毒素限量(1EU/ml)λυ:鲎试剂灵敏度的标示值(EU/ml)故检测大输液均应稀释后才能进行试验,如选用更高灵敏度的鲎试剂,则应增加样品的稀释倍数。
稀释用的细菌内毒素检查用水药典作了规定。
在4h内不得与鲎试剂产生凝集反应。
如果直接用供试品原液试验,就会使供试品的内毒素提高到0.5EU/ml或0.5EU/ml以下,这样,根据目前大输液的生产和贮藏条件,有些就会达不到,鲎法检出的阳性率会提高,兔法复核的机遇也将增多,势必会浪费人力物力。
1 目的规范操作人员检查细菌内毒素的方法,确保检验数据的准确性。
2 范围适用于本厂质监科化验室对注射用水的细菌内毒素检验。
3 责任化验员有责任按本操作规程进行正确操作,并对检验结果负责。
4 内容本法系利用鲎试剂与细菌内毒素产生凝集反应的机理,以判断供试品中细菌内毒素的限量是否符合规定的一种方法。
4.1仪器、用具4.1.1旋涡混合器、注射器(2.0ml、1.0ml)、注射针(6号、9号)、保鲜纸、试管(10×75 mm)、镊子(金属)、金属盒。
4.1.2用具除去外源性内毒素:将用具放入金属盒,在250℃干烤30分钟或180℃干烤120分钟。
4.2试剂4.2.1细菌内毒素国家标准品:系自大肠杆菌提取精制而成,用于标定、复核、仲裁鲎试剂灵敏度和标定细菌内毒素工作标准品的效价。
4.2.2细菌内毒素工作标准品:系以细菌内毒素国家标准品为基准标定其效价应不小于2EU,不大于50EU。
4.2.3细菌内毒素检查用水:系指与灵敏度为0.03EU/ml或更高灵敏度的鲎试剂在37℃±1℃条件下24小时不产生凝集反应的灭菌注射用水。
4.2.4鲎试剂:0.1ml/支(λ=0.5EU/ml)。
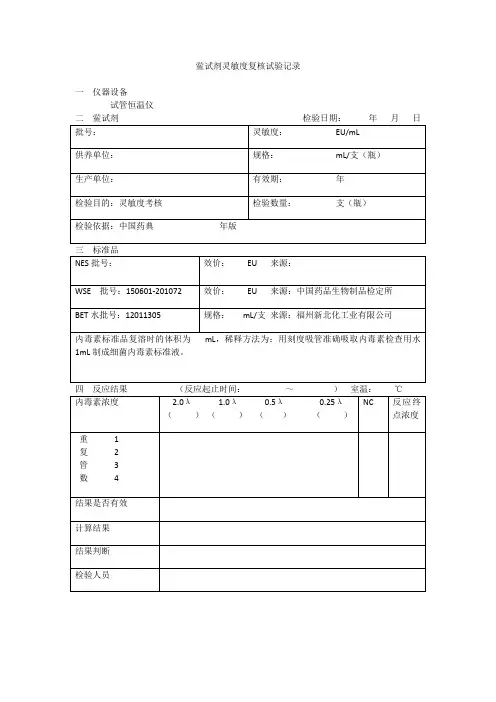
4.3鲎试剂灵敏度复核试验鲎试剂灵敏度的标示值(λ,此处λ=0.5EU/ml),将细菌内毒素国家标准品用细菌内毒素检查用水溶解,在旋涡混合器混合15分钟,然后制备成合适的稀释浓度,1EU/ml、0.5EU/ml、0.25EU/ml、0.125EU/ml,每稀释一步均应在旋涡混合器上混合30秒钟。
按检查法项下试验,每一浓度平行做4支,同时用细菌内毒素检查用水做2支阴性对照管,如最大浓度管均为阳性,最低浓度管均为阴性,阴性对照管均为阴性,按下式计算鲎试剂灵敏度的测定值λc。
λc=lg-1(∑X/4)式中X为反应终点浓度的对数值(lg)。
反应终点浓度是系列浓度递减的内毒素溶液中最后一个呈阳性结果的浓度。
当λc在0.5λ~2.0λ(包括0.5λ和2.0λ)时,方可用于细菌内毒素检查,并以λ为该批鲎试剂的灵敏度。

鲎试剂灵敏度复核SOP1.目的为规范鲎试剂标示灵敏度复核的操作,确保鲎试剂试验的准确性,特制定此操作规程。
2.范围本标准操作规程适用于新批号鲎试剂使用前对标示灵敏度的复核以及试验条件发生任何可能影响检验结果的改变时,对鲎试剂标示灵敏度的复核。
3.定义无4.职责4.1QC负责本规程的起草、修订、培训及执行。
4.2QA、QC组长、质量管理部经理负责本规程的审核。
4.3质量总监负责批准本规程。
4.4QA负责本规程执行的监督。
5.引用标准《中华人民共和国药典》2020年版二部6.材料见8.程序。
7.流程图无8.程序8.1试剂:细菌内毒素工作标准品、鲎试剂溶解水、待复核灵敏度的鲎试剂。
8.2仪器与设备:恒温水浴箱、烤箱、小试管、中管、半中管。
8.3操作步骤8.3.1试验前准备试验用吸管、小管等玻璃器皿应在试验前1~2天内包好,经200℃干烤2小时除内毒素备用。
8.3.2鲎试剂灵敏度复核根据鲎试剂灵敏度的标示值(λ),将细菌内毒素国家标准品或细菌内毒素工作标准品用细菌内毒素检查用水溶解,在漩涡混合器上混匀15分钟,然后制成2λ、λ、0.5λ和0.25λ四个浓度的内毒素标准溶液,每稀释一步均应在漩涡混合器上混匀30秒钟;取分装有0.1ml鲎试剂溶液的10mm×100mm试管18支,其中16管分别加入0.1ml不同浓度的内毒素标准溶液,每一个内毒素浓度平行做4管;另外2管加入0.1ml细菌内毒素检查用水作为阴性对照;将试管中溶液轻轻混匀后,封闭管口,垂直放入37℃±1℃的恒温器中,保温60分钟±2分钟。
8.4结果判定8.4.1将试管从恒温器中轻轻取出,缓缓倒转180°,若管内形成凝胶,并且凝胶不变形、不从管壁滑脱者为阳性;未形成凝胶或形成的凝胶不坚实、变形并从管壁滑脱者为阴性。
保温和拿取试管过程应避免受到振动造成假阴性结果。
8.4.2当最大浓度2λ管均为阳性,最低浓度0.25λ管均为阴性,阴性对照管为阴性,试验方为有效。

1目的建立细菌内毒素预试验操作规程,以指导检验人员的正确规范操作,为保证细菌内毒素检查凝胶法的准确性提供依据。
2适用范围适用于细菌内毒素检查法的“鲎试剂灵敏度复核试验”和“干扰试验”。
本规程参照《中国药典》2020年版四部-1143细菌内毒素检查法制定。
3职责进行细菌内毒素检查的操作人员严格按本规程进行操作。
4内容4.1基本原理4.1.1本法采用凝胶法,系通过鲎试剂与内毒素产生凝集反应的原理进行限度检测或半定量检测内毒素的方法。
4.1.2鲎试剂灵敏度复核试验:当使用新批号的鲎试剂或试验条件发生了任何可能影响检验结果的改变时,应进行鲎试剂灵敏度复核试验。
4.1.3干扰试验:当进行新药的内毒素检查试验前,或无内毒素检查项目的品种建立内毒素检查法时,须进行干扰试验;当鲎试剂、供试品的处方、生产工艺改变或试验环境中发生了任何有可能影响试验结果的变化时,须重新进行干扰试验。
4.1.4本试验操作过程应防止内毒素的污染。
4.1.5EU:内毒素单位,1EU与1个内毒素国际单位(IU)相当。
4.1.6λ:鲎试剂的标示灵敏度,单位:EU/ml。
4.1.7MVD:最大有效稀释倍数,指在试验中供试品溶液被允许达到稀释的最大倍数。
4.2材料准备4.2.1细菌内毒素工作标准品:应经细菌内毒素国家标准品为基准标定效价;4.2.2鲎试剂:当使用新批号前,按《细菌内毒素预试验SOP》进行灵敏度检查;4.2.3细菌内毒素检查用水(BET水):内毒素含量应<0.015EU/ml,且对内毒素试验无干扰作用;4.2.4供试品溶液:pH宜在6.0~8.0范围内,否则用已去除内毒素和干扰因子的酸、碱溶液或缓冲液调节。
4.3器具准备4.3.1移液枪、一次性除热源枪头(1000μL、200μL);4.3.2剪刀、砂轮片、试管架、除热源试管、医用胶布;4.3.3恒温培养箱(37℃±1℃)、旋涡混合器、洁净工作台。
4.4鲎试剂灵敏度复核试验4.4.1试验过程:4.4.1.1根据当批鲎试剂的标示灵敏度(λ),把细菌内毒素工作标准品用适量BET水充分溶解,避免产生气泡,在旋涡混合器上混匀15分钟。


鲎试剂灵敏度复核原理1 鲎试剂的灵敏度复核鲎试剂是一种病原体检测试剂,用于分析样品中是否存在鲎病毒。
它提供了有效、准确、可靠的检测,并且能够快速和准确地检测出鲎病毒的存在。
灵敏度复核是对鲎试剂灵敏度的一种检验。
本文将简要介绍鲎试剂灵敏度复核的原理。
2 鲎试剂灵敏度复核原理鲎试剂灵敏度复核是通过检测与水力均一系数和介质比容量(VCR)之间的相互关系来进行的。
水力均一系数(FDP)用来衡量样品的灵敏度,是一种量化的指标,它可以测量出检测样品中样品分子数量的值,它可以作为检测决策的依据。
介质比容量(VCR)指标可以衡量检测样品中DNA的质量,它可以用来测量检测分子的大小,它可以作为衡量检测样品的灵敏度的重要参数。
3 所用材料和实验方法用于鲎试剂灵敏度复核的材料包括:NIGP-9576流式细胞术引物、标准分子颗粒(SM)、即用型DNA表达质粒质粒(CTP)、库夫特—威尔斯质粒(KV)及它的完整酶切体(kVCF)、抗体制剂(AM)、核酸定量试剂盒(H∅QT)。
实验步骤主要包括:①用NIGP-9576流式细胞术引物按照要求(1∶15~2∶30)制备测定鲎病毒目标易位基因(TG)的反应混合液,加入与VCR和FDP相对应的原液;②进行表达质粒PCR,并且结合抗体进行抗体表达;③进行激活反应,并以KV或kVF为模版,分解样品;④将所有步骤重复10次,获得完整的酶的灵敏度复核试剂(V∅PoP);⑤最后,用H∅QT试剂盒进行检测,以获得定量的VCR和FDP值,便可得到灵敏度复核结果。
4 结论鲎试剂的灵敏度复核原理是通过VCR和FDP来衡量检测样品中分子数量和大小的,并用H∅QT试剂盒进行检测,以获取定性结果。
只有通过严格的技术测试才能确定样品中分子的灵敏度,确保安全操作。
USP这一章节的部分内容已与欧洲药典和日本药典协调一致,不一致的部分以符号*标出。
本章阐述了关于检查和定量供试品内毒素的方法。
本法利用鲎(Limuluspolyhemus或 Tachypleus tridentatus)血细胞提取物制备的用于内毒素检测的鲎试剂(LAL)检定内毒素。
*1该检查包括两种方法:一为凝胶法,系利用鲎试剂与内毒素产生凝集反应的原理;另一种为光度测定法,该法利用鲎试剂与内毒素反应过程中的光学变化来实现内毒素的测定,这种方法又可分为浊度法(基于形成凝胶的过程中,溶菌液的浊度变化)和显色法(得到的肽-呈色基团复合物断裂后,检测反应混合物的色度)。
检测时,可用其中任一种方法进行试验。
当测定结果可疑或有争议时,除非各论中另有规定,以凝胶法测定结果为准。
直接比较供试品溶液与标准内毒素溶液,判断凝胶法的反应终点。
内毒素含量以USP内毒素单位(USP-EU)表示。
[注-1USP EU相当于1个内毒素单位。
]LAL试剂专用于浊度检查法或显色法,因此,使用这两种方法进行检定时,必须符合它们各自的要求。
两种检查法都要求建立标准曲线,以检定供试品的内毒素含量。
主要的实验步骤有:在预定的时间内将内毒素和对照品分别与LAL试剂保温培养;读取相应波长处的吸光度等。
使用终点浊度法时,应在孵育时间结束时马上读数;对终点显色法,则要在孵育终止时添加酶反应-终止制剂,反应停止后,方可读数。
动态浊度法和动态显色法分析整个反应时间内吸光度的变化,并通过这些读数计算比值。
仪器所有玻璃器皿及由其他耐热材料制成的器皿需用已验证的工艺在热烘箱内进行去热原处理。
*2去热原时,常用的最小时间和温度设置分别为30分钟和250℃。
若使用塑料器械,如微孔板和微量进样器配套的吸头等,它们必须标明无内毒素并确对试验无干扰。
[注-本章内,“管”也包括任何其他反应容器,如微孔板的孔等。
]内毒素储备标准溶液和内毒素标准溶液的制备USP内毒素RS的效价规定为10000 USP内毒素单位(EU)/西林瓶。

错误!未找到引用源。
1.目的建立细菌内毒素检查法的操作标准,以免操作不当而造成损失及减少误差的来源。
2.范围本规程适用于各品种的细菌内毒素检查。
3.职责QC负责本规程的实施,QC主管对本规程的实施进行监督。
4.内容4.1实验材料及用具4.1.1天平(精度0.1mg);电热干燥箱(用于去除外源性内毒素,温度应能达到250℃);恒温水浴箱货适宜的恒温器(37℃±1℃);水银温度计或酒精温度计(精度1℃以下);旋涡混合器;鲎试剂(应有国家主管部门的批准文号);4.1.2细菌内毒素国家标准品或细菌内毒素工作标准品,除另有规定外,应使用由中国药品生物制品检定所统一发放的标准品;4.1.3细菌内毒素检查用水系指内毒素含量小于0.015EU/ml且对内毒素试验无干扰作用的灭菌注射用水。
4.1.4实验用具:移液管(或刻度吸管、微量加样器及无热原吸头)、凝集管(10mm ×75mm)、三角瓶、试管、试管架、洗耳球、封口膜、时钟、75%乙醇棉、剪刀、砂轮。
所用玻璃器皿须经250℃干烤30分钟以上。
若使用塑料器械、如微孔板和与微量加样器配套的吸头等,应选用标明无内毒素并且对试验无干扰的器械。
4.2实验准备4.2.1玻璃器皿的洗涤:将玻璃器皿放入铬酸洗液或其他热原灭活剂或清洗液中充分浸泡,然后取出将洗液空干,用自来水将残留洗液彻底洗净,再用蒸馏水反复冲洗三遍以上,空干后放入适宜的密闭金属容器中或用锡箔纸包好后再放入金属容器内,放置人电热干燥箱。
4.2.2玻璃器皿表面可能存在的外源性内毒素的去除:玻璃器皿置干燥箱后,将干燥箱调至250℃,待干燥箱温度升至设定的温度后开始计时,250℃干烤30分钟以上。
达到规定时间后,关断电源,待干燥箱温度自然降至室温。
在不打开金属容器的情况下,可在两天内使用;如果玻璃器皿用锡箔纸包装,在不打开包装的情况下可在两周内使用,否则须再次干烤除去可能存在的外源性内毒素。
4.2.3供试品溶液的制备:某些供试品需进行复溶、稀释或在水性溶液中浸提制成供试品溶液。
鲎试剂灵敏度复核excel计算公式以鲎试剂灵敏度复核Excel计算公式为标题鲎试剂灵敏度复核是在实验室中常见的质量控制程序之一,它用于检验鲎试剂的灵敏度是否符合规定的标准要求。
在进行鲎试剂灵敏度复核时,我们可以利用Excel软件来进行计算和分析。
本文将介绍如何使用Excel计算公式进行鲎试剂灵敏度复核。
我们需要准备一些实验数据,包括试剂的浓度和相应的反应结果。
假设我们有10个不同浓度的鲎试剂样本,我们可以将浓度和反应结果分别记录在Excel的两列中。
为了方便计算,我们可以将浓度列命名为“浓度”,将反应结果列命名为“反应结果”。
接下来,我们需要计算每个浓度对应的平均反应结果。
在Excel中,可以使用“平均”函数来实现。
我们可以在下方的空白单元格中输入“=AVERAGE(反应结果列的数据范围)”来计算平均值。
例如,如果反应结果列的数据范围是B2到B11,则我们可以输入“=AVERAGE(B2:B11)”。
计算平均反应结果后,我们还需要计算每个浓度对应的标准偏差。
标准偏差可以反映实验数据的离散程度,用于评估实验的稳定性。
在Excel中,可以使用“STDEV.S”函数来计算样本标准偏差。
我们可以在下方的空白单元格中输入“=STDEV.S(反应结果列的数据范围)”来计算标准偏差。
例如,如果反应结果列的数据范围是B2到B11,则我们可以输入“=STDEV.S(B2:B11)”。
计算平均反应结果和标准偏差后,我们可以根据实验要求来判断鲎试剂的灵敏度是否符合要求。
一般来说,如果平均反应结果在规定的范围内,并且标准偏差较小,则可认为鲎试剂的灵敏度符合要求。
否则,就需要进行进一步的调整或处理。
除了计算平均反应结果和标准偏差外,我们还可以使用其他的Excel 函数来进一步分析实验数据。
例如,我们可以使用“MAX”函数来计算反应结果的最大值,使用“MIN”函数来计算反应结果的最小值,以及使用“COUNT”函数来计算反应结果的个数。
精心整理文件编号:版本:鲎试剂灵敏度复核试验作业指导书1.0 目的对细菌内毒素检查试验所用的鲎试剂的灵敏度进行复核。
2.0 职责质量与法规部负责本规程的起草,QC及相关人员执行本规程。
3.0 范围适用于本公司细菌内毒素检查法。
4.0 参考文件2010年版《中国药典》二部附录ⅪE细菌内毒素检查法2010年版《中国药品检验标准操作规范》细菌内毒素检查法《细菌内毒素检查法及其应用》第一版,气象出版社5.06.06.16.2 料6.2.1 细菌内毒素工作标准品除另有规定外,使用由中国药品生物制品检定所统一发放的标准品。
6.2.2 细菌内毒素检查用水指内毒素含量小于0.015EU/ml(凝胶法)且对内毒素试验无干扰的灭菌注射用水。
6.2.3 鲎试剂规格一般选用0.25EU/ml。
7.0 操作步骤7.1 提前30min开启超净工作台,试验操作过程中不要开启风机,完毕后用75%乙醇擦拭超净工作台。
试验操作过程应防止微生物和细菌内毒素污染。
7.2 制备细菌内毒素标准溶液7.2.1 取细菌内毒素工作标准品1支,轻弹瓶壁使粉末落入瓶底,用砂轮在瓶颈上部划痕,用75%乙醇擦拭后启开,避免玻璃屑落入瓶内。
7.2.2 按照工作标准品说明书,加入细菌内毒素检查用水溶解。
封口膜封口,置漩涡混合器上混匀15min后用细菌内毒素检查用水进行稀释,制成2.0λ、1.0λ、0.5λ、0.25λ四个浓度的内毒素标准溶液。
每稀释一步均应在漩涡混合器上混匀30s。
注:在使用过程中,标准品溶液若静置超10min,必须重新混匀30s再重新使用。
7.2.3 试验举例设细菌内毒素工作标准品为100EU/支;待测鲎试剂λ=0.25EU/ml。
稀释过程如下:取细菌内毒素标准品100EU/支+1ml检查用水100EU/ml取0.2ml100EU/ml溶液+1.8ml检查用水10EU/ml取0.2ml10EU/ml溶液+1.8ml检查用水1EU/ml0.5EU/ml7.4浓度的第毒第毒第注:每次加样浓度必须是从高到低,加不同浓度的样品需更换新的移液器吸嘴。
培养基的灵敏度检查操作规程1 目的建立培养基灵敏度试验的操作规程,是为了规范、统一培养基灵敏度检测,确保微生物检测准确、安全进行。
2 适用范围本标准适用于培养基灵敏度检查。
3 责任者QC检验人员。
4 内容4.1 试验设备及用具:无菌培养皿、无菌刻度吸管或无菌注射器、无菌玻璃涂布器、酒精灯等。
4.2 使用的菌株:EP检测用ATCC的菌株:金黄色葡萄球菌Staphylococcus aureus ATCC 6538大肠埃希菌Escherichia coli ATCC 8739沙门氏菌Salmonella enterica subsp ATCC 14028黑曲霉Aspergillus niger ATCC 16404培养基灵敏度检测所用的菌株传代次数不得超过5代,并采用适宜的菌种保藏技术,以保证试验用菌株的生物学特性。
4.3 检测频率每批新买的培养基配制、灭菌后均应做灵敏度测试。
4.4 操作方法4.4.1 对照标准培养基用已经过试验证明合格的培养基或购买的有质量证书的成品平皿培养基作为对照标准培养基。
4.4.2 试验培养基将琼脂类的试验培养基加热融化后,冷却至45℃左右,以无菌的方式在无菌培养皿中注入15~20mL,备用。
液体类的培养基直接使用。
4.4.3 菌悬液的制备4.4.3.1 细菌菌悬液的制备取细菌的斜面培养物1耳匙接种在酪蛋白大豆消化肉汤培养基中30-35℃培养18-24h,取上述培养物用无菌水按10倍系列稀释,分别制成10-7-10-8的菌悬液备用。
4.4.3.2 霉菌菌悬液的制备取黑曲霉的斜面培养物加入无菌水4mL制成孢子悬液,取上述孢子悬液按10倍系列稀释,分别制成10-6-10-7的菌悬液备用。
4.4.4 培养基的生长促进实验4.4.4.1 琼脂类培养基的生长促进试验每株菌用2个试验培养基平皿,用表面涂布法在每个平皿表面接种0.1-0.5mL的菌悬液(约10-100CFU试验菌),同时用2个对照标准的培养基平皿做对照,接种相同量的菌悬液试验,在规定温度下培养,取出平皿点计菌落数。
正文
规范鲎试剂灵敏度复核的操作过程和要求,确保鲎试剂灵敏度复核结果的可靠性。
适用于质监处药理室对鲎试剂灵敏度的复核。
质监处药理室分析人员负责本规程的实施和完成相关记录。
USP版附录85;
CP版附录Ⅺ E 。
细菌内毒素检查法:本法系利用从鲎的变形细胞中提取的试剂来检测或量化由革兰阴性菌产生的细菌内毒素,以判断供试品中细菌内毒素的含量是否符合规定的一种方法。
细菌内毒素的量用内毒素单位(EU)表示。
鲎试剂:是鲎变形细胞的溶出物经提取而成。
鲎试剂灵敏度:在本检查法规定的条件下能检测出内毒素标准溶液或供试品溶液中的最低内毒素浓度,用EU/ml表示。
细菌内毒素检查用水(BET水):系指与灵敏度为0.03EU/ml 或更高灵敏度的鲎试剂在37 1 C 条件下24小时不产生凝集反应的灭菌注射用水。
细菌内毒素标准品:USP细菌内毒素参考标准品(RSE)或已标定的细菌内毒素工作标准品(CSE)。
反应终点浓度:是指系列递减的内毒素浓度中最后一个呈现阳性结果的浓度。
一、试剂、材料与仪器设备
试剂:
鲎试剂批号、灵敏度、规格、生产商。
细菌内毒素检查用水(BET水) 批号、规格、生产商。
细菌内毒素标准品批号、效价、生产商。
材料及仪器设备:
电热恒温水箱型号、设备编号。
电热鼓风干燥箱型号、设备编号。
漩涡混合器型号。
无热原试管规格:1075mm。
正文
无热原玻璃瓶规格:25ml。
无热原玻璃吸管规格:1ml、2ml、5ml、10ml。
试管架、无热原封口膜、洗耳球、酒精棉球、砂轮片。
二、试验准备
试验所用的容器、用具都必须经过处理,250C以上干烤至少1小时,以去除外源性内毒素。
试验操作过程应避免微生物的污染。
三、内毒素溶液配制
细菌内毒素标准品系列溶液配制取RSE或CSE 1瓶,按照使用说明书加入规定量的细菌内毒素检查用水使溶,并用漩涡混合器间歇混匀30分钟,然后用细菌内毒素检查用水进一步稀释至2.0、、 0.5、 0.25四个浓度的内毒素标准溶液,每一步稀释应混匀至少30秒钟。
四、试验过程
取被复核的鲎试剂原安瓿18支,分别用0.1ml细菌内毒素检查用水复溶。
其中16支加入0.1ml细菌内毒素系列标准溶液,每浓度溶液平行做4支;另外2支加入0.1ml 细菌内毒素检查用水作为阴性对照。
将安瓿中的溶液轻轻混匀后,用封口膜封闭管口,垂直放入371℃电热恒温水箱中,保温602分钟。
反应结果记录在鲎试剂灵敏度复核记录中。
五、试验结果观察和有效性
结果观察将安瓿从电热恒温水箱中轻轻取出,缓缓倒转180°时,管内凝胶不变形,不从管壁滑脱者为阳性,记录为(+);凝胶不能保持完整并从管壁滑脱者为阴性,记录为(–)。
保温和拿取安瓿过程应避免受到振动造成假阴性结果。
有效性对于细菌内毒素标准品系列溶液,如果最大浓度2.0管均为阳性,最低浓度0.25管均为阴性,阴性对照管均为阴性,试验方为有效。
六、灵敏度复核结果的计算
计算公式为:
c=lg-1(Σe/4)
正文
式中e为细菌内毒素标准品反应终点浓度的对数值。
合格标准当0.5≤c<1.3时,方可用于细菌内毒素检查,并以标示灵敏度为该批鲎试剂的灵敏度。
鲎试剂有效期按厂家规定执行。
鲎试剂应在2~10℃条件下保存。
当鲎试剂的批号改变时需进行鲎试剂灵敏度的复核。
.。