ISO13485:2016订单管理程序
- 格式:doc
- 大小:57.00 KB
- 文档页数:5
文件制修订记录对本公司产品的设计和开发的全过程进行控制,确保为本公司树立良好的形象,满足客户的需求和期望及有关法律、法规的要求。
2.0范围适用于本公司新产品的设计、开发全过程,包话引进产品的转化、定型产品生产过程的技术改进等。
3.0职责必要时技术质量部负责编制年度新产品开发大纲和老产品改进计划,负责设计和开发全过程的组织、协调、实施工作,进行设计和开发的策划、风险分析,确定设计开发的组织和技术的接口,确定设计输入、输出、评审、验证、确认、更改及外包设计的技术要求等。
3.1管理者代表负责审批年度新产品开发大纲、老产品改进计划、所需资源要求;下达设计开发任务书;负责批准设计开发风险分析报告、设计开发计划书、设计开发评审、设计开发验证资料。
3.2总经理负责批准试产报告。
3.3采购部负责新产品开发所需物料的采购。
3.4技术质量部负责新产品的检验和试验及产品型式试验。
3.5生产供应部负责新产品的加工试制和生产。
3.6销售部负责根据市场调研和分析,提供市场信息及新产品动向。
4.0工作程序4.1设计和开发的策划4.1.1技术质量部参与公司经营策划。
根据销售部已获得的订单、合同和对市场调查、分析、及国内外产品技术发展动向,生产技术部进行综合分析预测市场需求并编制年度新产品开发大纲和老产品改进计划报总经理批准后实施。
4.1.2管理者代表根据上述项目来源,确定项目负责人,编制《设计开发计划》。
计划内容包括:a)设计开发的输入、输出、评审、验证、确认等各阶段的划分和主要工作内容;b)各阶段人员职责和权限、进度要求和配合单位;c)资源配置需求,如人员、信息、设备、资金保证等及其它相关内容。
4.1.3设计开发策划的输出文件将随着设计开发的实际进展,在适当时予以修改。
4.1.4设计和开发不同组别的接口管理。
设计开发的不同组别可能涉及到公司不同部门和人员。
a)对于部门之间重要的设计开发信息沟通,由设计组负责人审批后发给相关部门。
一级文件页次Page 16 of 336
目标。
5.3.4总经理应将质量方针传达到管理、执行、验证和作业等层次,使全体员工正确理解并坚决执
行。
5.3.5本公司应持续地对质量方针进行适宜性评审,必要时可对其进行修改以适应本公司内、外
环境的变化,具体过程按《管理评审控制程序》执行。
5.3.6对质量方针的批准、发布、评审和修改都应实行控制,实行方法详见《文件、资料和记录管
理程序》。
5.4 策划
5.4.1 质量目标
5.4.2 质量目标设定原则:
a.质量目标的设定应体现质量方针的涵义;
b.质量目标应具有可测量性;
c.质量目标应分解到各部门形成子目标,通过完成子目标确保总的质量目标的完成。
5.4.3在建立体系之初,由总经理在总结公司历年经营情况的基础上,提出本年度的质量目标和
各部门的分解目标(具体见附录1),并在管理评审会上评审质量目标的完成情况。
此后
在每年的管理评审会上回顾上一年的质量目标。
5.4.4各部门对设定的质量目标的实现情况每半年进行回顾性总结。
5.4.5管理者代表如发现质量目标在实现过程中存在问题时,应组织有关部门或管理者代表进行分
析,采取相应的措施加以解决。
5.4.6质量管理体系策划
5.4.
6.1逻辑结构图:。


ISO13485-2016质量管理手册符合:YY/T 0287-2017 idt ISO 13485:2016《医疗器械质量管理体系用于法规的要求》国家药监局2014第64号令《医疗器械生产质量管理规范》质量方针和目标为改进公司产品质量,确保产品安全,尽力满足顾客等相关方的要求,提升顾客等相关方的满意度,以提高产品的市场占有率,从而保证公司利益的实现,公司制订了质量方针及目标,现予以公布,望在公司内得到充分沟通和理解,并采取措施予以执行。
1、质量方针:以人为本、绿色环保、诚信务实、拓展创新、精益求精2、质量目标:a、产品合格率≥98%b、顾客满意度≥80c、交期达成率≥98%3、管理承诺遵纪守法,为医疗器械生产企业提供卫生达标合格的包装材料。
总经理:2018年02月20日1、目的公司依据YY/T 0287-2017 idt ISO 13485:2016《医疗器械质量管理体系用于法规的要求》和GB/T 19001-2016/ISO 9001:2015《质量管理体系要求》标准,以及国家药监局2014第64号令《医疗器械生产质量管理规范》,结合公司的实际,建立并实施质量管理体系,以达到以下目的:a)证实公司有能力稳定地提供满足顾客和适用法律法规要求的产品;b)通过质量管理体系的有效应用,包括持续改进质量管理体系的过程以及保证符合适用的法律法规要求,旨在增强顾客满意。
c)展示质量体系所需的过程顺序和相互关系、引用的程序文件。
2、术语和定义本手册采用GB/T 19001-2016/ISO 9001:2015《质量管理体系基础和术语》、YY/T0287-2017 idt ISO13485:2016《医疗器械质量管理体系用于法规的要求》和GB/T ISO9001:2016/ISO 9001:2015《质量管理体系要求》标准中所确立的术语和定义。
3、质量管理体系覆盖的范围3.1本公司质量管理体系覆盖的范围:用于医疗器械包装灭菌纸塑袋产品的生产制造和销售服务;所涉及的部门是总经理、管理者代表、供销部、生产技术部(含仓库)、品质部、集团行政部、集团设备组。
文件制修订记录保证设备精度与性能,提高生产力,减少故障,提高产品质量。
2.0范围公司生产车间内所有生产设备。
3.0权责3.1设备维护组:负责设备保养、维修管理。
3.2有关部门给予配合。
4.0程序要求4.1新购设备4.1.1申请部门填写《请购单》或《固定资产申请书》,报运营经理、总经理批准后,采购部门依据《采购控制程序》进行购买设备。
4.1.2购买设备的型号以及参数必须与生产部共同确认。
4.1.3对于新进公司的设备,确定验收的项目和要求,由生产部门或设备维护组对设备进行初步检查,并填写《设备验收单》,如发现问题及时与供应商联系。
4.2设备台帐设备维护组负责制定《设备总览表》以及《设备履历表》并适时更新。
4.3关键设备识别4.3.1由试设备维护组根据设备技术资料及生产流程中设备使用情况判定是否属于关键设备并在《设备总览表》中进行注明。
4.3.2生产部根据《设备履历记录》确定适当的关键设备备件库存量,编制关键设备备件清单,当备件到达最低库存量时,设备维护组提出申购需求。
4.3.3设备维护组必须及时根据设备实际使用情况对《关键设备备件清单》进行更新。
4.4设备维护与保养4.4.1预防性维护方法可包括a)生产厂家建议的检查;b)统计过程控制数据与预防维护行动的相互关系;c)易损耗配件的重要特性;d)其它。
4.4.2预见性维护4.4.2.1试设备维护组根据所有设备的实际使用情况和其以往的维修状况结合其使用的频率和使用寿命,编制“设备预见性维护需求表”,报生产部经理审核,总经理批准后实施,并将维护结果填写在《设备履历记录》。
4.4.2.2设备维护组负责按《每日保养维护记录》、《年度保养维护记录》安排维修保养,包括日常点检、每月检查、每季检查、半年检查和每年检查。
4.4.3每日保养维护作业4.4.3.1设备维护组人员按照《每日保养维护记录》中需要检查的项目,每日实施点检保养并记录于《每日保养维护记录》内。
4.4.3.2每日保养中发现异常时,设备维护组进行故障排除,记录于《设备履历记录》内。
一级文件页次Page 16 of 336
目标。
5.3.4总经理应将质量方针传达到管理、执行、验证和作业等层次,使全体员工正确理解并坚决执
行。
5.3.5本公司应持续地对质量方针进行适宜性评审,必要时可对其进行修改以适应本公司内、外
环境的变化,具体过程按《管理评审控制程序》执行。
5.3.6对质量方针的批准、发布、评审和修改都应实行控制,实行方法详见《文件、资料和记录管
理程序》。
5.4 策划
5.4.1 质量目标
5.4.2 质量目标设定原则:
a.质量目标的设定应体现质量方针的涵义;
b.质量目标应具有可测量性;
c.质量目标应分解到各部门形成子目标,通过完成子目标确保总的质量目标的完成。
5.4.3在建立体系之初,由总经理在总结公司历年经营情况的基础上,提出本年度的质量目标和
各部门的分解目标(具体见附录1),并在管理评审会上评审质量目标的完成情况。
此后
在每年的管理评审会上回顾上一年的质量目标。
5.4.4各部门对设定的质量目标的实现情况每半年进行回顾性总结。
5.4.5管理者代表如发现质量目标在实现过程中存在问题时,应组织有关部门或管理者代表进行分
析,采取相应的措施加以解决。
5.4.6质量管理体系策划
5.4.
6.1逻辑结构图:。
文件制修订记录为加强不合格商品的管理,防止不合格商品对患者构成危害,特制定本控制程序。
2.0范围适用于对医疗器械的生产、养护及销售和服务实现过程中发生的不合格情况。
3.0权责3.1销售部门负责售后产品及服务的识别、处理,跟踪不合格产品及服务的处理结果;3.2质量部门负责生产产品不合格的控制和处理;3.3仓库负责库房内储存条件不合格控制处理;3.4总经理负责审批不合格品的处理方式。
4.0程序要求4.1不合格的分类a) 产品交付前不合格:产品采购到出库过程中发现的不合格现象;b) 产品交付后不合格:产品交付顾客后由顾客反馈的不合格现象;4.2不合格范围4.2.1采购无《医疗器械生产企业许可证》、《医疗器械经营许可证》、《营业执照》、《注册证》等或超出有效期的商品。
4.2.2包装破损、标识不清的;无产品合格证;产品不在有效期内;4.2.3产品质量不符合法定的质量标准或相关法律法规及规章的要求。
4.3交付前不合格品的确认4.3.1质量验收人员在验收的过程当中发现的外观质量、包装质量不符合要求的或通过质量复检确认为不合格的;4.3.2医疗器械监督管理部门的质量公报品种、通知禁售的品种,并经公司质量部核对确认的;4.3.3在保管养护过程中发现过期、失效、淘汰及其他有质量问题的医疗器械;4.4交付后不合格确认4.4.1通过顾客反馈确认交付后不合格;4.4.2在养护检查中发现不合格品时,按销售记录召回售出的不合格品;4.4.3药监部门公布的不合格品或厂家发布产品召回的产品;4.5不合格处理4.5.1交付前不合格的处理a)进货验证产品不合格处理采购产品在验收过程中发现不合格时,应上报质量部进行确认,经质量部确认后开立《不合格品处置单》,并将不合格品存放在不合格区,并将结果及时反馈给采购部门处理。
b)在库、出库不合格品处理在养护检查及出库复核中发现不合格品时,经质量部门确认后开立《不合格品处置单》同时通知销售部门停止销售,将不合格品移入不合格品区。

ISO13485-2016中文1. 引言一般要求:说明组织应遵循的质量管理体系的基本原则和要求,包括过程方法、风险管理、文件化、控制、评价等。
文件要求:说明组织应编制和控制的质量管理体系文件的类型、内容、格式、审批、发布、修改、保存等要求,包括质量手册、程序文件、记录等。
管理层承诺:说明组织高层管理人员应如何表达对质量管理体系的承诺,包括确立质量政策和目标、提供资源、促进沟通、参与审核等。
顾客关注事项:说明组织应如何确定和满足顾客的需求和期望,包括确定法规要求、确保顾客满意度、处理顾客投诉等。
质量政策:说明组织应如何制定和传达质量政策,以及质量政策应具备的特征,包括符合组织目标、符合法规要求、强调风险管理、提供改进机会等。
质量目标与计划:说明组织应如何制定和实施质量目标和计划,以及质量目标和计划应具备的特征,包括与质量政策一致、可测量、可实现、可跟踪等。
质量管理体系职责与权限:说明组织应如何确定和分配质量管理体系相关人员的职责和权限,以及如何确保他们的能力和意识,包括建立组织结构、制定职位描述、提供培训、评估效果等。
内部沟通:说明组织应如何在质量管理体系内部进行有效的沟通,以确保信息的及时、准确、完整的传递和反馈,包括建立沟通机制、确定沟通内容、选择沟通方式、记录沟通结果等。
管理评审:说明组织应如何定期对质量管理体系进行评审,以评估其适合性、充分性和有效性,以及提出改进措施,包括制定评审计划、确定评审输入、进行评审会议、记录评审输出等。
基础设施:说明组织应如何确定和提供质量管理体系所需的基础设施,包括建筑物、设备、工具、软件等,以及如何对其进行维护和控制,以保证其适用性和安全性。
工作环境:说明组织应如何确定和管理质量管理体系所需的工作环境,包括温度、湿度、光照、噪音等物理因素,以及心理因素,以保证其不会对产品和服务的质量产生不利影响。
产品实现计划:说明组织应如何制定和实施产品实现的计划,包括确定目标、过程、资源、风险等,并根据实际情况进行更新和调整。
![ISO13485-2016中文[1]](https://uimg.taocdn.com/1eb02918abea998fcc22bcd126fff705cc175cf7.webp)
ISO13485-2016中文1. 引言ISO13485标准是一套专用于医疗器械领域的质量管理体系标准,该标准突出关注医疗器械的安全有效,强调组织提供的医疗器械要满足顾客要求和法规要求。
ISO13485标准是应用于医疗器械领域的质量管理体系标准,该标准覆盖医疗器械产品和服务的全生命周期,适用于所有规模和类型的医疗器械组织,也可用于医疗器械产业链的供方和外部方。
ISO13485标准是基于以过程为基础的质量管理体系模式,总体结构为八章加两个附录,分别为:第一章:范围第二章:规范性引用文件第三章:术语和定义第四章:质量管理体系第五章:管理责任第六章:资源管理第七章:产品实现第八章:测量、分析和改进附录A:与ISO9001:2015之间的对应关系附录B:与其他标准之间的对应关系条款编号和名称条款要求(摘自ISO13485标准)条款说明(对条款要求进行解释和说明)条款实施(提供条款实施的方法、步骤、记录等)2. 范围2.1 条款要求本国际标准指定了一个质量管理体系,当一个组织需要证明其能够提供符合顾客及适用法规要求的医疗器械产品及相关服务时,可以按其规定建立、实施、维护及改进。
2.2 条款说明本条款确定了ISO13485标准适用的对象、目的和范围。
对象:本标准适用于提供医疗器械产品及相关服务的组织,包括医疗器械的设计、开发、生产、安装、销售、维修、回收等活动的组织,以及医疗器械产业链的供方和外部方。
目的:本标准的目的是帮助组织建立、实施、维护及改进一个质量管理体系,以证明其能够提供符合顾客及适用法规要求的医疗器械产品及相关服务。
范围:本标准覆盖了医疗器械产品及相关服务的全生命周期,包括从需求分析到废弃处理的各个阶段,以及与之相关的过程、资源、文件等要素。
2.3 条款实施为了按照本条款的要求建立、实施、维护及改进质量管理体系,组织应:确定自身是否属于本标准适用的对象,即是否提供医疗器械产品及相关服务。
确定自身提供的医疗器械产品及相关服务的类型、规格、等级、用途等信息。
文件控制程序(YY/T0287-2017 idt ISO13485-2016)1. 目的和适用范围1.1目的为确保质量管理体系的有效运行,特制定本程序,系统地设计、制定、审核、批准和发放质量管理体系文件,并确保本企业在生产和经营过程的所有场所使用的文件都是有效的版本。
1.2适用范围适用于本公司在生产和经营服务全过程中所有文件的管理。
1.3发放范围本公司各职能部门。
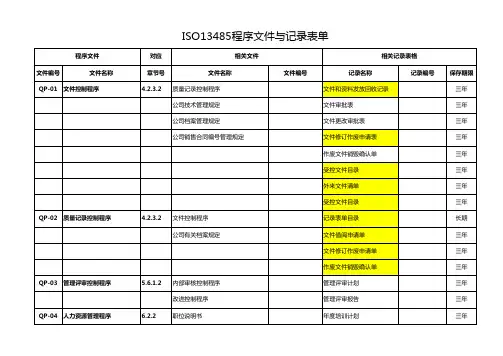
2.规范性引用文件质量手册文件分类规则文件命名规则文件编号规则文件编写规则记录控制程序GB/T19001-2016 idt ISO9001-2015质量管理体系要求YY/T0287-2017idt ISO13485-2016医疗器械质量管理体系用于法规的要求YY/T0316-2016医疗器械风险管理对医疗器械的应用医疗器械生产质量管理规范(总局公告2014年第64号)(2015年3月1日起施行)医疗器械生产质量管理规范附录无菌医疗器械(2015年第101号)(2015年10月1日起实施)医疗器械生产质量管理规范无菌医疗器械现场检查指导原则(食药监械监〔2015〕218号附件2)(2015年9月25日发布实施)GB/T 1.1-2009标准3.组织和职责3.1主责部门本程序的主责部门为办公室,主管领导为管理者代表。
——根据适用法律法规和行业标准要求并结合公司实际运营情况制定本程序;——指导文件编制、使用部门遵循本程序的规定;——组织对文件系统建立和评审工作;——对正式发布的各类文件进行登记和管理。
3.2相关部门各部门负责配合办公室按文件要求开展工作,正确使用现行有效的文件,及时提出文件修订、增补的申请。
4.步骤和方法4.1文件的设计文件使用部门根据外部环境的变化以及过程的控制需求,确定所需的文件。
根据文件从生成到文件作废和销毁,文件设计主要流程如附件流程图:在以下情况时,可能需要对文件需求进行重新识别a) 当外部环境发生变化时,如政策法规、标准规范、供方条件、顾客需求;b) 在文件的使用过程中,根据使用部门的实际需求反映。
Name Management Review ProcedureCode1/4Version A/0DocumentCode QM-COP-03Edited/RevisedDate2018.10.24IssuedDate2018.10.24 If this manual is printed, it is an UNCONTROLLED copy.1.0 PurposeThe purpose of this procedure is to define DaMei’s requirements for conducting Management Reviews.2.0 ScopeThis procedure is the primary document meeting the applicable regulatory requirements for conducting Management Reviews as defined in DaMei’sQuality System Manual (QM-A-01).References and RelationshipsISO 13485:201621 CFR 820.203.0 Responsibilities and AuthoritiesRole Responsibilities and AuthoritiesQuality Management / Management Representative Primary responsibility for maintaining this process and ensuring that Management Review meetings are scheduled, conducted and the minutes are recorded.Top Management The Management responsible for attending Management ReviewMeetings as identified in DaMei’sorganization chart. These managers areresponsible for participating in Management Reviews—including thepreparation of inputs to the Management Review and completion ofaction items resulting from Management Reviews. The organization chartshould also identify the following personnel:Management RepresentativeDeputy Management RepresentativeMost Senior Executive ManagerSecond Most Senior Executive ManagerIt is not recommended to have the same person hold two of the abovefour positions.4.0 Procedure1 Management Review meetings shall be scheduled at least once a year.2 The following inputs are required to the Management Review meeting for discussion:Quality policyQuality objectivesResults of audits – including internal, supplier, certification audits and FDA inspectionsCustomer feedback – including complaints and post-market surveillanceName Management Review ProcedureCode2/4Version A/0DocumentCode QM-COP-03Edited/RevisedDate2018.10.24IssuedDate2018.10.24 If this manual is printed, it is an UNCONTROLLED copy.Process performanceProduct conformitySupplier quality performanceStatus of corrective and preventive actionsFollow-up of action items from the previous Management Review(s)Changes that could affect the quality systemRecommendations for improvementNew and revised regulatory requirementsRisk management process (ISO 14971)Overall quality system effectiveness3 Management Review inputs shall be documented in a presentation slide deck using a controlledtemplate. The Management Representative shall assign responsibility for completing each slide of the presentation as an action item in the previous Management Review. These assignmentsshall be documented in the meeting minutes. Each input slide shall be provided to theManagement Representative at least 10 calendar days prior to the planned review date, and the Management Representative shall combine the slides into a draft presentation andelectronically deliver the draft presentation to Top Management at least 7 calendar days prior to the planned review date. Any necessary corrections to slides should be communicated to theManagement Representative as soon as possible so that corrections can be communicated toTop Management prior to the planned review date.4 During the Management Review, Top Management shall review the Quality Policy to ensure it:is appropriate to DaMei’spurpose,includes a commitment to comply with requirements and to maintainthe effectiveness of thequality management system,provides a framework for establishing and reviewing qualityobjectives,is communicated and understood within the organization, andis reviewed for continuing suitability during at least one Management Review meeting eachyear.5 During the Management Review meeting, the Management Representative is assigned the roleof scribe to record notes about the discussions.6 Top management shall ensure that quality objectives, including thoseneeded to meetrequirements for product, are established for all functions (i.e., departments) and all levelswithin the organization. The qualityobjectives shall be measurable and consistent with thequality policy. The status of quality objectives shall be reviewed during management reviewsand when one objective is met, Top Management shall determine if the objective shall be。
文件制修订记录通过建立《风险管理控制程序》,评价公司所生产产品的采购、储存、运输、销售等过程中的风险,并对风险加以控制,保证公司活动的安全性。
2.0范围适用于公司所生产产品的采购、储存、运输、销售等过程。
3.0权责3.1最高管理者:为风险管理活动分配充分的资源和有资格能胜任的人员。
规定风险管理的职责和权限,确定风险管理小组成员。
主持每年的风险管理活动评审。
批准《风险管理活动记录》。
3.2管理者代表:负责制组织管理风险活动。
3.3业务部门:负责将顾客服务相关信息反馈给质量部提供参考。
3.4 售后服务部:负责收集相关产品的风险信息。
4.0程序要求4.1风险管理过程概述:风险管理过程包括风险分析、风险评价、风险控制、综合剩余风险评价以及风险/受益分析。
应记录风险管理活动结果并纳入相关文档。
风险管理过程见图1。
4.2风险管理活动4.2.1风险管理策划应包括:a)风险管理活动范围:判定和描述医疗器械经营活动和适用于计划每个要素的阶段;b)职责和权限的分配;c)风管理活动的评审要求;d)决定风险可接受准则,包括在损害发生概率不能估计时的可接受风险的准则;e)验证活动;f)相关的生产和生产后信息的收集和评审的有关活动。
4.2.2风险管理活动输出:风险管理小组应对风险管理活动进行评审,必要时可补充确定评审人员。
评审内容包括:对风险管理计划实施情况的评审;对综合剩余风险是否可接受的判断;是否已有适当的方法获得相关生产和生产后信息。
上述评审的结果填写《风险管理活动记录》,该报告最终由最高管理者审批通过,经营活动风险控制的最终结论依据。
4.2.3风险管理资料输入(信息收集):公司建立收集和评审医疗器械信息的系统时,尤其应当考虑:a)由医疗器械的操作者、使用者或负责医疗器械安装、使用和维护人员所产生信息的收集和处理机制;b)新的或者修订的标准。
另外,也应当将最新技术水平因素和对其应用的可行性考虑在内。
并应注意该系统不仅应当收集和评审本企业类似产品的相关信息(及内部信息),也应当收集和评审市场上其它类似医疗器械产品的公开信息(及外部信息)。
文件制修订记录为确保与质量管理体系有关的各部门、岗位所使用的文件为现行有效版本,防止因误用失效文件影响质量管理体系的正常运行的情况发生,特制订本规程。
2.0范围适用于质量管理体系运行过程中所涉及各类文件的起草、修订、审核、批准、更改、作废等各环节。
3.0权责3.1 总经理负责质量手册、程序文件的批准;3.2 管理者代表负责质量手册、程序文件的审核;3.3各部门负责编制质量管理体系相关的程序文件。
3.4质量部负责文件的发放、回收、处置管理,以及外来文件的收集保管。
4.0程序要求4.1文件管理工作流程。
4.2文件分类与保管4.2.1 质量手册、程序文件、管理制度、操作规程以及外来文件,质量记录等质量体系文件均由质量部控制,各相关部门保管下发的本部门使用的文件及已填报的相关记录。
4.2.2 文件分类按4.2文件要求控制。
4.2.3 公司外来文件,包括与质量管理体系相关的法律法规文件等,由质量部按本程序相关条款执行。
4.3文件的编号4.3.1 质量管理体系文件的编号:4.3.1.1质量手册编号说明Xxx-QM-XXXX编制年份文件层次,代表质量手册公司名称简称4.3.1.2 程序文件编号说明Xxx-QP XX-XXXX编制年份程序文件标准章节号(4.2.3、4.2.4…..)文件层次,代表程序文件公司名称简称4.3.1.3 规范类文件编码说明Xxx-WI- HR XX-XXXX编制年份文件序列号(01、02…..)部门英文缩写,代表人力资源部文件层次,代表工作指导文件公司名称简称4.3.1.4 质量记录编号说明Xxx-QR- QP/HR XX –XX序列号(01、02…)程序文件序号/工作指导文件序号(01、02…)程序文件或部门英文缩写文件层次,代表工作指导文件公司名称简称部门代码:生产部SC、人力资源部HR、质量部QM、市场部MD、销售部SD、售后服务部AS、商务部MC、采购部PD 、仓库WD。
4.3.2 外来文件不做编号,以文件名称或标准号作为原始编号,登记时记录接收和分发日期以区分新旧版本,为识别有效版本。
管理体系受控文件 未经批准不得扩散
文件编号 QP-10
质量管理体系文件
版 本 A0
程序文件
主控部门 业务部
订单管理程序
程序文件修订记录
№ 修订说明 版本 修订日期 修订人 批准
1 新制订 A/0 2016-11-1
编制: 审核: 批准:
日期: 日期: 日期:
生效日期: 2016年11月1日
文件受控盖章处
管理体系受控文件 未经批准不得扩散
有限公司
编号:QP-10
版本:A0
页码:第 1 页 共 4 页
1、目的
为了对在与顾客有关的过程实施系统管理,对与产品有关的要求进行确定及评审,使其规
定得合理、明确,并形成文件,确保本公司产品要求和服务与顾客需求一致,让顾客满意,特
编制本程序文件。
2、范围
适用于本公司与产品有关的要求的确定,在销售合同签订前与产品有关的要求的评审及相
关活动。适用于本公司顾客订单接受、评审、生产跟进至交货安排全过程的管理。
3、职责
3.1 业务部:负责与顾客沟通,收集顾客信息及与产品有关的要求的确定;负责组织销售订单
与产品有关的要求的评审,并在评审通过后传达、跟进生产进度,及时与顾客沟通订单及产品
情况;在订单发生变更后,组织订单变更的评审,并确保相关文件的变更及发放至相关部门及
人员。
3.2 工程部:参与新产品、新工艺以及重要合同的评审。
3.3 生产部:按实际需要参与合同评审;负责按订单要求组织生产,确保按时交货。
3.4 品管部:按实际需要参与合同评审;负责生产过程产品的监视和测量,对顾客投诉的分析、
调查、处理和纠正和预防措施的跟踪验证。
3.5 采购部:按实际需要参与合同评审;负责评估物料采购要求及采购周期是否满足出货要求。
3.6 总经理:按实际需要参与合同评审;负责订单评审表、订单变更评审表、销售订单、出货
单的批准。
4、定义
4.1 要求:明示的、通常隐含的或必须履行的需求或期望。
5、内容
5.1与产品有关的要求的确定
5.1.1 业务部针对与产品有关的要求的确定,须确定以下方面:
a) 顾客合同/订单规定的要求,包括对交付及交付后活动的要求;
b) 顾客合同/订单虽然没有明示,但规定的或已知的预期用途所必需满足的要求;
c) 与医疗器械产品有关的适用的法规要求;
d) 任何为保证医疗器械规定的性能和安全使用所需的用户培训;
e) 本公司确定的任何附加要求(如产品技术要求、产品规范、检验标准等)。
5.1.2 业务部接收顾客订单,或将顾客要求在产品销售合同/订单上予以记录和明确。
5.2 与产品有关的要求的评审
管理体系受控文件 未经批准不得扩散
有限公司
编号:QP-10
版本:A0
页码:第 2 页 共 4 页
5.2.1 订单评审的范围包括:
a) 书面的销售合同、订单;
b) 口头、电话、传真订货要求;
c) 质量/技术协议;
d) 相关订货要求的变更等。
e) 有些情况下,如网上销售等,对每一个订单进行正式评审是不可能的,因此取而代之的产品
有关信息,如产品目录、产品广告内容需要进行评审。
5.2.2业务员与顾客进行充分的沟通,详细了解顾客要求,并索取与产品有关的文件。若顾客的
某些要求未以书面形式明确规定时,业务人员应予以适当记录并得到顾客确认。顾客的要求包
括:
a) 产品名称、规格、数量;
b) 交付方式和交货期;
c) 技术/质量要求;
d) 价格和付款方式;
e) 服务或其他相关要求等。
5.2.3在同意按要求向顾客供货前(如:接受订单),业务部应对顾客的相关要求进行评审,以
确保:
a) 产品要求得到规定并形成文件。
b) 与以前表述不一致的合同或订单的要求已予解决。合同/订单评审过程中的任何不明确,由业
务部人员与顾客沟通,在完全了解顾客要求后再进行合同评审,评审合格后顾客原合同/订单回
传顾客,以示顾客的合同/订单生效。
c) 满足适用的法规要求。
d) 任何依据产品要求的识别的用户培训是可获得的或预期可获得的;
c) 公司有能力满足顾客订单的要求。
5.2.4通过评审确定公司可以满足顾客要求时,业务部应将顾客订单的内容转化成《销售订单》
交相关部门执行;否则,业务部不得接受顾客订单。
5.3 订单评审记录
5.3.1业务部人员对已收到的合同/订单首先进行分类:
◇ 若为本公司已生产过产品,相关产品技术文件齐全(如产品技术要求、产品规范、检验标准
等),则由业务部人员核查产品成品库存情况,确认是否具备发货条件;若需重新按排生产,则
业务部人员与生产部进行评审,确定生产交期,并在合同/订单上签字回复顾客。
◇ 若顾客合同/订单的要求为新产品,业务部组织工程部、生产部、品管部、采购部等相关部
门参加合同/订单评审,并经总经理批准后生效。需进行样品确认时,则业务部人员须将顾客的
打样要求通知生产部进行打样,业务部人员跟进打样结果,经顾客确认样品符合要求后进行批
量生产。