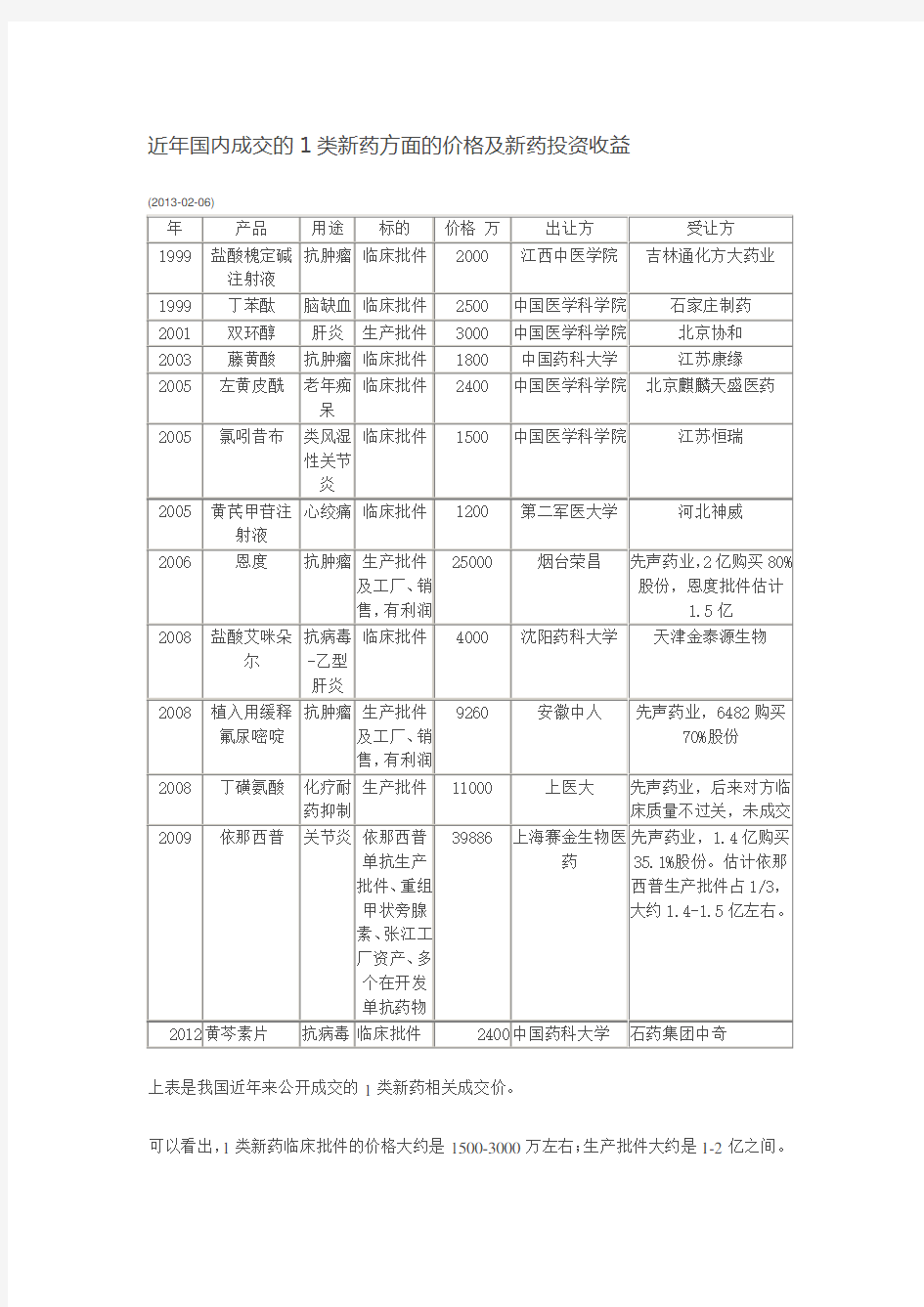

近年国内成交的1类新药方面的价格及新药投资收益
(2013-02-06)
上表是我国近年来公开成交的1类新药相关成交价。
可以看出,1类新药临床批件的价格大约是1500-3000万左右;生产批件大约是1-2亿之间。
关键是,新药投资收益如何?
好比说有一个一类新药,过去的4年投入现金715万,已经拿到临床批件:
- 可以现在转让临床批件,2000-4000万;
- 继续做完1-3期临床,还需要5年,投入2400万,届时生产批件价值1-2亿。
新药的投资收益
新药的投资收益与什么因素有关呢?
- 直接投入,一般是委托的CRO合同研究费用,上表中的费用即为直接的CRO费用
- 溢价管理费:研发有不同的组织形式,
1) 高效率的就只有项目管理人员,他们不具体做瓶瓶罐罐的试验,全部是委托;
2)另一种是人员比较多的那种,什么部门多有,也外包,也自己弄一点,人比较多,依托于大公司的研究部门往往是这样的,这样的研究部门人员多,资产大,效率不高。
对于第一种,一个项目增加50%的管理或执行费用就已经很不错了。例如上面这个项目,如果做到生产批件总投入是3325万,如果额外投入50%管理费就是1670万,这也是相当可观的。
对于第二种,加上一些失败的项目、加上人头费、折旧、各种运营费用、再加上地效率,通常要乘以2.5倍的系数。
根据上述假设,实际费用投入按照直接费用投入的2.5倍和1.5倍、按照转让生产批件和临床批件、及按照不同价格估算年均投资收益率:
如果生产批件按照1.3亿转让,收益率在11-22%左右;临床批件按照3000万转让,收益率在10%-33%之间。如果卖不到这样的价格,收益率就要大大降低了。
新药研发的收益率,在于项目成功率、费用、时间、项目本身的价值:
- 项目成功:项目按照既定价格、时间成交,就是成功。项目的失败将大大降低整体收益率;尽管一个项目的平均年收益率高达30、50%,但是,如果如果只能每三个或者两个项目成功一个,那么收益率也将大大降低。因此,对于不能成功的项目要及时终止,越早越好。
- 项目费用:要采取一切措施最大限度地降低费用:不要做无效、过头的工作;同样质量的工作要采用更低的人力和方案;要充分利用社会资源,要权衡外包CRO和自己实施的总体成本(包括设备、折旧、资金成本);要有让项目经理处处主动降低成本的考核机制。
- 时间:时间的作用主要是资金成本。因为资金的成本要计算复利,因此,时间对成本的影响是呈指数增长的,足见时间对收益率的影响非常大。例如,某个历史10年的项目,在总的费用和项目转让价格确定的情况下,其年收益率为34%,如果缩短1年则收益率提升到38%;缩短两年则提升到44%;缩短3年则提升到52%;缩短4年则提升到64%,收益率翻倍。当然,项目不能无限度地缩短;但是需要避免不必要的延误、补充资料,项目延长则意味着收益率呈指数降低。良好的项目时间管理和精细的方案以避免不必要的延误,是提高收益率的关键。
- 项目本身的价值:项目本省的价值无疑是项目收益率的最大影响因素。在项目立项的时候就要考虑到潜在项目受让方的出价能力。
创新药研究和发展工作是个有较高技术、智力、资金门槛的工作,是个高风险的工作,理应对应着高的收益。其核心是如何发现潜在优秀项目(项目可行性、潜在价值)、如何以高于同行的效率(费用和时间)进行组织和实施
国家一类新药汇总 ★双环醇片(Bicyclol) 商品名“百赛诺”,由中国医学科学院、中国协和医科大学药物所开发的产品,是我国第一个具有自主知识产权一类抗肝炎合成创新药物。1996年12月经卫生部批准进入临床试验,2001年9月获得原SDA颁发的新药证书及生产批准文,由北京协和药厂生产上市。目前已在世界16个国家地区获得20年发明专利保护,国内享有12年的行政保护期。 本品是在降酶药物联苯双酯和五味子基础上,经过化学合成筛选的药物,其良好的双重机制,能清除自由基作用以保护细胞膜,并且能保护肝细胞核DNA免疫受损,减少细胞凋亡的发生。临床显示对乙肝病毒有较好的抑制作用,安全性好,毒性小,无致突变性不良反应,适用于轻、中度慢性肝炎。临床显示远期疗效优于联苯双酯,已成为抗慢性病毒性肝炎的首选药物之一。 ★爱普列特 爱普列特是一种新型的5α-还原酶选择性抑制剂,1993年7月由中国药科大学、中科院上海有机化学研究所、扬州制药厂共同协作开发,1996年,爱普列特已批准进入临床研究阶段,1999年8月获得原SDA颁发的原料药及片剂新药证书与生产批件,2001年获得转正,现已成为联环药业的主导产品,享受12年的独家生产保护期。爱普列特能选择性抑制5α-还原酶,其抑制作用的选择性优于非那雄胺,不良反应小,是安全有效的药物,也是竞争性较强的治疗药品。 该药可抑制睾酮的转化过程,使前列腺体内双氢睾酮含量下降,导致增生的前列腺体萎缩,从而达到改善良性前列腺增生病人排尿困难等症状,具有安全可靠性。该药已列入《国家基本医疗保险药品目录》。 ★恩必普(简称NBP) 由中国医学科学院药物研究所和石家庄制药集团有限公司共同开发的,主要成分为丁苯酞,是我国心脑血管领域首个拥有自主知识产权的一类新药,获得两项国家专利。“恩必普”适用于缺血性脑卒中(俗称脑中风)的治疗,主要成分丁苯酞与从芹菜籽中提取的芹菜甲素结构相同。 “恩必普”按现行的注册管理办法可划分为1.1类,其原料药化学名丁苯酞,油状,最初从南方生的水芹菜及其籽中提取分离而得,现已实现化学合成。适用于缺血性脑卒中,石家庄制药集团恩必普药业生产,目前已上市。 ★血卟啉注射液 血卟啉是我国首创的一种光敏剂,属于一类新药,是治疗恶性肿瘤的新药,被誉为“光化导弹”。重庆华鼎现代生物制药于2003年获得原SDA颁发的生产批文,独家生产上市,商品名“喜泊分”。 药物通过静脉滴注进入人体后,随着血液循环到达恶性肿瘤组织并聚集和潴留在里面,然后用特定波长的激光照射这些肿瘤,其药物发生光化反应,产生一种单价氧,直接杀死恶性肿瘤组织,全过程对正常人体组织没有损伤。无论对原发癌症或复发癌症,可达到治愈或好转的目的,同时减少患者致残的等痛苦同时,该药也用于癌症的定位诊断和治疗,具有选择性强,对正常组织基本无损伤,副反应小的特点,临床用于癌症的定位诊断率达91%。 ★甘氨双唑钠 一类新药抗癌增敏剂甘氨双唑钠(CMNa)是我国自行设计、研制,具有自主知识产权的创新药物,2002年广州莱泰制药获得原SDA颁发的一类新药证书和生产批文,独家生产上市,商品名“希美纳”。
新药的自主研发过程 新药的自主研发过程指的是从新化合物的发现到新药成功上市的过程,其中包括以下四个步骤: ——通过计算机药物分子设计或通过植物、动物、矿物、微生物、海洋生物等各种途径获取新的化学物质,然后将这些物质在特定的体外或体内药理模型上进行筛选评价,以发现具有新颖结构类型和显著药理特性的先导化合物。 ——合成一系列与先导化合物结构类似的物质,进行构效关系研究,以优化化合物的治疗指数,选择一个最佳化合物作为临床候选药物。 ——提出新药临床研究申请,并严格遵循GCP进行Ⅰ、Ⅱ、Ⅲ期临床试验,如果证明新药是安全、有效、稳定的,则可申请注册。 ——新药的注册申请。 与新药自主研发相比,一种难度和风险都较小的研发方式——模仿创新已日益受到业内人士的关注。 模仿创新是一种渐进性创新活动。它以市场上已获得成功的率先研究者的创新思路和创新行为为榜样,并以其创新产品为示范,跟随它的思路,充分吸收率先者的成功经验和失败教训,并在此基础上对率先创新进行改进和完善,进一步开发和生产富有竞争力的产品参与竞争。 A. 相对于率先创新而言,模仿创新的优势主要体现在以下三方面: 第一,由于有现成的成功范例,模仿创新者可以从中学习到先进的方法并吸取失败的教训,因此降低了技术难度。 第二,曼斯费尔德曾对美国化工、医药、电子等行业的48项产品的率先创新和模仿创新的成本进行了比较,结果发现,模仿创新相对于率先创新而言,成本约为前者的65%,耗时约为前者的70%,研发成本较低。 第三,模仿创新在其产品市场占有率上与率先创新者基本相当。也有学者认为模仿创新者在市场份额、市场引导性方面具有一定的优势,主要原因可能是由于率先者产品的不完善及市场开发具有一定的难度影响了其一部分利益市场。 模仿创新之所以具有如此多的优势,其根本原因在于率先创新者的一部分利益溢出:如新思想对跟进者的启迪、创新中的经验教训以及对新市场的开辟等。模仿创新者可以无偿地拥有这一部分利益溢出,促进其自身创新活动的进行。 对于新药研发而言,模仿创新是在别人专利药物的基础上,以已知药物结构作为先导化合物进行化学结构修饰和改造,并通过系统的临床前及临床研究,获取自己的专利药,它不同于完全照抄他人化学结构的仿制药。同时,这种方法无需经历发现先导化合物这一过程,而且有可供借鉴的药理评价体系,目的性强,投资少,周期短,成功率高,目前已得到广泛的使用。据统计,1975年~1994年间,全世界共上市1061个新化学实体,其中属于模仿创新的共802个,占总数的76%。由此可见,模仿创新是国际上流行的一种后发优势明显的新药研发方法。 B、研发难度降低 新药研发是一项涉及到化学、生物学、药学、生理学、医学和经济学等学科的复杂的系统的创新活动。作为一项创新活动,它具有一定的累积性,也就是说:
中国新药上市为什么慢 2014-06-10 09:51 改革开放后,全世界绝大部分创新产品,只要不违反中国法律,均可以在第一时间自由地进入中国市场造福国人,只是在部分领域有配额和关税等方面的限制而已,但药品是个例外。 丘××,今年80岁,4年前被湖南长沙湘雅医院诊断为前列腺癌晚期,医生采用了常规的激素疗法,但病人出现了耐受,治疗失败。之后又采用常规化疗,依然没有效果,病情逐渐恶化,医生束手无策,病人似乎只能回家等待奇迹了。 一次闲聊中,医生提到美国刚刚批准了一种名为阿比特龙(Abiraterone)的新药,专门对付这种情况。阿比特龙是由美国美洲狮(Cougar)公司于上世纪90年代发现的,Ⅱ期临床试验证明此药对于激素治疗和常规化疗失败的前列腺癌疗效明显。2008年该公司开始了Ⅲ期临床试验,两年后研究人员对试验数据进行中期分析,发现疗效十分明显,试验组和对照组差异巨大。美国FDA得知情况后破例给予了优先审批通道,仅4个月即于 2011年4月批准了该药在美国上市。 最新的临床试验数据显示,阿比特龙的总体生存期比常规经典药物改善了将近5个月。要知道,此类病人的生存期通常仅有10个月左右,这就意味着病人的生存期提高了将近50%,这在目前已被批准的治疗前列腺癌的药物中是史无前例的。 2009年,著名的强生公司(Johnson & Johnson)将美洲狮公司买下,并于2012年正式向中国药监部门提交了上市申请,但截至发稿日,仍在等待药品审评中心进行技术审评。 与此同时,丘××的家人没有放弃,一直在积极寻找治疗机会。他们原打算从美国购药,但对方不但要求医院开出相应的处方,还要求病人去美国接受检查。考虑到路途遥远,以及不菲的开销,家属没有同意。此时传来利好消息,香港政府于2012年批准了阿比特龙,于是病人家属带着医生处方去香港医院买药。虽然海关的清关手续繁杂,还要交22%的进口税,甚至曾经遇到过被退关的情况,但不管怎样药终于买回来了。服药后丘老先生的病情得到了控制,各项指标均有大幅度改善,至今仍然健在。 丘老先生及其家人算是幸运的。很多像他这样的患者找不到境外买药的渠道,还有很多患者费尽精力去境外购药,结果买到的却是假药,最后人财两空。所以说,只有想办法让新药及早获取在我国的上市许可,才能真正解决像丘××这样的病人所面临的窘境。 这种案例并不罕见,很多人都听说过甚至经历过类似的事情。通常情况下,一种外国商品被限制进口,主要原因是为了保护国内的相关产业,但药是一种很特殊的商品,
化学药品注册分类 一、注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 中药、天然药物注册分类 本附件中的中药是指在我国传统医药理论指导下使用的药用物质及其制剂。 本附件中的天然药物是指在现代医药理论指导下使用的天然药用物质及其制剂。 一、注册分类及说明 (一)注册分类 1.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。 2.新发现的药材及其制剂。 3.新的中药材代用品。 4.药材新的药用部位及其制剂。 5.未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。 6.未在国内上市销售的中药、天然药物复方制剂。 7.改变国内已上市销售中药、天然药物给药途径的制剂。 8.改变国内已上市销售中药、天然药物剂型的制剂。 9.仿制药。 (二)说明
17款1类新药获批临床,PD-1、THR-β、KRAS 截止2020年8月14日,中国国家药监局药品审评中心(CDE)数据显示,共计有2537项临床试验申请通过“默示许可”。上周新增62项临床试验默示许可,其中有33项1类创新药申请(不含补充申请),涉及17款药物,来自恒瑞医药、辉瑞(Pfizer)、信达生物、诺华(Novartis)、赫普化医药等公司。 1、药物名称:HPN-01肠溶胶囊药物机制/靶点:未披露公司:赫普化医药适应症:治疗非酒精性脂肪性肝炎(NASH) 2、药物名称:ABI-H2158片药物机制/靶点:HBV核心抑制剂类药物公司:Assembly Biosciences、艾杉贝瑞生物适应症:慢性乙型肝炎 3、药物名称:脯氨酸恒格列净片药物机制/靶点:钠-葡萄糖转运蛋白2(SGLT2)抑制剂公司:恒瑞医药适应症:联合厄贝沙坦治疗合并高血压的2型糖尿病肾病 4、药物名称:NHWD-870 HCl片药物机制/靶点:BET抑制剂公司:恒雅医药、文达医药适应症:暂定难治或复发非霍奇金淋巴瘤、皮肤/黏膜黑色素瘤、非小细胞肺癌、小细胞肺癌 5、药物名称:PF-06801591注射液药物机制/靶点:PD-1公司:辉瑞
适应症:高危非肌层浸润性膀胱癌 6、药物名称:信迪利单抗注射液药物机制/靶点:PD-1公司:信达生物适应症:晚期或转移性鳞状非小细胞肺癌 7、药物名称:Finerenone片药物机制/靶点:盐皮质激素受体拮抗剂公司:拜耳医药(Bayer)适应症:治疗左心室射血分数≥40%的心力衰竭 8、药物名称:雷珠单抗注射液药物机制/靶点:VEGF公司:诺华制药适应症:用于治疗中重度至重度增殖性糖尿病视网膜病变(PDR)9、药物名称:ASC41片药物机制/靶点:THR-β激动剂公司:甘莱制药适应症:治疗成人非酒精性脂肪性肝炎 10、药物名称:注射用重组人源化抗HER2单抗-AS269偶联物(ARX788)药物机制/靶点:HER2公司:浙江医药、新码生物适应症:HER2阳性表达的晚期乳腺癌 11、药物名称:AST-3424注射液药物机制/靶点:广谱靶向药公司:艾欣达伟适应症:适用于复发或难治性急性淋巴细胞白血病患者。 12、药物名称:BI 1701963片药物机制/靶点:泛KRAS抑制剂公司:
新药临床研究与新技术的应用 一、单选题(每题只有一个最佳选项,请选出最符合题意的答案) 1. 临床适应性设计的类型主要包括 ( B )种 A8条B 10条C 12条D 6条 2. 临床适应性设计的类型不包括( C ) A 适应性随机化 B 样本量重估 C、适应性生物等效 D 适应性治疗转换 3.0 期试验研究三种类型不包括( B ) A 用于评价分子靶向抗肿瘤药物对人体肿瘤和(或)替代组织的药效学作用,验证非临床研究模型中发现的作用机制 B 用修正的药理学与毒理学研究结果来确定临床起始剂量或剂量爬坡 C 比较研究两种以上作用于相同靶点的结构类似物的异同 D 研究适用于开发新型的显影探针或显影技术,用于评价药物在人体内的生物分布、组织结合及靶向作用等 4.ADMET(或ADMETox) 实验, 不包含下列哪项( D) A 吸收实验 B 分布实验 C 代谢实验 D 药效实验 5. 粉末喷射给药系统的主要制约因素是所用剂量必须小于( B) A 5mg B 6mg C 7mg D 8mg 二、多选题(每题有一个或一个以上最佳选项,请选出最符合题意的答案) 1.新药临床研究的适应性设计与传统设计比较具有( ABCD )等优点。 A 设计灵活 B 节约成本 C 缩短研发时间 D 加快新药上市 2. 通过 0 期试验,新药开发者可以实现以下目标( ABCD ) A 验证临床前研究中发现的药物作用机制是否在人体上同样适用 B 提供重要的药代动力学(pharmacokinetics,PK)信息 C 从候选药物中确定最有希望的先导化合物 D 探索药物的生物分布特征。
3. 自动化分析在AD METox实验中的应用( ABD ) A 离子通道药物 B 小肠上皮细胞系对药物的吸收与排泄 C 靶向药物筛选 D P45O酶代谢和细胞凋亡 4. 智能化筛选包含(ABCDE) A 微流体操作系统 B 智能化数据 C 信号检测 D 微实验室系统 E 体内检测 5. 新型给药系统(DDS) ( ABCDE ) A 缓控释给药系统 B 靶向给药系统 C 纳米给药系统 D 透皮给药系统 E 生物黏附给药系统 我国药品价格管制政策 一、单选题(每题只有一个最佳选项,请选出最符合题意的答案)” 1、药品价格管制的主体是( C ) A 药品监督管理局 B 发展改革委员会 C 政府 D企业 2、以下哪种价格规制方法是传统规制阶段的主要方法( A ) A成本加成 B价格上限 C利润分享 D成本调整契约 3、我国的国家零售指导价属于( B ) A投资回报率管制 B价格上限管制 C利润分享计划 D成本调整契约管制 4、医院制剂的原料损耗,西药最高不得超过( D ) A 20% B 15% C 16% D 5% 5、我国的医院药品加成政策最早在设立于( A )年 A 1954 B 1996 C 2000 D 2006 二、多选题(每题有一个或一个以上最佳选项,请选出最符合题意的答案) 1、药品价格管制的理论依据是( ABC ) A外部性 B垄断性 C信息不对称 D政府意志
jjswcqz资料 本新药上市申请临床评价指南,旨在为评价首个在中国注册上市的新处方药提供一个模板化的科学指导。 本模板是结构化的。它把新药上市所必须开展的有效性、安全性评价的循证逻辑链加以结构化,并在各部分的导语中把各个结构单元采集、分析数据和相关信息的要求以及相关的关系加以明确。同时,指南对评价中所使用的相关术语和标准的定义也提出了明确的要求。 本模板是在参考了ICH的相关指南和欧美有关评价机构的临床审评指南后撰写的。申请人可以按照ICH的CTD文档的要求提供相关的临床研究资料。了解并理解这一模板,有助于申请人理解评价机构在分析、采信申请人所提供的临床研究数据和资料时,所遵循的评价逻辑。对于临床研究者和其他从事临床研究的人员而言,了解这一模板对于设计某一临床研究方案、系统地构建或完善某一新药的临床开发路径也是有益的。 本模板对于评价机构的临床审评遵循质量、效率、清晰、一致、透明和可预见的《审评质量管理规范》(Good Review Practice)是十分重要的。新药的临床研发与评价是一个十分复杂的系统。对这一复杂的系统,必须有伦理的要求、科学条件的定义、相关专业的要求及逻辑结构的限定,才能克服临床研发进程中出现的偏倚,规避以个体化经验去理解和评价药物的有效性、安全性的风险。 本模板是结构化的。它提供了不同疾病领域在探索其未被满足的临床需求时所遵循的共同原则。各疾病领域可保留模版适度的灵活性,为拓展和完善与适应症匹配的临床评价模板和/或要点提供了空间。 本模板对评价中所使用的数据源提出了要求。由于临床研究的全球化以及研究进程中分布的不均衡,全面、科学、准确地分析评估不同的数据源对审评提出了更高的要求。 临床审评的学习、培训与实践,应建立在本模板以及其所约定内容的概念、方法的准确理解和掌握之上。
国家级一类新药,国家科委生命科学技术发展中心1035工程重大项目成果,国家自然科学基金重大项目成果绿色植物性药品,具有出色的安全性单一体结构,同时具有多种药理作用,全面治疗缺血性脑卒中明显减少梗塞后神经功能缺失、改善患者生活能力状态。 全面的药理作用缩小脑梗塞面积、改善神经功能缺失改善局部脑血流量,改善脑微循环改善脑缺血能量代谢耗竭减轻脑缺血所致脑水肿抗血栓形成和抗血小板的聚集改善脑缺血记忆障碍有针对性的适应症:急性缺血性脑卒中显著的临床疗效改善脑缺血区微循环,增加局部脑血流;明显减轻缺血性脑水肿;改善能量代谢;明显缩小脑缺血的梗塞面积;无需分期,全程安全应用。 【英文名】Butylphthalide Soft Capsules 【汉语拼音】Dingbentai Ruanjiaonang 【主要成分】本品成份为丁苯酞,其化学名称:消旋-3-正丁基苯酞(简称丁苯酞或记作NBP)。 【化学结构式】※[图片:5169.BMP]※ 【分子式】C12H14O2 【分子量】190.24 【性状】本品为软胶囊,内容物为淡黄色或黄色油状液体。 【药理毒理】药理作用: 丁苯酞为人工合成的消旋正丁基苯酞,与自芹菜籽中提取的左旋芹菜甲素的结构相同。临床研究结果显示,丁苯酞(与丹参注射液静脉滴注联合应用)对急性缺血性脑卒中患者中枢神经功能的损伤有改善作用,可促进患者功能恢复。动物药效学研究显示,丁苯酞可阻断缺血性脑卒中所致脑损伤的多个病理环节,具有较强的抗脑缺血作用,可明显缩小大鼠局部脑缺血的梗塞面积,减轻脑水肿,改善脑能量代谢和缺血脑区的微循环和血流量,抑制神经细胞凋亡,并具有抗脑血栓形成和抗血小板聚集的作用。丁苯酞可能通过降低花生四烯酸含量,提高脑血管内皮NO和PGI2的水平,抑制谷氨酸释放,降低细胞内钙浓度,抑制自由基和提高抗氧化酶活性等机制发挥上述药效作用。毒理研究重复给药毒性:大鼠经口给药 120mg/kg、250mg/kg、500mg/kg,连续6个月,血糖(各剂量组)、胆固醇(高、中剂量组)明显高于对照组,停药后可恢复正常。犬经口连续给药6个月,剂量分别为80、500mg/kg/天,高剂量组动物体重增加缓慢,肝脏明显增大,肝细胞空泡样肿大,血液碱性磷酸酶活性明显增加,停药后上述表现恢复正常;小剂量组动物仅出现唾液分泌增加。遗传毒性:丁苯酞Ames试验、中国仓鼠肺细胞(CHL)染色体畸变试验及小鼠微核试验结果均为阴性。生殖毒性:一般生殖毒性试验中,雌、雄动物经口给药(雌性动物为交配前给药二周,受孕后继续给药15天,雄性动物连续给药8周)80mg/kg、200mg/kg和500mg/kg,结果对亲代动物生育力无明显影响,未表现出明显胚胎毒性和致畸作用,仅高剂量组动物摄水量明显增加,给药后前几天出现流涎、爬伏症状。围产期生殖毒性试验中,经口给药80mg/kg、200mg/kg和500mg/kg,结果高剂量组动物出现妊娠期延长趋势,其中一只孕鼠难产(剖检为死胎),少数动物无乳汁分泌,并出现仔鼠(4日龄)存活率下降,仔鼠(4日至3周龄)体重明显下降;高剂量组F1 代大鼠斜板试验和悬垂试验分数降低(反映协调平衡能力),F2代仔鼠骨骼发育有一定延迟;中、低剂量组未见明显影响。 【药代动力学】1、人体药代动力学中国健康男性受试者单次口服不同剂量丁苯酞软胶囊的I期药代动力学研究: 受试者 分为3组,分别口服丁苯酞软胶囊100mg、200mg、400mg,于给药前(0小时)和给药后0.25、0.5、0.75、1、1.5、2、2.5、3、4、6、8、12、24采集血样进行血药浓度测定,结果:100mg、200mg剂量组中,均有一些受试者在末端的一个或多个采样点样品的血药浓度在检测限以下。在进行T1/2统计时,100mg组、200mg组、400mg组分别有3个、8个、12个完整受试者。丁苯酞血浆浓度平均达峰时间分别为0.88,1.25和1.25小时;平均峰浓度分别为78.7±115.8,204.7±149.0和 726.6±578.7ng/ml;平均消除半衰期分别为12.46±2.50,11.84±4.09,7.52±1.32小时;平均AUC0-t分别为93.2±114.0,323.8±201.0,1314.2±965.7ng·hr/ml。餐后给予200mg丁苯酞,达峰时间从约1小时推迟至约4小时;达峰浓度从204.7ng/ml 降至67.0ng/ml;平均AUC0-t和AUC0-∞分别从323.8ng·hr/ml和460.5ng·hr/ml减少到136.8ng·hr/ml和193.6ng·hr/ml。空 腹和餐后给予200mg丁苯酞在Tmax、Cmax、AUC0-∞均有统计学显著差异(P<0.05),说明食物影响丁苯酞的吸收。中国健康男性受试者多次口服丁苯酞软胶囊的I期药代动力学研究: 每天口服四次丁苯酞软胶囊,每次200mg,共服药13次,结果显示:
中国百强药企的新药研发思路 内容来源:医药经济报 导读: 制药行业的产品力主要源于创新和研发的积累和突破。除了华药、哈药、石药这样的传统大型国有制药企业,以及华润、复星这样的资本巨鳄,大部分制药企业的发展都离不开其核心品种的成功。比如复方丹参滴丸之于天士力,心血康之于地奥。 在全球范围内,近年来发展最快的制药企业如Roche、BMS、Celgene、Gilead、Biogen等,年均研发投入都不低于营销收入的15%。对比中国制药企业,虽然有所进步,但整体而言研发投入依然远低于西方水平。据CFDA南方医药经济研究所的报告,2012年制药行业的平均研发投入仅占销售收入的1.62%。相对于行业平均水平,百强企业的研发投入强度明显较高,一些以创新为核心的企业,研发投入强度达到了6%以上。这样也形成了一个良性循环:百强企业因为收入较高,可以提供较高研发投入以提升企业的产品力,产品力的提升则能带来更高的销售收入。对于中小企业而言,多数由于经费有限,研发投入不可能太高,那就必须把有限的经费用在刀刃上。借鉴百强企业的新品研发思路,无疑有利于选择合适的研发方向。不过,相比于研发投入,企业似乎更有兴趣将投入用于销售。笔者查看48家在内地上市的百强企业发现,总体来看,销售费用占营业收入的比例高达23.2%,研发投入仅占3.9%。上市企业由于信息披露较为公开,更具参考价值。根据CFDA南方医药研究所的中国制药工业百强榜,上市公司已经占据了绝大多数。因此,我们筛选出部分国内上市公司、核心业务为药品(不含原料药)、研发投入强度超过6%的百强企业。根据公开数据,梳理其新药研究进展,分析其研发思路。 海正药业 研发投入强度:14.9% 特点:以西方投入水平支持转型 海正药业曾是一家优秀的外向型原料药企。不过近年来,海正逐步向“研发营销型”转型。其2013年研发投入强度14.9%,接近西方药企水平。较高的研发投入增强了产品竞争力,2013年海正制剂业务销售额增长50%,达到15亿元。
新药上市申请临床评价指南(草案) 本新药上市申请临床评价指南,旨在为评价首个在中国注册上市的新处方药提供一个模板化的科学指导。 本模板是结构化的。它把新药上市所必须开展的有效性、安全性评价的循证逻辑链加以结构化,并在各部分的导语中把各个结构单元采集、分析数据和相关信息的要求以及相关的关系加以明确。同时,指南对评价中所使用的相关术语和标准的定义也提出了明确的要求。 本模板是在参考了ICH的相关指南和欧美有关评价机构的临床审评指南后撰写的。申请人可以按照ICH的CTD文档的要求提供相关的临床研究资料。了解并理解这一模板,有助于申请人理解评价机构在分析、采信申请人所提供的临床研究数据和资料时,所遵循的评价逻辑。对于临床研究者和其他从事临床研究的人员而言,了解这一模板对于设计某一临床研究方案、系统地构建或完善某一新药的临床开发路径也是有益的。 本模板对于评价机构的临床审评遵循质量、效率、清晰、一致、透明和可预见的《审评质量管理规范》(Good Review Practice)是十分重要的。新药的临床研发与评价是一个十分复杂的系统。对这一复杂的系统,必须有伦理的要求、科学条件的定义、相关专业的要求及逻辑结构的限定,才能克服临床研发进程中出现的偏倚,规避以个体化经验去理解和评价药物的有效性、安全性的风险。 本模板是结构化的。它提供了不同疾病领域在探索其未被满足的临床需求时所遵循的共同原则。各疾病领域可保留模版适度的灵活性,为拓展和完善与适应症匹配的临床评价模板和/或要点提供了空间。 本模板对评价中所使用的数据源提出了要求。由于临床研究的全球化以及研究进程中分布的不均衡,全面、科学、准确地分析评估不同的数据源对审评提出了更高的要求。 临床审评的学习、培训与实践,应建立在本模板以及其所约定内容的概念、方法的准确理解和掌握之上。
一、背景资料 GCP:Good Clinical Practice的缩写,中文意为“药物临床试验质量管理规范”。我国现行的GCP法规于2003年8月6日发布,并于2003年9月1日起正式施行。 1、GCP诞生的历史背景 GCP的概念产生于70年代,主要源于: ?滥用人类受试者 ?随意进行临床试验 ?不重视受试者的权益 ?发生严重不良事件 让我们来了解两起著名的严重不良事件 1938年的万灵磺胺事件 1937年秋天,美国田纳西州的马森吉尔药厂,未经有关政府部门批准,采用工业溶剂二甘醇代替酒精,生产出一种磺胺酏剂,用于治疗感染性疾病。到这一年9-10月间,美国南方一些地方开始发现患肾功能衰竭的病大量增加。调查证明这种情况与该公司生产的磺胺酏剂有关,共发现358名病人,死亡107人。 20世纪最大的药物灾难:60年代的“反应停”事件 20世纪50年代后期原联邦德国格仑南苏制药厂生产了一种声称治疗妊娠反应的镇静药Thalidomide (又称反应停、沙利度胺、肽咪哌啶酮)。实际上这是一种10 0%的致畸胎药。该药出售后的6年间,先后在原联邦德国、澳大利亚、加拿大、日本以及拉丁美洲、非洲的共28个国家,发现畸形胎儿12000余例(其中西欧就有6 000~8000例,日本约有1000例)。患儿无肢、短肢、肢间有蹼、心脏畸形等先天性异常,呈海豹肢畸形(phocornelia)。 严重不良事件的发生,使世界各国政府认识到有必要通过立法,要求药品上市前必须进行临床试验,以评价其安全性和有效性。 2、GCP的制订 国际上: ?1980年美国最早提出GCP ?1981年欧共体制定GCP ?1992年欧共体颁发GCP指南 ?1994年WHO-GCP指南发布 ?1996年ICH-GCP发布 国内:
新药临床研究的基本要求 从1999年5月开始实施的《新药审批办法》,到2002年12月实施的《药品注册管理办法》(试行),都规定了,在我国,新药需要经过国家药品监督管理局批准后方可进行临床研究或生产上市。其宗旨就是为了保证人民用药的安全有效。当完成临床研究后,结合临床研究期间对药学及药理毒理工作完善的情况,经过审评认为可以保证临床用药的安全有效性后,国家药品监督管理局将发给申报单位新药证书及生产,许可生产上市。本人结合新药研发的规律、临床研究的系统性,将对临床研究的基本要求,临床试验的目的、内容、设计和结果评价进行系列阐述。 新药临床研究的基本要求包括了,新药临床研究前需具备的药学及药理毒理研究工作的基础,临床研究方案的设计,以及实施临床研究的所要遵循的法律、法规等要求。 一个新的化合物是否能开发为药品上市,要经历许多阶段,其中必须经过临床研究来确定其疗效和安全性,同时临床研究也是在整个开发过程中所占时间最长、资金最多,对一个新药的上市起决定性的作用。通过小人群的研究,保证更大人群应用的安全、有效,避免药害事件的发生。因此临床研究在新药开发中占有非常中要的地位。 进行临床研究一定要具备充分的药学工作及药理毒理工作的基础。 药学工作是新药研究的最基本工作,在进行临床研究以前首先应该作到新药化学结构或组分确证,制备工艺相对稳定,制剂处方固定,质控方法可行,以便对产品质量进行控制,保证试制样品质量的一致性和进入临床试验后的安全性,同时必须具有一定的化学稳定性,以保证在临床研究期间的稳定。由于药物开发的阶段性,药学方面的研究工作是不可能在临床前彻底完成的。对这一阶段存在的问题如何看待,需要综合考虑。在此阶段存在的问题如果不会影响到临床用药的安全有效性,那么这些完善工作可以在临床研究期间完成,甚至在产品上市后进一步研究完善。 作为一个化合物如果要作为药物用于人类,必须要有一定的动物研究的结果,才符合伦理学的原则。因此动物研究的目的就是为保证化合物初次用于人体的安全有效性,动物试验应显示主要的药效作用和毒性以及药代动力学特性,并根据动物试验的结果能够为临床试验推荐适应症、计算进入人体试验的安全剂量。应该重点关注动物的毒性实验结果,毒性试验的资料应显示药物对靶器官毒性作用的性质、剂量依赖性、毒性与药物体内暴露程度(药物在靶器官的浓度和作用时间)的关系以及靶器官毒性的潜在可逆性,这些资料对估算人体试验的最初剂量或确定潜在临床不良反应的监测指标都很重要。虽然在临床试验初期,非临床资料量相对有限,可能并不完全,但这些资料应充分阐明在其所支持的不同阶段临床研究情况下药物可能出现的潜在毒性特征。也就是为临床试验提示需要关注的注意观察不良反应的脏器器官。 由于临床研究方案设计的好坏往往决定了临床试验的结果是否能够或足以评价新药的安全有效性,所以应对方案设计引起足够的重视。临床研究方案是临床研究的重要文件,应该由申办者与主要研究者共同讨论制定。方案的制定必须符合我国的相关法规。 临床研究必须有明确的目的,方案中必须阐明试验设计方法、病例数、适应症、用法用量、对照药的合理性、疗程、疗效判断标准等,还应包括病例观
2015 年美国FDA批准上市的45种新药【小分子药物&生物制品】 导读 2015年,FDA一共批准了45种新药,这是自1996年以来被批准最多的一年,1996年共有53种新药获得了FDA的批准。按照治疗领域分类,其中有14款为抗肿瘤药物,9种心血管系统药物、3种抗精神药物、2种抗病毒药物、1种抗生素、1种抗真菌药物、1种治疗糖尿病药物、1种呼吸系统药物及13种其他治疗药物。按照药物的类型分,其中13种为生物制品,32种为化学小分子药物。 2015年,FDA一共批准了45种新药,这是自1996年以来被批准最多的一年,1996年共有53种新药获得了FDA的批准。
按照治疗领域分类,其中有14款为抗肿瘤药物(只有3个药物未被授予突破性治疗药物资格或一些其它的用来加快审评程序的资格),9种心血管系统药物、3种抗精神药物、2种抗病毒药物、1种抗生素、1种抗真菌药物、1种治疗糖尿病药物、1种呼吸系统药物及13种其他治疗药物。 其中有21个属于罕见病药物,它们中有12个新分子实体药物,9个生物制品类药物。这21个罕见病药物的获批再次见证了《罕见病药物法案》对全球制药巨头增强罕见病药物的研发兴趣和动力起到了积极作用。 按照药物的类型分,其中13种为生物制品,32种为化学小分子药物。以下是2015年FDA 的45种新药介绍(按照批准时间点排序)。 1.Savaysa(edoxaban)——抗凝血药物 2015年1月8日,FDA批准了第一三共株式会社的抗凝血药物Savaysa(edoxaban),化合物专利为WO 03000657A1(2002 年3 月20日),用于降低非心脏瓣膜病引起的房颤患者卒中和危险血栓(系统性栓塞)风险,其预期销售峰值将达2.2亿美元。 2.Cosentyx(secukinumab)——银屑病
[作者简介] 谭芬来,男,博士,主要从事分子医学研究工作。联系 电话:(010)67869692,E 2mail:tanfenlai@hot m ail .com 。 ?新药述评? 国家一类新药盐酸埃克替尼的药理与临床评价 谭芬来1 ,张 力2 ,赵 琼3 ,刘东阳4 ,胡云雁1 ,刘 勇1 ,丁列明1 ,胡 蓓4 ,王印祥 1 (1浙江贝达药业,杭州311100;2北京协和医院呼吸科,北京100730; 3浙江大学医学院附属第一医院呼吸科,杭州310003;4北京协和医院临床药理基地,北京100730) [摘要] 盐酸埃克替尼(icotinib hydr ochl oride,克美纳T M ,浙江贝达药业有限公司生产,规格:100,125,150,200mg )是一种新型口服表皮生长因子受体酪氨酸激酶抑制剂(EGFR 2TKIs ),临床用于治疗既往接受过1种或2种化疗的局部晚期或转移的非小细胞肺癌。文中对盐酸埃克替尼在中国的I ~II a 期临床试验进行 了总结,并对其药理作用、药动学、临床和安全性评价进行了综述。 [关键词] 盐酸埃克替尼;非小细胞肺癌;表皮生长因子受体酪氨酸激酶抑制剂;药理学;药动学 [中图分类号]R983;R979.1 [文献标志码]A [文章编号]1003-3734(2009)18-1691-04 Pharmacology and cli n i ca l eva lua ti on of i coti n i b hydrochlor i de T AN Fen 2lai 1 ,ZHANG L i 2 ,Z HAO Q i ong 3 ,L IU Dong 2yang 4 ,HU Yun 2yan 1 , L IU Yong 1 ,D ING L ie 2m ing 1 ,HU Bei 4 ,WANG Yin 2xiang 1 (1Zhejiang B eta Phar m a,Hangzhou Zhejiang 311100,Ch ina;2D epart m ent of R espiratory,Peking U nion M ed ical College Hospita l,B eijing 100730,Ch ina;3D epart m ent of R espiratory,The F irst A ffiliated Hospital of College of M edicine,Zhejiang U niversity,Hangzhou 310003,China ;4C linical Pha r m acology R esearch Center , Peking U nion M edica l College Hospital,B eijing 100730,China ) [Abstract] I cotinib hydr ochl oride is a novel oral ep ider mal gr owth fact or recep t or 2tyr osine kinase inhibit or (EGFR 2TKI ).It is indicated f or the treat m ent of patients with l ocally advanced or metastatic non 2s mall cell lung cancer (NSCLC )p revi ously treated with one or t w o che motherapy .Several phase I and II a trials in both healthy vol 2unteers and NSCLC patients have been conducted in China recently .I n this revie w we summarized the brief results fr om these trials,the phar macol ogy and phar macokinetics of icotinib hydr ochl oride . [Key words] icotinib hydr ochl oride;non 2s mall cell lung cancer (NSCLC );ep ider mal gr o wth fact or recep 2t or 2tyr osine kinase inhibit or (EGFR 2TKI );phar macol ogy;phar macokinetics 肺癌是全世界范围内各种恶性肿瘤中导致死亡 最常见的疾病。据世界卫生组织估计,我国每年新发现的肺癌患者超过40万人,其中约317000(80%)为非小细胞肺癌(NSCLC ),死亡病例约272000例。以铂类为基础的化疗是目前全世界范 围内普遍采用的一线治疗方案。一线化疗后复发或 进展的患者需进行二线治疗。目前国际上普遍推荐的二线化疗方案有多西紫杉醇(docetaxol,泰索帝)、 培美曲塞(pe metrexed,力比泰)或表皮生长因子受体酪氨酸激酶抑制剂(Ep ider mal Gr owth Fact or Re 2cep t or 2Tyr osine Kinase I nhibit ors,EGFR 2TKIs ),如吉 非替尼(gefitinib )和厄洛替尼(erl otinib )。其中多西紫杉醇、培美曲塞属于细胞毒类,不良反应较大;厄洛替尼和吉非替尼属靶向抗癌药,不良反应明显低于多西紫杉醇和培美曲塞。 盐酸埃克替尼(icotinib hydr ochl oride,商品名CONMANAT M ,克美纳 T M ,浙江贝达药业有限公司生 产,规格:100,125,150,200mg )是一种口服EGFR 2TKIs,属国家化药“1.1”类新药,化学结构和阿斯利
新药临床研究分期 新药的临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ、期。 Ⅰ期临床试验:在新药开发过程中,将新药第一次用于人体以研究新药的性质的试验,称之为Ⅰ期临床试验.即在严格控制的条件下,给少量试验药物于少数经过谨慎选择和筛选出的健康志愿者(对肿瘤药物而言通常为肿瘤病人),然生仔细监测药物的血液浓度\排泄性质和任何有益反应或不良作用,以评价药物在人体内的性质.Ⅰ期临床试验通常要求健康志愿者住院以进行24小时的密切监护.随着对新药的安全性了解的增加,给药的剂量可逐渐提高,并可以多剂量给药.通过Ⅰ期临床试验,还可以得到一些药物最高和最低剂量的信息,以便确定将来在病人身上使用的合适剂量.可见,Ⅰ期临床试验的目的是通过初步的临床药理学及人体安全性评价试验。观察人体对于新药的耐受程度和药物代谢动力学,为制定给药方案提供依据。Ⅱ期临床试验:通过Ⅰ期临床研究,在健康人身上得到了为达到合理的血药浓度所需要的药品的剂理的信息,即药代动力学数据.但是,通常在健康的人体上是不可能证实药品的治疗作用的. 在临床研究的第二阶段即Ⅱ期临床试验,将给药于少数病人志愿者,然后重新评价药物的药代动力学和排泄情况.这是因为药物在患病状态的人体内的作用方式常常是不同的,对那些影响肠、胃、肝、和肾的药物尤其如此。以一个新的治疗关节炎的止通药的开发为例。Ⅱ期临床研究将确定该药缓解关节炎病人的疼通效果如何,还要确定在不同剂量时不良反应的发生率的高低,以确定疼痛得到充分缓解但不良反应最小的剂量。可以说,Ⅱ期临床试验是对治疗作用的初步评价阶段。Ⅱ期临床试验一般通过随机盲法对照试验(根据具体目的也可以采取其他设计形式),对新药有效性及安全性作出初步评价,推荐临床给药剂量。 III期临床试验:在Ⅰ、Ⅱ期临床研究的基础上,将试验药物用于更大范围的病人志愿者身上,遵循随机对照原则,进行扩大的多中心临床试验,进一步评价药物的有效性和耐受性(或安全性),称之为Ⅲ期临床试验。Ⅲ期临床试验可以说是治疗作用的确证阶段,也是为药品注册申请获得批准提供依据的关键阶段,该期试验一般为具有足够样本量的随机化盲法对照试验。临床试验将对试验药物和安慰剂(不含活性物质)或已上市药品的有关参数进行比较。试验结果应当具有可重复性。可以说,该阶段是临床研究项目的最繁忙和任务最集中的部分。除了对成年病人研究外,还要特别研究药物对老年病人,有时还要包括儿童的安全性。一般来讲,老年病人和危重病人所要求的剂量要低一些,因为他们的身体不能有产地清除药物,使得他们对不良反应的耐受性更差,所以应当进行特别的研究来确定剂量。而儿童人群具有突变敏感性、迟发毒性和不同的药物代谢动力学性质等特点,因此在决定药物应用于儿童人群时,权衡疗效和药物不良反应应当是一个需要特别关注的问题。在国外,儿童参加的临床试验一般放在成人试验的Ⅲ期临床后才开始。如果一种疾病主要发生在儿童,并且很严重又没有其他治疗方法,美国食品与药品管理局允许Ⅰ期临床试验真接从儿童开始,即在不存在成人数据参照的情况下,允许从儿童开始药理评价。我国对此尚无明确规定。 Ⅳ期临床试验:新药上市后监测。在广泛使用条件下考察疗效和不良应(注意罕见不良应)。
中国一类新药汇总 按: 二年前,根据自己平时的工作和信息积累摘编整理了一份中国新药汇总, 没想到这份很不完整的新药名录被到处转载,臭名远扬. 暂且不说这份资料有权威性和准确性的问题, 这只是鄙人的工作笔记.对多数人而言没有多大价值. 但可气的是好几个网站,包括生物谷竟然声称是该网站的原创作品,一经转贴就成了版权所有, 真让人怀疑该网站究竟有多少作品是原创,有多少真假李逵活跃于各网站豪取掠夺. 知识是全人类的,信息有许多共享的. 在尊重知识产权和版权及客观事实的基础上,许多东西是可以交换或互惠交易的. 今天再次把原贴公布于众,一则希望不断有更新,二则以正视听,让兄弟网站的网友了解并非自我标榜的原创作品都是物真价实. 中国一类新药汇总 介绍部分国内研究开发的一类新药, 还有20多种正在临床试验的新药,( NCE 或中药), 希望各位添加补充其他新药信息. ★今又生(P53注射液) 全球第一个基因治疗产品(P53注射液),据了解,已有5000多人接受过治疗,但尚未在国际知名杂志发表临床试验论文(综述除外)它的研制过程: 第一阶段:Ⅰ期临床阶段。1998年3月9日,深圳市赛百诺基因技术有限公司成立;1998年12月28日,国家药品监督管理局批准重组人p53腺病毒注射液进入Ⅰ期临床试验。 第二阶段:Ⅱ期临床阶段。2000年10月7日,国家药品监督管理局批准重组人p53腺病毒注射液进入Ⅱ期临床试验;2000年12月,基因治疗制品生产厂房在深圳建成;2002年7月3日,获得国家知识产权局颁发的生产工艺发明专利;2002年11月22日,获得广东省药品监督管理部门颁发的药品生产许可证。 第三阶段:获得新药证书和正式上市阶段。2003年10月16日,重组人p53腺病毒注射液经国家食品药品监督管理局批准,获得新药证书。2004年1月20日,经批准获得准字号生产批文。 ★双环醇片(百赛诺Bicyclol) 商品名“百赛诺”,是由中国医学科学院、中国协和医科大学药物所开发的产品,是中国第一个具有自主知识产权一类抗肝炎合成创新药物。1996年12月经卫生部批准进入临床试验,2001年9月获得原SDA颁发的新药证书及生产批准文,由北京协和药厂生产上市。目前已在世界16个国家地区获得20年发明专利保护,国内享有12年的行政保护期。本品是在降酶药物联苯双酯和五味子基础上,经过化学合成筛选的药物,其良好的双重机制,能清除自由基作用以保护细胞膜,并且能保护肝细胞核DNA免疫受损。临床显示对乙肝病毒有较好的抑制作用,安全性好,毒性小,无致突变性不良反应,适用于轻、中度慢性肝炎。临床显示远期疗效优于联苯双酯,有望成为抗慢性病毒性肝炎的首选药物之一。