PrimerBLAST 操作说明
- 格式:ppt
- 大小:2.50 MB
- 文档页数:55

序列比对,绝大多数战友都会想到BLAST,但BLAST的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多.如果把BLAST的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST的使用。
所以我在这里也就“画龙点睛"——以比对核酸序列为例来给大家介绍一下BLAST的使用,也算是BLAST的入门课程吧。
请看帖的战友好好体会,如果你用心看,在看帖完毕之后BLAST的基本使用(包括其他序列的比对)应该没有问题了。
一、打开BLAST 页面,http://www。
ncbi.nlm.nih。
go/BLAST/ 打开后如图所示:(缩略图,点击图片链接看原图)对上面这个页面进行一下必要的介绍:BLAST的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST.相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST的三条途径。
第一部分BLAST Assembled Genomes就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面.第二部分Basic BLAST包含了5个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST是一些特殊目的的BLAST,如IgBLAST、SNP等等,这个时候你就需要在Specialized BLAST部分做出适当的选择了。
总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST 途径。
下面以最基本的核酸序列比对来谈一下BLAST的使用,期间我也会含沙射影的说一下其他序列比对的方法.二、点击Basic BLAST部分的nucleotide blast链接到一个新的页面.打开后如图所示:screen.width-333)this。

序列比对,绝大多数战友都会想到BLAST,但BLAST的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多。
如果把BLAST的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST的使用。
所以我在这里也就“画龙点睛”——以比对核酸序列为例来给大家介绍一下BLAST的使用,也算是BLAST的入门课程吧。
请看帖的战友好好体会,如果你用心看,在看帖完毕之后BLAST的基本使用(包括其他序列的比对)应该没有问题了。
一、打开BLAST页面,http://www.ncbi.nlm.nih.go/BLAST/ 打开后如图所示:(缩略图,点击图片链接看原图)对上面这个页面进行一下必要的介绍:BLAST的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST。
相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST的三条途径。
第一部分BLAST Assembled Genomes就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面。
第二部分Basic BLAST包含了5个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST是一些特殊目的的BLAST,如IgBLAST、SNP等等,这个时候你就需要在Specialized BLAST部分做出适当的选择了。
总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST途径。
下面以最基本的核酸序列比对来谈一下BLAST的使用,期间我也会含沙射影的说一下其他序列比对的方法。
二、点击Basic BLAST部分的nucleotide blast链接到一个新的页面。
打开后如图所示:screen.width-333)this.width=screen.width-333" width=640 height=462 title="Click to iew full 2.JPG (849 X 613)" border=0 align=absmiddle> 介绍一下上述页面:Enter Query Sequence部分是让我们输入序列的,你可以直接把序列粘贴进去,也可以上传序列,还可以选择你要比对的序列的范围(留空就代表要比对你要输入的整个序列)。

BLAST使用方法BLAST(Basic Local Alignment Search Tool)是一种用于比较生物学序列的工具,可以在数据库中查找类似序列,并计算它们之间的相似度。
BLAST可用于寻找相似的基因、蛋白质序列、DNA序列等,以及用于确定序列的功能和进化关系。
本文将介绍BLAST的使用方法。
2. 准备序列:在使用BLAST之前,你需要准备你想要比较的序列。
可以是DNA序列、蛋白质序列或其他生物学序列。
可以从公共数据库如NCBI的GenBank中获取序列,也可以使用你自己的实验数据。
3.选择数据库:BLAST使用数据库来存储和检索序列。
常见的数据库包括NCBI的NT数据库(核苷酸数据库),NR数据库(非冗余蛋白质数据库)等。
根据你的研究需要,选择适合你的数据库。
你也可以建立自己的数据库,将实验室内部的数据添加到其中。
4.运行BLAST:使用BLAST的命令行接口或网页界面,输入你的序列和数据库信息,运行BLAST。
下面是使用命令行接口运行BLAST的示例:`$ blastn -query sequence.fasta -db nt -out result.txt`在这个命令中,`blastn`是BLAST程序的名称,`sequence.fasta`是包含你的序列的FASTA文件,`nt`是数据库的名称,`result.txt`是结果输出的文件。
如果使用网页版BLAST,你只需将序列和数据库信息输入网页表单,点击运行即可。
5.解析结果:BLAST运行完成后,会生成一个结果文件,其中包含比对结果和相似度分数。
你可以使用BLAST提供的工具来解析和可视化这些结果,以便进一步分析。
结果中通常包括比对的相似度分数、比对的位点、比对的长度、匹配的碱基或氨基酸序列等。
通过分析结果,你可以确定序列的功能和进化关系,或者寻找可能的同源序列。
6.参数调整:BLAST提供了许多参数用于调整比对过程和结果的特性。

blast用法BLAST(Basic Local Alignment Search Tool)是一种常用的生物信息学工具,用于在数据库中搜索和比对生物序列(如DNA、RNA、蛋白质等)。
以下是使用BLAST的基本步骤和用法:1. 准备输入序列:首先,准备待查询的序列数据。
可以是DNA序列、蛋白质序列或其他类型的生物序列。
2. 选择BLAST程序:根据要比对的序列类型,选择合适的BLAST程序。
常见的BLAST程序包括blastn(用于DNA比对)、blastp(用于蛋白质比对)、blastx(用于DNA与蛋白质相互比对)等。
3. 选择数据库:确定要在哪个数据库中进行比对。
BLAST提供了多个数据库选项,如NCBI提供的nr数据库(非冗余蛋白质序列数据库)。
4. 运行BLAST:使用命令行或图形界面工具,输入BLAST命令或设置相应的参数进行比对。
例如,可以使用以下命令运行blastp程序进行蛋白质比对:```blastp -query input.fasta -db database -out output.txt```其中,`input.fasta`是输入序列文件,`database`是要比对的数据库,`output.txt`是输出结果文件。
5. 解析和分析结果:BLAST运行完成后,会生成比对结果文件。
可以使用相应的工具或脚本来解析、过滤和分析结果,以获取所需信息(如相似性、E值、比对长度等)。
6. 结果解释和进一步分析:根据比对结果,可以进一步解释和分析序列的功能、同源性等信息。
可以使用其他生物信息学工具和数据库来进一步研究和验证结果。
需要注意的是,BLAST具有多个参数和选项,可以根据具体的研究目的和需求进行调整和优化。
建议参考相关的文档、教程或使用BLAST 提供的帮助命令(如`blastn -help`)来了解更多详细的用法和参数设置。

BLAST使用方法一、BLAST的安装和准备工作2.获取待比对的序列文件,可以是FASTA格式的DNA或蛋白质序列。
二、BLAST的常用参数和选项1. Program:指定使用哪种BLAST程序(如BLASTn、BLASTp等)。
2. Database:指定使用哪个数据库进行比对。
3. Query:指定待比对的序列文件。
4. E-value:期望值。
一种描述比对结果误差率的指标,值越小表示结果越可信。
通常情况下,E-value小于0.01被认为是显著结果。
5. Word size:BLAST在比对时使用的核心词的长度。
长度越大表示查全率(sensitivity)越高,但速度会减慢。
6. Gap open:允许在比对过程中插入空位(如插入一个碱基)。
Gap open参数定义了开放一个空位的惩罚分数。
7. Gap extension:允许空位的延伸。
Gap extension参数定义了延伸一个空位的惩罚分数。
三、使用BLAST进行比对1.命令行方式:-打开命令行界面,并定位到BLAST软件的安装目录。
- 输入命令,指定BLAST程序、数据库、查询文件和其他参数。
例如:blastn -db nt -query query.fasta -out output.txt -evalue 0.01-运行命令,BLAST将开始进行比对并生成结果文件。
2.网页方式(以NCBIBLAST为例):- 打开NCBI网站的BLAST页面()。
-选择需要使用的BLAST程序(如BLASTn、BLASTp等)。
-上传待比对的序列文件,或者粘贴序列文本到输入框中。
-选择适当的数据库和其他参数。
-点击“BLAST”按钮,等待比对完成。
四、解读BLAST结果1. E-value:表示在随机比对中获得与查询序列相似度更高的结果的期望概率。
E-value越小表示比对结果越显著。
2. Bitscore:用于表示比对结果的质量。
Bitscore越高表示比对结果越可信。
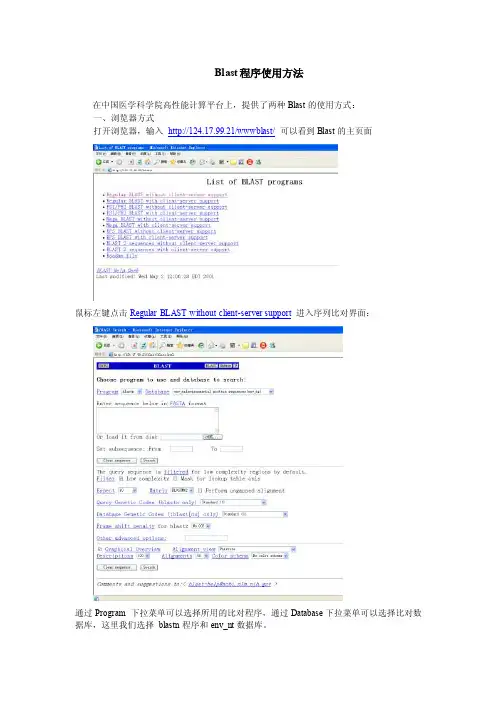
Blast程序使用方法在中国医学科学院高性能计算平台上,提供了两种Blast的使用方式:一、浏览器方式打开浏览器,输入http://124.17.99.21/wwwblast/可以看到Blast的主页面鼠标左键点击Regular BLAST without client-server support进入序列比对界面:通过Program 下拉菜单可以选择所用的比对程序,通过Database下拉菜单可以选择比对数据库,这里我们选择blastn程序和env_nt数据库。
接下来在文本框中输入需要比对的序列:设置完成后,鼠标左键点击“Search”按钮,开始搜索过程。
搜索结束后,得到类似下图的比对结果,完整结果参看BLASTN result.doc。
二、命令行方式LoadLever作业脚本方式提交作业,用户只需要修改输入序列和需要比对的数据库即可。
[loadl@f01n01 /gpfs/home/loadl/lltest]$ vi blast.cmd#!/bin/sh# @ error = /gpfs/home.AIX/loadl/lltest/blast.$(Hostname).$(jobid).err# @ output = /gpfs/home.AIX/loadl/lltest/blast.$(Hostname).$(jobid).out# @ requirements = (Pool == 1)# @ queue/gpfs/application/blast/bin/blastall -p blastp -d /gpfs/tmp/benchmarks/blast/swissprot -i /gpfs/tmp/benchmarks/blast/short100运行如下命令提交作业:[loadl@f01n01 /gpfs/home/loadl/lltest]$ llsubmit blast.cmdllsubmit: The job "f01n01.171" has been submitted.作业成功提交后可以看到系统返回的作业编号"f01n01.171"。

Primer-BLAST是NCBI的引物设计和特异性检验工具。
Primer-Blast介绍Primer-BLAST,在线设计用于聚合酶链反应(PCR)的特异性寡核苷酸引物。
Primer-BLAST可以直接从Blast主页(/)找到,或是直接用下面的链接进入:/tools/primer-blast/这个工具整合了目前流行的Primer3软件,再加上NCBI的 Blast进行引物特异性的验证。
Primer-BLAST免除了用另一个站点或工具设计引物的步骤,设计好的引物程序直接用Blast进行引物特异性验证。
并且,Primer-BLAST能设计出只扩增某一特定剪接变异体基因的引物–an important feature for PCR protocols measuring tissue specific expression(注:没办法准确的翻译,只好作罢,汗!)。
Primer-BLAST有许多改进的功能,这样在选择引物方面比单个的用 Primer3和NCBI BLAST更加准确。
Primer-BLAST的输入Primer-BLAST界面包括了Primer3和BLAST的功能。
提交的界面主要包括三个部分:target template(模板区), the primers(引物区), 和specificity check(特异性验证区)。
跟其它的BLAST一样,点击底部的“Advanced parameters”有更多的参数设置。
在“PCR Template”下面的文本框,输入目标模板的序列,FASTA格式或直接用Accession Number。
如果你在这里输入了序列,是用于引物的设计。
Primer-BLAST 就会根据你输入的序列设计特异性引物,并且在目标数据库(在specificity check区选择)是唯一的。
引物(Primers)如果你已经设计好了引物,要拿来验证引物的好坏。
可以在Primer Parameters 区填入你的一条或一对引物。

序列比对,绝大多数战友都会想到BLAST,但BLAST的使用确实又是一个很大的难题,因为他的功能比较强悍,里面涉及到的知识比较多,而且比对结束后输出的结果参数(指标)又很多。
如果把BLAST的使用详细的都讲出来,我想我发帖发到明天也发不完,更何况我自己也不是完全懂得BLAST的使用.所以我在这里也就“画龙点睛”——以比对核酸序列为例来给大家介绍一下BLAST的使用,也算是BLAST的入门课程吧.请看帖的战友好好体会,如果你用心看,在看帖完毕之后BLAST的基本使用(包括其他序列的比对)应该没有问题了。
一、打开BLAST页面,http://www.ncbi.nlm。
nih.go/BLAST/ 打开后如图所示:(缩略图,点击图片链接看原图)对上面这个页面进行一下必要的介绍:BLAST的这个页面主体部分(左面)包括了三部分:BLAST Assembled Genomes、Basic BLAST、Specialized BLAST.相信大家可以看懂这三个短语的意思,我就不多说了;我要说的是,可以认为这是三种序列比对的方法,或者说是BLAST的三条途径。
第一部分BLAST Assembled Genomes就是让你选择你要比对的物种,点击相应物种之后即可进入比对页面。
第二部分Basic BLAST包含了5个常用的BLAST,每一个都附有简短的介绍。
第三部分Specialized BLAST是一些特殊目的的BLAST,如IgBLAST、SNP等等,这个时候你就需要在Specialized BLAST部分做出适当的选择了.总之,这是一个导航页面,它的目的是让你根据自己的比对目的选择相应的BLAST途径。
下面以最基本的核酸序列比对来谈一下BLAST的使用,期间我也会含沙射影的说一下其他序列比对的方法。
二、点击Basic BLAST部分的nucleotide blast链接到一个新的页面。
打开后如图所示:screen.width-333)this.width=screen.width—333" width=640 height=462 title="Click to iew full 2.JPG (849 X 613)” border=0 align=absmiddle> 介绍一下上述页面:Enter Query Sequence部分是让我们输入序列的,你可以直接把序列粘贴进去,也可以上传序列,还可以选择你要比对的序列的范围(留空就代表要比对你要输入的整个序列)。

blast使用指南Blast使用指南Blast(Basic Local Alignment Search Tool)是一种常用于生物信息学研究中的序列比对分析工具。
它可以根据输入的查询序列,在数据库中搜索相似序列,并给出比对结果。
本文将为大家提供一份Blast使用指南,帮助大家更好地使用Blast进行序列比对分析。
一、什么是Blast?Blast是一种基于局部比对算法的工具,它可以在大规模的数据库中快速搜索相似的序列。
通过比对查询序列和数据库中的序列,Blast 可以找到相似度较高的序列,从而推测它们之间的功能和结构的相似性。
二、Blast的使用步骤1. 准备查询序列在使用Blast之前,首先需要准备查询序列。
查询序列可以是DNA 序列或蛋白质序列,可以通过实验测序或从已有的数据库中获取。
确保查询序列的准确性和完整性非常重要,因为查询序列的质量将直接影响到Blast的结果。
2. 选择合适的Blast程序和数据库Blast有多个版本和程序可供选择,根据具体的研究目的和需求,选择合适的Blast程序和数据库非常重要。
常用的Blast程序包括Blastn(用于DNA序列比对)、Blastp(用于蛋白质序列比对)等。
数据库则可以选择NCBI的nr数据库、UniProt数据库等。
3. 运行Blast程序在选择好Blast程序和数据库后,可以通过命令行或图形界面来运行Blast程序。
对于初学者来说,推荐使用图形界面,因为图形界面更直观、易于操作。
在运行Blast程序时,需要输入查询序列文件和选择合适的参数设置,如比对算法、期望阈值、返回结果的数量等。
4. 解读Blast结果Blast运行完毕后,会生成一个结果文件。
这个结果文件包含了查询序列和数据库中相似序列的比对结果。
通过分析比对结果,可以了解到查询序列与数据库中序列的相似性程度、可能的功能和结构等信息。
需要注意的是,Blast结果并不是绝对准确的,需要结合实验数据和其他信息进行综合分析。

BLAST的使用方法与参数选项调整
BLAST(Basic Local Alignment Search Tool)是一种在生物信息学中广泛使用的序列比对工具,可以用于搜索数据库中的序列,并找到与之相似的序列。
以下是BLAST的基本用法:
1.选择BLAST软件:BLAST包括多种算法,如BLASTN、BLASTP、BLASTX、
TBLASTN和TBLASTX等。
根据需要搜索的序列类型,选择相应的BLAST 算法。
2.准备查询序列:将要搜索的序列准备妥当,可以是DNA、蛋白质或RNA
序列。
3.选择数据库:选择将要搜索的数据库,可以是核酸数据库或蛋白质数据库
等。
4.运行BLAST:在相应的BLAST软件中输入查询序列和选择数据库,运行
BLAST程序。
5.结果解析:解析BLAST结果,包括匹配的序列、匹配的位置、匹配的得分
和E值等。
需要根据E值和得分来判断匹配的可靠性和相似性。
在BLAST的使用过程中,还有一些参数和选项可以调整,例如设置期望值阈值、最大目标序列数目等。
这些参数和选项可以通过查阅BLAST的官方文档或相关教程来了解和使用。
Primer-Blast:在线引物设计工具生物信息学引物设计用于PCR 聚合酶链式反应⏹PCR:Polymerase Chain Reaction⏹PCR 是一种用于放大扩增特定的DNA片段的分子生物学技术,它可看作是生物体外的特殊DNA复制,PCR的最大特点,是能将微量的DNA大幅增加。
Why PCR?⏹在某个特定物种内1.qPCR用于测定mRNA表达量:mRNA/cDNA水平2.基因克隆:基因水平——引物设计的要求:引物在该基因组内或cDNA库中具备特异性⏹在一个已知基因序列的基因组A内设计引物,然后在另一个还未基因注释的(近缘)基因组B内扩增引物——引物设计的要求:(1)引物在该A基因组内或cDNA 库中具备特异性,并且(2)引物在A和B内是保守的,所以设计的引物在A基因组内往往处于基因的编码区域(编码区域在不同物种间比非编码区更保守)How to do PCR?变性退火延伸How: 引物设计原则⏹退火温度(Tm):两个引物的Tm值相差不能大于5℃,扩增产物与引物的Tm值相差不能大于10℃决定引物退火温度Tm值的最主要因素是引物长度Tm = 4*(G+C)+ 2*(A+T)⏹碱基组成(G+C含量):40%~60%,4种碱基要分布均匀⏹引物长度⏹不能有大于3bp的方向重复序列或自身互补序列存在⏹一个引物的3’末端序列不能结合到另一个引物的任何位点上⏹不要有局部的GC rich或AT rich(特别是3’端),避开T/C或A/G的连续结构较基础全面的引物设计解说Primer Design/GenWeb/Molecular/seq_anal/primer_design/primer_design.htm常用引物设计软件⏹Primer Premier 6 (P6)⏹Beacon Designer 8NCBI Primer-Blasthttps:///tools/primer-blast/index.cgi?LINK_LOC=BlastHome⏹NCBI Primer-Blast: Finding primers specific to your PCR template (using Primer3 and BLAST)⏹能在线设计引物,并验证设计好的引物。
Primer Premier 4.10Primer Premier4.0是由加拿大的Premier公司开发的专业用于PCR或测序引物以及杂交探针的设计,评估的软件,和Plasmid Premier2.02一起是该公司推出的最新的软件产品。
其主要界面同样也是分为序列编辑窗口(Genetank),引物设计窗口(Primer Design),酶切分析窗口(Restriction Sites)和纹基分析窗口(Motif)。
这里我们主要介绍其引物设计功能,其他功能的介绍请参看Plasmid Premier2.02。
打开程序首先进入的是序列编辑参看,与Plasmid Premier相比,其多了一个语音校正的功能,即在输入序列的时候,程序自动将碱基读出,以便用户进行校正,保证输入的正确和快速。
点击该界面的按钮即可进入到程序的引物设计窗口。
该界面共分为四层,最上面一层左面是5个控制按钮,用于实现引物设计中的各种功能,包括引物自动寻找,寻找结果查看和引物编辑;右边是观察两个引物在模板上结合位置的直观图以及对正链还是负链引物进行选择;第二层是显示模板和引物序列及二者间的配对情况的显示;第三层是显示两个引物的各种参数,包括给引物的打分,引物以及产物的起始位置、长度、Tm值、GC%消光系数、简并性;最后一层是给出的有关于引物的二聚体结构、发卡结构、错配情况和引物间二聚体结构的预测,左边是显示是否存在以上各种对PCR扩增有影响的结构,右边显示的是这些结构的位置,结构细节和稳定能,利用这些参数可以对引物作出可靠的评价。
下面是根据模板序列寻找引物的界面,在该界面中可以设定所要搜索的引物的类型,包括PCR引物,测序引物和杂交探针以及引物所在的链;另外也能设定搜索引物的范围,以及最终PCR产物的长度和引物的长度等。
并通过来点击按钮来设置一些搜寻参数:这些参数包括引物的Tm值,GC比,有简并性碱基,3’端稳定性,引物的稳定性,重复序列,二聚体/发卡结构和与模板及可能的杂质DNA(需要从另外的序列文件中读入)之间的错配情况,这些参数的设定可以根据要求变化,程序本身根据一定的标准分成从极高严谨性到极低严谨性5个档次。
PCR引物设计及软件使用技巧PCR引物设计及软件使用技巧PCR(聚合酶链反应)是一种重要的分子生物学技术,在基因测序、基因突变检测、基因定量等领域具有广泛的应用。
而PCR引物的设计是PCR反应成功与否的关键因素之一。
本文将介绍PCR引物设计的原理和方法,并向读者介绍一些常用的PCR引物设计软件及其使用技巧。
一、PCR引物设计的原则和策略PCR引物设计的目标是选取一对特异性的引物,使其能在目标DNA序列的两侧结合并扩增出特定的DNA片段。
PCR引物的设计应遵循以下原则和策略:(一)特异性PCR引物应与目标DNA序列特异结合,避免与其他非目标DNA 序列结合产生非特异性扩增。
为了确保特异性,引物的设计中应避免高度保守的序列,尽量选择低度保守区域。
(二)长度和GC含量PCR引物的长度通常应在18-30个碱基对之间,过短会降低特异性,过长可能会导致扩增效率降低或产生非特异性扩增。
另外,引物的GC含量应在40%-60%之间,过高或过低都会影响扩增效果。
(三)避免二聚体和内外引物二聚体PCR引物设计时应避免引物之间以及引物与内外引物之间形成二聚体。
二聚体会影响引物的特异性和扩增效率,甚至导致PCR反应失败。
因此,引物设计时可以利用一些在线工具进行二聚体分析。
(四)避免引物间的交叉杂交和截断引物引物间的交叉杂交会导致非特异性扩增,而截断引物会导致扩增结果缺失或产物截断,因此引物设计时应避免以上情况的发生。
(五)引物间距和末尾相对于PCR引物的设计目标,引物间距相对固定,一般为100-500bp之间。
此外,引物的末端设计也要考虑,如添加限制性酶切位点、引入位点或尾部,以方便后续的克隆操作。
二、PCR引物设计的方法PCR引物设计可以采用多种方法,如序列比对、限制性酶切位点分析、引物簇设计等。
下面介绍一些常用的PCR引物设计方法。
(一)序列比对法序列比对法是一种简单易行的PCR引物设计方法。
通过将目标序列与参考序列进行比对,找出保守区域来设计引物。