苯甲醇苯甲酸水杨酸的毛细管电泳分离
- 格式:pdf
- 大小:255.48 KB
- 文档页数:8


毛细管电泳法分离测定食品中山梨酸、苯甲酸、糖精胡美珍;王文铮;张燕琴【摘要】采用毛细管电泳-紫外检测法,考察了波长,电压,缓冲试剂、浓度、pH对山梨酸、苯甲酸、糖精分离的影响,得到了优化的实验条件.以硼砂20mmol/L(pH=7.5)为运行缓冲溶液,20kV为分离电压,检测波长为230nm的电泳条件下,进样时间为10s,山梨酸、苯甲酸、糖精可在12min内实现分离.山梨酸在5mg/L~50mg/L,苯甲酸在5mg/L~50mg/L,糖精在15mg/L~150mg/L范围内呈良好线性关系,迁移速度、峰面积相对标准偏差均小于4.5%,(n=5).用上述方法对实际样品进行测定,回收率在95%以上.【期刊名称】《上海师范大学学报(自然科学版)》【年(卷),期】2004(033)003【总页数】4页(P63-66)【关键词】毛细管电泳-紫外检测法;添加剂;山梨酸;苯甲酸;糖精【作者】胡美珍;王文铮;张燕琴【作者单位】上海师范大学,生命与环境科学学院,上海,200234;上海师范大学,生命与环境科学学院,上海,200234;上海师范大学,生命与环境科学学院,上海,200234【正文语种】中文【中图分类】O6570 引言毛细管电泳(简称CE),是80年代初发展起来的一种新型高效的分离技术[1].它具有高效、快速、样品用量少、测定成本低等优点,自其产生以来,迅速渗透到与分析化学相关的各个学科.如:医药、环境、食品等各个领域.苯甲酸、山梨酸、糖精是常用的食品添加剂.国家规定限量使用.所以测定食品中苯甲酸、山梨酸、糖精的含量十分重要.目前上述三种添加剂,大都采用紫外分光光度法、薄层层析法、气相色谱法和高效液相色谱法等[2~4].本文用CE法分离测定上述三种添加剂,方法简便,灵敏度和重现性均符合食品分析的要求,样品前处理测定步骤较紫外光度法、薄层层析法简单,测定成本较HPLC低,有很好的实用价值.1 实验部分1.1 主要仪器与试剂仪器高效毛细管电泳仪、ACS2000紫外检测器(北京彩陆科学仪器有限公司)(中科院研究生院应用化学研究所); 毛细管总长度:60cm, 有效长度:54cm, 直径:50μm; CQ50超声波清洗器(上海超声波仪器厂);pHs-3C 型酸度计(上海华侨仪表厂);过滤器Ø25(上海市医工院);0.22μm 滤膜(上海兴亚净化材料厂);注射器5mL试剂硼砂(优级纯)、磷酸二氢钠、磷酸氢二钠、三(羟甲基)氨基甲烷(简称Tris)、山梨酸、苯甲酸、糖精均为分析纯,样品为汽水、番茄沙司,蜜饯,水为二次蒸馏水.1.2 实验方法1.2.1 标准样品的制备参照国标法,准确称取山梨酸、苯甲酸、糖精.分别溶解、定容,使其质量浓度均为1.0mg/mL.以0.22μm滤膜过滤备用.1.2.2 样品的制备汽水,经超声波超声,除去二氧化碳,0.22μm滤膜过滤,待用.番茄沙司、蜜饯:准确称取2g左右样品置于具塞小量筒中,加H3PO4数滴,准确加入乙醚10mL,连同量筒称其总量,猛烈振摇2min,再次称重,如重量减少,加入乙醚补充至原有重量,混匀.取乙醚5mL,置于烧杯中,经40℃水浴加热,将乙醚挥发后,用硼砂缓冲液定容至10 mL.经0.22μm滤膜过滤,待用.1.2.3 电泳条件以硼砂20mmol/L(pH=7.5)为电泳缓冲液,在20kV恒压下进行电泳分离,在230nm波长处检测,电动进样,进样时间10s.毛细管使用前用0.10mol/L的NaOH,水和电泳缓冲液分别冲洗30min.每次电泳后,用上述溶液各冲洗2min.2 结果与讨论2.1 测定波长的选择据有关资料[3,4]报道苯甲酸的最大吸收波长为227nm,山梨酸为250nm,糖精为207nm,综合考虑以230nm为测定波长.2.2 分离电压的选择分别配制山梨酸、苯甲酸、糖精浓度均为10mg/L的混合标准溶液.在波长为230nm,进样时间为10s,缓冲液为硼砂30mmol/L(pH=7.5)的电泳条件下,改变电压(10~25kV),并观察分离情况.结果表明,电压在10~20kV范围内各组分的迁移时间随着分离电压增高,迁移时间减小,峰高增大,但分离度有所降低.电压增高,电流增大,产生焦耳热,使谱线变宽.综合考虑,实验选用20kV作为分离电压.2.3 缓冲试剂的选择分别配制浓度均为30mmol/L的硼砂、磷酸体系(磷酸二氢钠和磷酸氢二钠混合液)、Tris的缓冲体系,调节pH到7.5,进行试验.用山梨酸、苯甲酸、糖精浓度均为10mg/L的混合标准溶液试验.电泳条件:波长为230nm,进样时间为10s,电压20kV.结果表明,山梨酸、苯甲酸、糖精在在硼砂体系中分离情况最好,选择硼砂体系作为缓冲体系.2.4 缓冲液浓度选择以硼砂为缓冲液,分别配制20,30,40mmol/L硼砂溶液,pH均调节为7.5,按上述电泳条件进行.结果表明,硼砂浓度为20mmol/L时,山梨酸、苯甲酸、糖精可在12min内得到很好的分离,峰面积、峰高和峰形较30 mmol/L,40 mmol/L好,故选择20 mmol/L硼砂作为运行缓冲溶液.2.5 缓冲液pH的选择以20mmol/L硼砂溶液为缓冲介质,分别调节溶液的pH为7.5,8.0,8.5,9.0.在上述试验确定的条件下,观察上述3种组分的迁移行为,实验结果表明在pH为7.5时山梨酸、苯甲酸、糖精迁移时间差别最大,而且其中糖精的峰高较其他pH值时高,故缓冲液pH选择7.5.2.6 优化条件下的电泳图配制30 mg/L的山梨酸、30 mg/L的苯甲酸和90 mg/L的糖精混合标准溶液,按上述确定的分离条件进行,分离情况,见图1.2.7 工作曲线及检测限在上述优化条件下,对一系列山梨酸、苯甲酸、糖精的标准混合样品进行分析测定,测定其峰面积与浓度的关系,所得山梨酸、苯甲酸、糖精的线性范围相关系数及检测限.结果见表1.1-山梨酸;2-苯甲酸;3-糖精图1 混合标准液电泳图表 1 山梨酸、苯甲酸、糖精的线性范围及检测限被测物线性范围mg/L相关系数γ检测限mg/L山梨酸5.0^500. 99583.0苯甲酸5.0^500.99504.0糖精15^1500.99535.0*检测限按3倍噪声计算2.8 精密度试验配制浓度分别为30 mg/L的山梨酸、苯甲酸和90 mg/L的糖精的标准混合液平行进样5次,测得山梨酸迁移时间的RSD为1.9%,峰面积的RSD为4.5%;苯甲酸的迁移时间的RSD为2.2%,峰面积的RSD为4.2%;糖精的迁移时间的RSD 为3.8%,峰面积的RSD为3.3%.2.9 实际样品的测定取汽水、番茄沙司、蜜饯,经预处理后,按上述确定的电泳条件进行分离测定,结果见表2:表 2 实际样品测定结果(n=3)样品名称组分实际含量 (mg/L)添加量(mg/L)测得量(mg/L)回收率(%)汽水苯甲酸81.4440119.495.0番茄沙司苯甲酸82.2080169.4109山梨酸68.3580149.2101蜜饯苯甲酸107.080183.295.0糖精104.040145.11032.10 结论本法选用20mmol/L(pH=7.5)的硼砂作为缓冲溶液,20kV为采样和分离电压,10s进样,检测波长为230nm的电泳条件下,,山梨酸、苯甲酸、糖精可在12min内得到分离.本法线性关系良好(R>0.995),迁移时间和峰面积相对标准偏差均小于4.5%,实际样品测定的回收率在95%以上,方法简单、快速,相对于其他分析方法,具有预处理简便,检测成本低等特点.由此可见,毛细管电泳法在食品分析领域有良好的应用前景.参考文献:[1] 陈义. 毛细管电泳技术及应用[M]. 北京:化学工业出版社,2000.[2] 胡慰望,谢笔钧.食品化学[M].北京:科学出版社,1992.[3] 国家技术监督局. 中华人民共和国国家标准[S].1996.[4] 黄伟坤.食品检验与分析[M]. 北京:中国轻工业出版社,1993.。
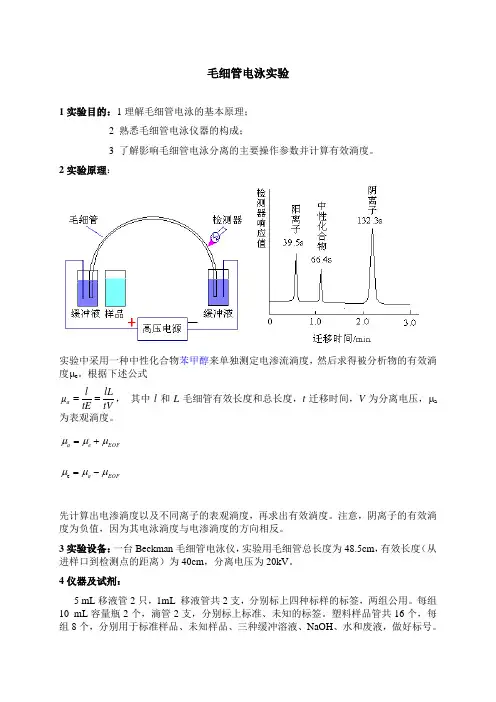
毛细管电泳实验1实验目的:1理解毛细管电泳的基本原理; 2 熟悉毛细管电泳仪器的构成;3 了解影响毛细管电泳分离的主要操作参数并计算有效淌度。
2实验原理:实验中采用一种中性化合物苯甲醇来单独测定电渗流淌度,然后求得被分析物的有效淌度μe 。
根据下述公式tV lL tE l a ==μ, 其中l 和L 毛细管有效长度和总长度,t 迁移时间,V 为分离电压,μa 为表观淌度。
EOF e a μμμ+= EOF a μμμ-=e先计算出电渗淌度以及不同离子的表观淌度,再求出有效淌度。
注意,阴离子的有效淌度为负值,因为其电泳淌度与电渗淌度的方向相反。
3实验设备:一台Beckman 毛细管电泳仪,实验用毛细管总长度为48.5cm ,有效长度(从进样口到检测点的距离)为40cm ,分离电压为20kV 。
4仪器及试剂:5 mL 移液管2只,1mL 移液管共2支,分别标上四种标样的标签,两组公用。
每组10 mL 容量瓶2个,滴管2支,分别标上标准、未知的标签。
塑料样品管共16个,每组8个,分别用于标准样品、未知样品、三种缓冲溶液、NaOH 、水和废液,做好标号。
滴瓶一共5个,分别装三种缓冲液(buffer)、1mol/L的NaOH和乙醇。
镊子、洗瓶、吸耳球、试管架、塑料样品管架、废液烧杯每组一个。
剪刀一把,记号笔一支,滤纸。
标样:苯甲醇、苯甲酸、水杨酸、对氨基水杨酸,均溶于二次水中,浓度1.00 mg/mL,作为标准品。
缓冲溶液(buffer):10 mmol/L NaH2PO4-Na2HPO4 1:1缓冲溶液(NaH2PO4和Na2HPO4 各5mMol/L);20 mmol/L NaH2PO4-Na2HPO41:1缓冲溶液(NaH2PO4和Na2HPO4 各10mMol/L),。
1mol/L NaOH溶液,二次去离子水。
5实验步骤:1.仪器的预热和毛细管的冲洗:在实验教师的指导下,打开仪器和配套的工作站。

毛细管电泳法分离水杨酸、苯甲酸及阿司匹林中的含量测定毛细管电泳法分离水杨酸、苯甲酸及阿司匹林中的含量测定毛细管电泳又称高效毛细管电泳( High Performance Capillary Electrophoresis, HPCE) 是一种仪器分析方法。
通过施加10-40kV 的高电压于充有缓冲液的极细毛细管,对液体中离子或荷电粒子进行高效、快速的分离。
现在,HPCE 已广泛应用于氨基酸、蛋白质、多肽、低聚核苷酸、DNA 等生物分子分离分析,药物分析,临床分析,无机离子分析,有机分子分析,糖和低聚糖分析及高聚物和粒子的分离分析。
人类基因组工程中DNA 的分离是用毛细管电泳仪进行的。
毛细管电泳较高效液相色谱有较多的优点。
其中之一是仪器结构 简单(见图1)。
它包括一个高电压源,一根毛细管,紫外检测器及计算机处理数据装置。
另有两个供毛细管两端插入而又可和电源相连的缓冲液池。
high-v oltagepower supply BufferV ialBuffer V ial Detector Recording dev icecapillaryElectrode Electrode图1 CE 仪器组成示意图毛细管中的带电粒子在电场的作用下,一方面发生定向移动的电泳迁移,另一方面,由于电泳过程伴随电渗现象,粒子的运动速度还明显受到溶液电渗流速度的影响。
粒子的实际流速 V 是电泳流速度 Vep 和渗流速度 Veo 的矢量和。
即:V = Vep + Veo (1)电渗流是一种液体相对于带电的管壁移动的现象。
溶液的这一运动是由硅/水表面的Zeta 势引起的。
CE 通常采用的石英毛细管柱表面一般情况下(pH>3)带负电。
当它和溶液接触时,双电层中产生了过剩的阳离子。
高电压下这些水合阳离子向阴极迁移形成一个扁平的塞子流,如图2。
毛细管管壁的带电状态可以进行修饰,管壁吸附阴离子表面活性剂增加电渗流, 管壁吸附阳离子表面活性剂减少电渗流甚至改变电渗流的方向。


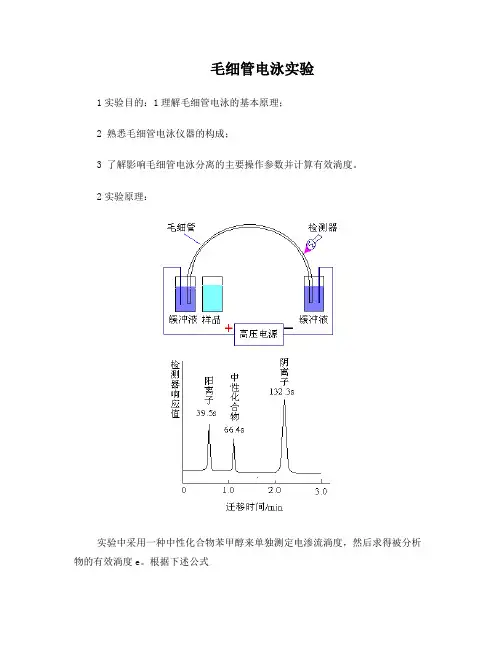
毛细管电泳实验1实验目的:1理解毛细管电泳的基本原理;2 熟悉毛细管电泳仪器的构成;3 了解影响毛细管电泳分离的主要操作参数并计算有效淌度。
2实验原理:实验中采用一种中性化合物苯甲醇来单独测定电渗流淌度,然后求得被分析物的有效淌度e。
根据下述公式,其中l和L毛细管有效长度和总长度,t迁移时间,V为分离电压,a为表观淌度。
先计算出电渗淌度以及不同离子的表观淌度,再求出有效淌度。
注意,阴离子的有效淌度为负值,因为其电泳淌度与电渗淌度的方向相反。
3实验设备:一台Beckman毛细管电泳仪,实验用毛细管总长度为48.5cm,有效长度(从进样口到检测点的距离)为40cm,分离电压为20kV。
4仪器及试剂:5 mL移液管2只,1mL 移液管共2支,分别标上四种标样的标签,两组公用。
每组10 mL容量瓶2个,滴管2支,分别标上标准、未知的标签。
塑料样品管共16个,每组8个,分别用于标准样品、未知样品、三种缓冲溶液、NaOH、水和废液,做好标号。
滴瓶一共5个,分别装三种缓冲液(buffer)、1mol/L的NaOH和乙醇。
镊子、洗瓶、吸耳球、试管架、塑料样品管架、废液烧杯每组一个。
剪刀一把,记号笔一支,滤纸。
标样:苯甲醇、苯甲酸、水杨酸、对氨基水杨酸,均溶于二次水中,浓度1.00 mg/mL,作为标准品。
缓冲溶液(buffer):10 mmol/L NaH2PO4-Na2HPO4 1:1缓冲溶液(NaH2PO4和Na2HPO4 各5mMol/L);20 mmol/L NaH2PO4-Na2HPO4 1:1缓冲溶液(NaH2PO4和Na2HPO4 各10mMol/L),。
1mol/L NaOH溶液,二次去离子水。
5实验步骤:1.仪器的预热和毛细管的冲洗:在实验教师的指导下,打开仪器和配套的工作站。
工作温度设置为30℃,不加电压,冲洗毛细管,顺序依次是:1 mol/L NaOH溶液5 min,二次水5 min,10 mmol/L NaH2PO4-Na2HPO4 1:1缓冲溶液5 min,冲洗过程中出口(outlet)对准废液的位置。

分离苯甲酸和苯甲醇是一个常见的化学反应,它能够将两种化合物分开,以获得单一的化合物。
在本文中,我们将介绍如何分离苯甲酸和苯甲醇的方法。
首先,将苯甲酸与水混合,将其加热至沸点,使苯甲酸熔化,苯甲醇会从溶液中挥发。
然后,将溶液分离成两部分,其中一部分是苯甲醇,另一部分是苯甲酸溶液。
最后,将苯甲酸溶液加热,使其蒸发掉,从而得到纯粹的苯甲酸。
其次,也可以采用水洗法来分离苯甲酸和苯甲醇。
首先,将苯甲酸与水混合,加热至沸点,使苯甲酸熔化,并将苯甲醇从溶液中挥发出来。
然后,将混合溶液放入另一个容器中,放入相同的量的水,搅拌均匀,使苯甲酸和苯甲醇分离开来,最后,将分离出的苯甲酸浓缩,得到纯粹的苯甲酸。
最后,也可以用碱溶液来分离苯甲酸和苯甲醇。
将苯甲酸和碱溶液混合,在加热的情况下搅拌,使苯甲酸熔化,苯甲醇会从溶液中挥发出来。
然后,将混合溶液放入另一个容器中,加入适量的水,搅拌均匀,使苯甲酸和苯甲醇分离开来,最后,将分离出的苯甲酸浓缩,得到纯粹的苯甲酸。
以上就是分离苯甲酸和苯甲醇的三种方法。
它们都很有效,但是要根据具体情况选择合适的方法,以获得最佳的结果。


毛细管电泳手性分离流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!Download tips: This document is carefully compiled by theeditor. l hope that after you downloadthem,they can help yousolve practical problems. The document can be customized andmodified afterdownloading,please adjust and use it according toactual needs, thank you!毛细管电泳手性分离流程简述如下:①样品准备:选取需要分离的对映体混合物,适当稀释并去蛋白、过滤,确保样品纯净。
②缓冲液配制:根据目标化合物性质,配制合适的pH值和离子强度的缓冲液,加入手性添加剂如环糊精、手性离子对等。
③毛细管填充:将配好的缓冲液注入毛细管内,通过电渗作用使缓冲液均匀分布。
④电场建立:在毛细管两端施加恒定电压,形成稳定电场,驱动样品组分在毛细管中迁移。
⑤样品注入:通过压力或电渗作用将样品引入毛细管,注意控制注入体积,避免过载。
⑥分离进行:在电场作用下,样品中各组分依据电荷、大小及与手性添加剂的相互作用差异被分离。
⑦检测记录:使用紫外/可见光吸收检测器、激光诱导荧光检测器等监测分离后的组分,记录色谱图。
⑧数据分析:分析色谱图,确定对映体的分离度、保留时间和峰面积,评价分离效果。
⑨优化条件:根据分离效果,调整缓冲液组成、电场强度、温度等参数,以优化分离条件。
⑩结果验证与应用:重复实验验证分离稳定性,将优化的方法应用于实际样品的手性分析中。
此流程展示了毛细管电泳手性分离技术的基本步骤,是分析化学中分离手性化合物的重要手段。

毛细管电泳分离混合样品
摘要:区带电泳
1、试药与仪器:苯甲酸钠、吡啶、苯酚及三者的混合样品
2、方法和结果:
2.1原理
在电场作用下,毛细管柱中出现电泳现象和电渗流现象。
毛细管壁带负电,靠外层带正电,加高压电场下形成向负极移动的电渗流,电泳是正、负电粒子自身的差速迁移,因此:
正离子:本身迁移速度+电渗流速度,最快,在负极最先流出。
中性离子:本身无电泳现象,受电渗流影响,在正离子后流出。
阴离子:电渗流速度-本身迁移速度,最慢,在负极最后流出。
在PH=5.8时,吡啶为正离子,苯酚为中性,苯甲酸钠为负离子。
本实验主要看分离电压对迁移时间、分离度的影响。
采用PH=5.8的缓冲盐,估计出峰顺序为吡啶、苯酚、苯甲酸钠。
2.2方法:254nm下分离混合样品
先采用20KV的电压分离,迁移时间太长。
改用30KV的电压分离,分离效果较好,记录出峰时间t R分别为吡啶
7.218min,苯酚10.237min,苯甲酸钠21.597min。
3、讨论:
毛细管电泳时,分离电压小会使分析时间长,进样前需用水冲洗后用缓冲液冲洗,冲洗2~3min,使毛细管柱充满缓冲溶液;设定方法时,将工作站INI 下进行初始化。
毛细管电泳法测定食品中8种添加剂
杨桂君;高文惠
【期刊名称】《食品科学》
【年(卷),期】2010(031)018
【摘要】为建立毛细管电泳法同时测定亮蓝、香兰素、山梨酸、苯甲酸、日落黄、新红、苋菜红、柠檬黄8种食品添加剂的分析方法.采用毛细管区带电泳-紫外检测法,以磷酸-硼砂缓冲体系为运行缓冲液,于240nm波长处对饮料、果冻、蜜饯样品进行检测,一次进样分析在8min内完成.结果表明:该方法的线性范围为2.5~
1000μg/mL,线性相关系数在0.9986~0.9998之间,甲均回收率在85.2%~100.3%之间,相对标准偏差(RSD)≤6.98%(n=5),检测限为0.25~10μg/mL.该方法操作方便,检测快速,在食品添加剂的检测上取得了令人满意的效果.
【总页数】4页(P377-380)
【作者】杨桂君;高文惠
【作者单位】河北科技大学生物科学与工程学院,河北省,石家庄,050018;河北科技大学生物科学与工程学院,河北省,石家庄,050018
【正文语种】中文
【中图分类】TS202.3;O657.8
【相关文献】
1.毛细管电泳在食品添加剂检测中的应用 [J], 韩双艳;崔堂兵;林小琼;吴虹
2.毛细管电泳在食品添加剂检测中的应用 [J], 高文惠;裴红;杨桂君
3.高效毛细管电泳在食品添加剂中应用的研究进展 [J], 田益玲;陈冠华;祝彦忠
4.毛细管电泳技术在食品添加剂分析中的应用 [J], 王茜;王广增
5.氢化物发生-原子荧光光度法测定食品和食品添加剂中的砷、汞和铅 [J], 陈艳晶;李佳华;晏廷照
因版权原因,仅展示原文概要,查看原文内容请购买。
毛细管电泳法分离检测饮料中的防腐剂王钰;杨思文;代语林;邓宁;王燕;何建波;朱燕舞【摘要】采用高效毛细管电泳法,同时在线分离检测对羟基苯甲酸甲酯、山梨酸钾和苯甲酸3种防腐剂.考察了运行缓冲液的浓度、pH值、分离电压、进样时间等因素对电泳分离的影响.在检测波长为230nm时,获得了3种防腐剂分离检测的最优化实验条件为:20mmol/L pH 9.2的硼砂缓冲溶液,分离电压为22kV,进样时间15s.在最优化实验条件下,3种防腐剂检测的线性相关系数均大于0.98,检出限为1.0×10-2g/kg.利用此方法检测了市售3种饮料中所含防腐剂的种类和含量,各种防腐剂的回收率在88%~102%之间.【期刊名称】《中国酿造》【年(卷),期】2014(033)004【总页数】5页(P124-128)【关键词】毛细管电泳;防腐剂;分离;检测【作者】王钰;杨思文;代语林;邓宁;王燕;何建波;朱燕舞【作者单位】合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009;合肥工业大学化学工程学院,安徽合肥230009【正文语种】中文【中图分类】O657.8食品是人类生存的基本物质保障,现代化的食品生产要求食品能够方便贮存和运输,防腐剂既能使食品保存期延长,也能防止食品腐败变质,因此被广泛应用于食品生产中。
但是食品防腐剂的过量使用会对人体健康造成较大的危害,伤害人体正常机能,降低人体免疫力。
我国明确规定了防腐剂使用标准,但仍然有一些商家没有按照标准合理使用防腐剂。
2007年国家质检总局对北京等13个省、直辖市96家企业生产的117种产品进行抽查,抽查中发现部分碳酸饮料中防腐剂的含量超标[1]。
非水体系毛细管电泳-紫外检测法测定苯甲酸和苯甲醛李利军X1,黄文艺2,程昊2,孔红星1,吴健玲2(1.广西工学院生物与化学工程系,柳州545006; 2.广西大学化学与化学工程学院,南宁530005)摘要:建立了用非水相体系高效毛细管电泳-紫外检测法同时测定苯甲酸和苯甲醛的新方法,考察了运行电压、非水相介质和电解质等因素的影响,在25e下,以V(乙腈)B V(碳酸丙烯酯)=1B1的混合液为溶剂,缓冲体系中含15mmol P L十六烷基三甲基溴化铵体积分数1%乙酸,重力进样30s,运行电压20kV,毛细管总长45cm有效长度30c m,<75L m,检测波长285nm。
苯甲酸线性范围为5~40L g P mL,线性方程为:y=13.473Q+13.336,相关系数r=0.9985,检出限为0.92L g P mL,RSD为318%。
苯甲醛的线性范围75~1125L g P mL线性方程为:y=512449Q+564101,相关系数r=019997,检出限为15160L g P m L,RSD为315%。
已用于经空气氧化后的苯甲醛中苯甲酸和苯甲醛的测定。
关键词:苯甲酸;苯甲醛;非水相体系;毛细管电泳;紫外检测中图分类号:O657.8文献标识码:A文章编号:1000-0720(2006)07-056-04苯甲醛是一种广泛应用于合成染料、香料和药物等精细化学品的中间体。
在合成中发挥着越来越重要的作用。
在苯甲醛的合成过程中,通常会有少量的副产物苯甲酸生成。
测定合成过程中苯甲酸和苯甲醛的量,据此来选择适当的投料比,尽量避免副产物苯甲酸的生成,以获得满意的苯甲醛产率。
因此,建立能同时快速、准确、简便测定苯甲醛和苯甲酸的方法具有一定实际意义。
测定苯甲醛的方法主要有气相色谱法[1]、分光光度法[2]、极谱法[3]和荧光光度法[4]等。
这些方法大多数采用将苯甲醛衍生后间接进行测定,因此样品处理较繁琐。
而且由于苯甲醛不溶于水而苯甲酸溶于水,能同时对苯甲醛和苯甲酸进行测定的方法较少。
毛细管电泳中常用的检测方法毛细管电泳中常用的检测方法毛细管电泳(C E ) 以其高效、快速的分离, 成为一种令人瞩目的分析手段。
为了便于热量散失和进行柱上检$lJ , 采用了极小内径的毛细管(≤50 umi.d.) , 这样允许进样量就很小(10 一9 g )。
如此小的进样量要求有高灵敏的检测方法, 才能进行定性、定量分析。
紫外吸收法:一般, 常用于C E 的检测器是市售的紫外和荧光检测器。
当采用紫外一可见吸收法时, 石英毛细管壁的内涂层常常用有机溶剂溶解或灼烧而刮去, 使出现一个“小窗口” , 作为柱上检测的流通池。
内径很小的毛细管使得流通池的长度也相应很小, 检测灵敏度相当有限, 尤其在生物样品分析中常常需要先行样品预富集然后再检测, 以提高灵敏度。
在使用长方形徽面的毛细管进行电泳分离分析时,柱上检测的光学流通池长度明显增大, 使检测灵敏度提高7 1 5 倍。
采用高能量的光源, 如氨灯、激光等, 可较大地提高检测灵敏度, 但费用较高, 又因可供选择使用的人射光波长范围较窄, 限制了它的应用。
荧光检测法:荧光检测的灵敏度比紫外吸收法高几个数量级。
对于有适当的激发荧光和发射荧光的供试品, 采用激光诱导荧光检测法和光电倍增管, 可使检测灵敏度大大提高, 而对于绝大多数无自然荧光的化合物, 则必须进行柱前或柱后衍生化, 才能进行荧光检测。
间接检测法:对供试品进行衍生化的操作繁琐, 且易引入误差, 对于被测浓度极低的生物样品更是如此。
于是, 间接检测法应运而生。
间接检测就是在电泳缓冲液中加入具有检测响应的检测剂, 如发色团、荧光物质等, 作为本底响应,以产生基线信号。
供试品进样后, 供试品离子与反电荷的检测剂离子形成离子对, 或置换了检测剂的同电荷离子, 分别产生正峰和负峰,使基线信号发生改变而被检测。
在间接荧光法中, 被分析物能吸收荧光辐射或使本底荧光淬灭, 本底信号因此降低或消失而进行检测。
这种方法要求供试品必须能吸收荧光或淬灭荧光。
苯甲醇、苯甲酸、水杨酸的毛细管电泳分离一、实验目的1.进一步理解毛细管电泳的基本原理;2.熟悉毛细管电泳仪器的构成;3.了解影响毛细管电泳分离的主要操作参数。
二、实验原理1.电泳淌度毛细管电泳(CE )是以电渗流 (EOF)为驱动力,以毛细管为分离通道,依据样品中组分之间淌度和分配行为上的差异而实现分离的一种液相微分离技术。
离子在自由溶液中的迁移速率可以表示为:ν = µE (1)式中ν是离子迁移速率,µ为电泳淌度,E 为电场强度。
对于给定的荷电量为q 的离子,淌度是其特征常数,它由离子所受到的电场力(F E )和通过介质所受到的摩擦力(F F )的平衡所决定。
F E = qE (2)对于球形离子:F F = -6πηr ν (3)式中η为介质粘度,r 为离子的流体动力学半径。
在电泳过程达到平衡时,上述两种力方向相反,大小相等:qE = -6πηr ν (4)将式(4)代入式(1),得:r 6q πηµ= (5)因此,离子的电泳淌度与其荷电量呈正比,与其半径及介质粘度呈反比。
带相反电荷的离子其电泳淌度的方向也相反。
需要指出,我们在物理化学手册中可以查到的离子淌度常数是绝对淌度,即离子带最大电量时测定并外推至无限稀释条件下所得到的数值。
在电泳实验中测定的值往往与此不同,故我们将实验值称为有效淌度(µe )。
有些物质因为绝对淌度相同而难以分离,但我们可以通过改变介质的pH 值,使离子的荷电量发生改变。
这样就可以使不同离子具有不同有效淌度,从而实现分离。
下文中所提到的电泳淌度除特别说明外,均指有效淌度。
2.电渗流和电渗淌度电渗流(EOF )是CE 中最重要的概念,指毛细管内壁表面电荷所引起的管内液体的整体流动,来源于外加电场对管壁溶液双电层的作用。
在水溶液中多数固体表面根据材料性质的不同带有过剩的负电荷或正电荷。
就石英毛细管而言,表面的硅羟基在pH 大于3以后就发生明显的解离,使表面带有负电荷。
为了达到电荷平衡,溶液中的正离子就会聚集在表面附近,从而形成所谓双电层,如图1所示。
这样,双电层与管壁之间就会产生一个电位差,叫做Zeta 电势。
但毛细管两端施加一个电压时,组成扩散层的阳离子被吸引而向负极移动。
由于这些离子是溶剂化的,故将拖动毛细管中的体相溶液一起向负极运动,这便形成了电渗流。
需要指出,很多非离子型材料如聚四氟乙烯和聚丙烯等材料表面也可以产生电渗流,原因可能是其表面对阴离子的吸附。
电渗流的大小可用速率和淌度来表示:()E EOF ηεξν/=(6) 或者 ηεξµ/=EOF (7)式中νEOF 为电渗流速率,µEOF 为电渗淌度,ξ为Zeta 电势,ε为介电常数。
Zeta 电势主要取决于毛细管表面电荷的多寡。
一般来说,pH 越高,表面硅羟基的解离程度越大,电荷密度越大,电渗流速率就越大。
除了受pH 的影响外,电渗流还与表面性质(硅羟基的数量、是否有涂层等)、溶液离子强度有关,双电层理论认为,增加离子强度可以使双电层压缩,从而降低Zeta 电势,减小电渗流。
此外,温度升高可以降低介质粘度,增大电渗流。
电场强度虽然不影响电渗淌度,但却可改变电渗流速率。
显然,电场强度越大,电渗流速率越大。
由上可知,电渗流的方向一般是从正极到负极,然而,在溶液中加入阳离子表面活性剂后,由于毛细管表面强力吸附阳离子表面活性剂的亲水端,而阳离子表面活性剂的疏水端又会紧密结合一层表面活性剂分子,结果就形成了带负电的表面,双电层Zeta 电势的极性发生了反转,最后使电渗流的方向发生了变化。
在分析小分子有机酸时,这是常用的电渗流控制技术。
电渗流的一个重要特性是具有平面流型。
由于引起流动的推动力在毛细管的径向上均匀分布,所以管内各处流速接近相等。
其优点是径向扩散对谱带扩展的影响非常小,如图2所示。
与此形成鲜明对照的是高压泵驱动的抛物线流型(如在HPLC 中),由于管内径向上各处的流速不同,使得谱带峰形变宽。
这也是与HPLC 相比,CE 具有更高分离效率的一个重要原因。
电渗流的另一个重要优点是可以使几乎所有被分析物向同一方向运动,而不管其电荷性质如何。
这是因为电渗淌度一般比离子的电泳淌度大一个数量级,故当离子的电泳淌度方向与电渗流方向相反时,仍然可以使其沿电渗流方向迁移。
这样,就可在一次进样分析中,同时分离阳离子和阴离子。
中性分子由于不带电荷,故随电渗流一起运动。
如果对毛细管内壁进行修饰可以降低电渗流,而被分析物的淌度则不受影响。
在此情况下,阴阳离子有可能向不同的方向迁移。
3.毛细管电泳的仪器及操作图3所示为CE 仪器示意图。
其组成部分主要是高压电源、缓冲液瓶(包括样品瓶)、毛细管和检测器。
下面分别简要讨论之。
高压电源是为分离提供动力的,商品化仪器的输出直流电压一般为0~30kV,也有文献报道采用60kV以至90kV电压的。
大部分直流电源都配有输出极性转换装置,可以根据分离需要选择正电压或负电压。
缓冲液瓶多采用塑料(如聚丙烯)或玻璃等绝缘材料研制成,容积为1~3mL。
考虑到分析过程中正负电极上发生的电解反应,体积大一些的缓冲液瓶有利于pH的稳定。
进样时毛细管的一端伸入样品瓶,采用压力或电动方式将样品加载到毛细管入口,然后将样品瓶换为缓冲液瓶,接通高压电源开始分析。
4.毛细管电泳的分离模式CE有6种常用的分离模式,其分离依据及应用范围如表1 所示。
其中毛细管区带电泳(CZE)、胶束电动毛细管色谱(MEKC)和毛细管电色谱(CEC)最为常用。
本实验的内容为CZE。
表1 6种CE分离模式的分离依据及应用范围分离模式 分离依据 应用范围毛细管区带电泳(CZE) 溶质在自由溶液中的淌度差异 可解离的或离子化合物、手性化合物及蛋白质、多肽等毛细管胶束电动色谱(MECC) 溶质在胶束与水相间分配系数的差异中性或强疏水性化合物、核酸、多环芳烃、结构相似的肽段毛细管凝胶电泳(CGE) 溶质分子大小与电荷/质量比差异蛋白质和核酸等生物大分子毛细管等电聚焦(CIEF) 等电点差异 蛋白质、多肽毛细管等速电泳(CITP) 溶质在电场梯度下的分布差异(移动界面)同CZE,电泳分离的预浓缩毛细管电色谱(CEC) 电渗流驱动的色谱分离机制 同HPLC毛细管区带电泳(CZE)是最简单的CE模式,因为毛细管中的分离介质只是缓冲液。
在电场的作用下,样品组分以不同的速率在分立的区带内进行迁移而被分离。
由于电渗流的作用,正负离子均可以实现分离。
在正极进样的情况下,正离子首先流出毛细管,负离子最后流出。
中性物质在电场中不迁移,只是随电渗流一起流出毛细管,故得不到分离。
在CZE中,影响分离的因素主要有缓冲液的种类、浓度和pH值、添加剂、分析电压、温度、毛细管的尺寸和内壁改性等。
缓冲液种类的选择主要考虑其pKa值要与分析所用pH匹配,另外,有的缓冲液与样品组分之间有特殊的相互作用,可提高分析选择性。
比如,分析多羟基化合物时,多用硼酸缓冲液,因为硼酸根可与羟基形成络合物,有利于提高分离效率。
增大缓冲液的浓度一般可以改善分离,但电渗流会降低,因而延长了分析时间,过高的盐浓度还会增加焦耳热。
缓冲液的pH 主要影响电渗流的大小和被分析物的解离情况,进而影响被分析物的淌度,是CZE 分析中最重要的操作参数之一。
缓冲液添加剂多为有机试剂,如甲醇、乙腈、尿素、三乙胺等,其作用主要是增加样品在缓冲液中的溶解度,抑制样品组分在毛细管壁的吸附,改善峰形。
提高分析电压有利于提高分离效率和缩短分析时间,但可能造成过大的焦耳热。
温度的变化可以改变缓冲液的粘度,从而影响电渗流。
毛细管内径越小,分离效率越高,但样品容量越低;增加毛细管长度可提高分离效率,但延长了分析时间。
有时为了改善分离,要对毛细管内壁进行改性,比如采用涂层技术。
5.毛细管电泳的基本参数CE 中的分析参数可以用色谱中类似的参数来描述,比如与色谱保留时间相对应的有迁移时间,定义为一种物质从进样口迁移到检测点所用的时间,迁移速率(ν)则是迁移距离(l ,即被分析物质从进样口迁移到检测点所经过的距离,又称毛细管的有效长度)与迁移时间(t )之比:t l=ν (8)因为电场强度等于施加电压(V)与毛细管长度(L)之比:L V E = (9)就CE 的最简单的模式—毛细管区带电泳(CZE )而言,结合式(1),可得:tV lL tE l a ==µ (10)在毛细管区带电泳(CZE )条件下测得的淌度是电泳淌度与电渗流淌度的矢量和,我们称之为表观淌度µa ,即:EOF e a µµµ+= (11)实验中可以采用一种中性化合物,如二甲亚砜或丙酮等,来单独测定电渗流淌度,然后求得被分析物的有效淌度。
例如,图4是一个混合物的分离结果,其中三个峰分别为阳离子(t = 39.5s )、中性化合物(t = 66.4s )和阴离子(t = 132.3s )。
实验用毛细管总长度为48.5cm ,有效长度(从进样口到检测点的距离)为40cm ,施加电压为20kV 。
根据上述公式,我们便可以计算出电渗淌度以及不同离子的表观淌度和有效淌度。
电渗淌度:11231046.14.66200005.4840−−−⋅×=××=s V cm EOF µ阳离子:11246.25.39200005.4840−−⋅=××=s V cm a µ 1123101−−−⋅×=−=s V cm EOF a e µµµ阴离子:11241033.73.132200005.4840−−−⋅×=××=s V cm a µ11241087.5−−−⋅×−=−=s V cm EOF a e µµµ注意,阴离子的有效淌度为负值,因为其电泳淌度与电渗淌度的方向相反。
三、实验设备每四人一组共用一台Capel-103RT 电泳仪。
四、实验仪器及试剂5 mL 移液管2只,1mL 移液管共2支,分别标上四种标样的标签,两组公用。
每组10 mL 容量瓶2个,滴管2支,分别标上标准、未知的标签。
塑料样品管共16个,每组8个,分别用于标准样品、未知样品、三种缓冲溶液、NaOH 、水和废液,做好标号。
滴瓶一共5个,分别装三种缓冲液(buffer)、1mol/L 的NaOH 和乙醇。
镊子、洗瓶、吸耳球、试管架、塑料样品管架、废液烧杯每组一个。
剪刀一把,记号笔一支,滤纸。
标样:苯甲醇、苯甲酸、水杨酸、对氨基水杨酸,均溶于二次水中,浓度1.00 mg/mL ,作为标准品,混合稀释作为标样。