Chip-Seq项目结题报告
- 格式:pdf
- 大小:4.52 MB
- 文档页数:23
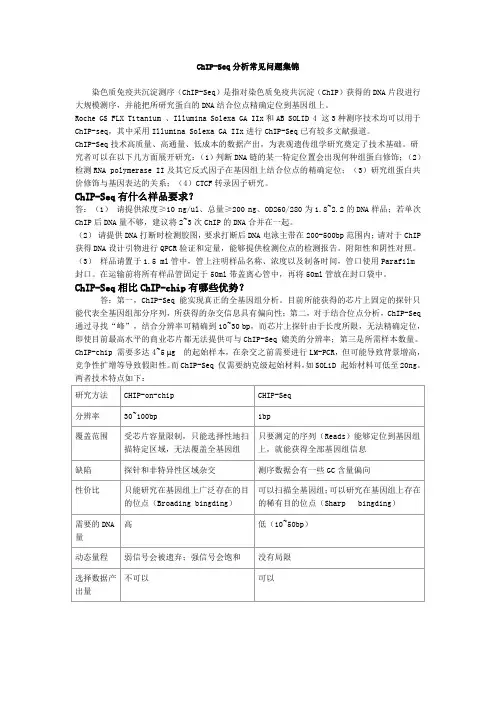
3 / 231 生物实验流程染色体免疫共沉淀(ChIP, chromatin immunoprecipitation)是一种用于研究蛋白质与DNA的体内相互作用的经典实验技术。
采用特异性抗体将目的蛋白进行免疫沉淀,由此可以把目的蛋白所结合的基因组DNA片段也富集下来。
通过与高通量测序技术的结合,对ChIP后的DNA产物进行测序分析,从全基因组范围内寻找目的蛋白的DNA结合位点,以高效率的测序手段得到高通量的数据结果。
1.1 ChIP免疫沉淀实验流程目前主要有两种不同的ChIP实验方法,大致流程如下(均以细胞样品的处理过程为例):Cross-liking Chromatin Immunoprecitation (X-ChIP)1.甲醛处理细胞,使 DNA-protein 的相互结合作用被交联固定。
2.裂解细胞,得到全细胞裂解液。
3.超声处理,将基因组 DNA 打断至 100-500bp。
4.抗体免疫沉淀:在细胞裂解液中加入一抗和 beads,并进行孵育。
5.采用合适的实验条件进行洗脱,并解交联。
6.通过 qPCR 对 ChIP 结果进行验证。
7.准备好的 ChIP 后的 DNA 样品可以用于 ChIP Sequencing 建库。
Native Chromatin Immunoprecipitation (N-ChIP)1.通过非变性的方式得到核裂解液。
2.微球菌核酸酶(Micrococcal nuclease)消化染色质,得到单核小体或核小体寡聚体。
3.抗体免疫沉淀:在细胞裂解液中前后加入一抗和 beads,并进行孵育。
4.DNA 分离。
5.通过 qPCR 对 ChIP 结果进行验证。
6.准备好的 ChIP 后的 DNA 样品可以用于 ChIP Sequencing 建库。
1.2 ChIP文库构建流程文库构建流程主要有以下步骤:1.DNA片段末端修复、3’端加A碱基,连接测序接头(详细步骤请参考Illumina公司Paired-End DNA SamplePrep kit)。
2.PCR扩增及DNA产物的片段大小选择(一般为100-300bp,包括接头序列在内)。
3.合格的文库用于上机测序。
流程如下:4 / 232 信息分析流程由Illumina测序产生的数据通过质量控制以及过滤,借助比对工具与参考基因组比对。
提取比对上唯一位置的序列,结果以bed文件存放,用bed文件做后续信息分析,包括read的分析和peak扫描。
在全基因组范围对peak进行扫描,对于扫描到的peak,对其相关联的基因进行分析,包括GO以及Pathway富集分析。
另外对于多样品,还可以做样品间差异Peak的鉴定。
信息分析流程如下:5 / 23二、分析结果2.1 数据过滤质控测序完成后,对原始数据进行去接头、去除低质量数据等处理,得到可用数据并统计产量。
一条序列如符合以下任一条件则会被作为不合格序列去除:1.序列含有adapter接头;2.N碱基含量超过10%序列长度;3.质量值低于20的碱基含量超过50%序列长度。
数据产出统计样品名序列长度序列总数碱基总数%GC Q20Q30 Sample1------Sample2------6 / 232.2 与参考序列比对2.2.1 比对结果统计将可用数据与所选参考基因组序列进行比对,设定允许不超过2个碱基的错配,其中比对到基因组上唯一位置的序列(唯一比对序列)将用于后续的信息分析。
比对结果统计样品名总序列数比对序列数比对率(%)唯一比对序列数唯一比对率(%) Sample1-----Sample2-----结果文件列表比对结果2.2.2 基因组测序深度累积分布以比对后得到的唯一比对序列为分析对象,分析其在参考基因组上的覆盖分布,统计基因组位点的深度信息,得到基因组上测序深度统计结果。
Sample1Sample1Sample1基因组测序深度累积分布图Sample27 / 23Sample2Sample2基因组测序深度累积分布图2.2.3 基因测序深度分布以比对后得到的唯一比对序列为分析对象,分析其在基因本体区间及上下游2k区间内的深度分布,得到基因及上下游区间深度分布结果。
Sample1Sample18 / 23Sample2Sample2Sample2基因及上下游测序深度分布图2.3 Peak 分析2.3.1 Peak扫描基于一定的分析模型在全基因组范围进行peak(ChIP Sequencing富集区域)扫描,得到peak在基因组上的位置信息,peak区域序列信息等。
peak结果以wiggle文件格式存放,可上传至UCSC查看,详细步骤参见帮助文档“UCSC Genome Browser使用说明”。
Peak信息统计样品名Peak数Peak总长度Peak平均长度Peak总序列深度Peak平均序列深度基因组比例(%) Sample1------Sample2------结果文件列表Peak统计2.3.2 Peak长度分布peak的长度是peak区间的重要信息之一,分析结果根据peak结果绘制得到每一个样品的peak长度分布。
以所有peak为分析对象进行绘图,x轴为peak的长度,y轴为特定长度peak分布数值。
Sample19 / 23Sample1peak 长度分布示意图Sample2Sample2 peak 长度分布示意图2.3.3 Peak 深度分布peak 区域所含序列数也是peak 区间的重要信息之一。
分析结果根据peak 结果绘制得到每一个样品的peak 所含序列10 / 23Sa mple1Sample 2以所有peak为分析对象进行绘图,x轴为peak的区域内序列数,y轴为特定序列数下peak的累积分布比例值。
Sample1Sample1Sample1 peak深度分布示意图Sample2Sample211 / 232.4 Peak 注释2.4.1 peak在基因功能元件上的分布以饼图表示Peak在基因的exon、intron、upstream、downstream、intergenic等功能元件的分布特征。
Sample1Sample1Sample1 peak基因功能元件分布示意图Sample212 / 23Sample2Sample2 peak基因功能元件分布示意图2.4.2 Peak 相关基因分析通过找到与目的蛋白相结合区域(Peak区域)在基因组上的定位及其与那些基因有关,从一定程度上表示了目的蛋白或特定组蛋白修饰可能调控的靶基因区域。
结果文件列表Sample1Sample22.4.3 peak相关基因的GO功能显著性富集分析Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,提供了一套动态更新的标准词汇表(controlled vocabulary)来全面描述生物体中基因和基因产物的属性。
GO总共有三个ontology(本体),分别描述基因的分子功能(molecular function)、所处的细胞位置(cellular component)、参与的生物过程(biological process)。
GO的基本单位是term(词条、节点),每个term都对应一个属性。
Sample113 / 23Sample1 peak相关基因GO富集示意图Sample2Sample2 peak相关基因GO富集示意图结果文件列表Sample1Sample22.4.4 peak相关基因的Pathway功能显著性富集分析在生物体内,不同基因相互协调行使其生物学,基于 Pathway 的分析有助于更进一步了解基因的生物学功能。
KEGG 是有关 Pathway 的主要公共数据库,Pathway 显著性富集分析以 KEGG Pathway 为单位,应用超几何检验,找出与整个基因组背景相比,在 peak相关基因中显著性富集的 Pathway。
结果文件列表Sample1Sample22.5 鉴定样品间差异Peak基于MAnorm工具,对两个样品进行差异分析,确定存在样品间差异修饰的区间。
结果文件列表14 / 232.6 样品间差异Peak注释2.6.1 差异Peak的基因功能元件分布以饼图表示Peak在基因的exon、intron、upstream、downstream、intergenic等功能元件的分布特征。
Sample1-vs-Sample2Sample1-vs-Sample2Sample1-vs-Sample2 peak基因功能元件分布示意图2.6.2 差异Peak相关基因分析结果文件列表Sample1-vs-Sample22.6.3 差异Peak相关基因的GO功能显著性富集分析结果文件列表Sample1-vs-Sample22.6.4 差异Peak相关基因的Pathway功能显著性富集分析结果文件列表Sample1-vs-Sample22.7 Motif分析基因表达起始于多种蛋白因子结合于特异的非编码DNA序列,非编码区域的主要研究方向之一即是Motif研究。
基因表达调控机制研究是生物学研究的重点内容,鉴定DNA调控元件尤其是DNA Motif,对于基因表达调控机制研究具有重要意义。
15 / 23Sample1_Motif_mastSample2_Motif_scanSample2_Motif_mast三、帮助文档3.1 生物信息分析3.1.1 原始序列数据(Raw data)测序得到的原始图像数据经base calling转化为序列数据,我们称之为raw data或raw reads,结果以FASTQ文件格式存储,包含reads的序列以及reads的测序质量。
在FASTQ格式文件中每个read由四行描述,如下:@A80GVTABXX:4:1:2587:1979#ACAGTGAT/1NTTTGATATGTGTGAGGACGTCTGCAGCGTCACCTTTATCGGCCATGGT+BTTMKZXUUUdddddddddddddddddddddddddddadddddd^WYYU每个序列共有4行,第1行和第3行是序列名称(有的fq文件为了节省存储空间会省略第三行“+”后面的序列名称),由测序仪产生;第2行是序列;第4行是序列的测序质量,每个字符对应第2行每个碱基,第四行每个字符对应的ASCII值减去64,即为该碱基的测序质量值,比如c对应的ASCII值为99,那么其对应的碱基质量值是35。
从Illumina GA Pipeline v1.5开始,碱基质量值范围为2到41。
表1 为Illumina HiSeq TM 2000测序错误率与测序质量值简明对应关系。
如果测序错误率用E表示,碱基质量值用sQ表示,则有下列关系:Table 1. Illumina HiSeq TM 2000测序错误率与测序质量值简明对应关系Sequencing error rate Sequencing quality value Character 5%13M1%20T0.1%30^3.1.2 数据质控和过滤为了保证数据质量,要在信息分析前对原始数据进行质控和过滤。