
2006年新车间杆菌肽洗涤过滤干燥机
清洗验证方案
文件编号:三合一清洗AmB-PV-F
1 目的
通过清洗验证,验证杆菌肽洗涤过滤干燥机清洗操作的适用性及有效性,确认按照清洗操作规程操作,设备上的杆菌肽的残留达到了规定的清洁限度要求,不会对下批或更换品种后的生产造成交叉污染。
2 系统简介
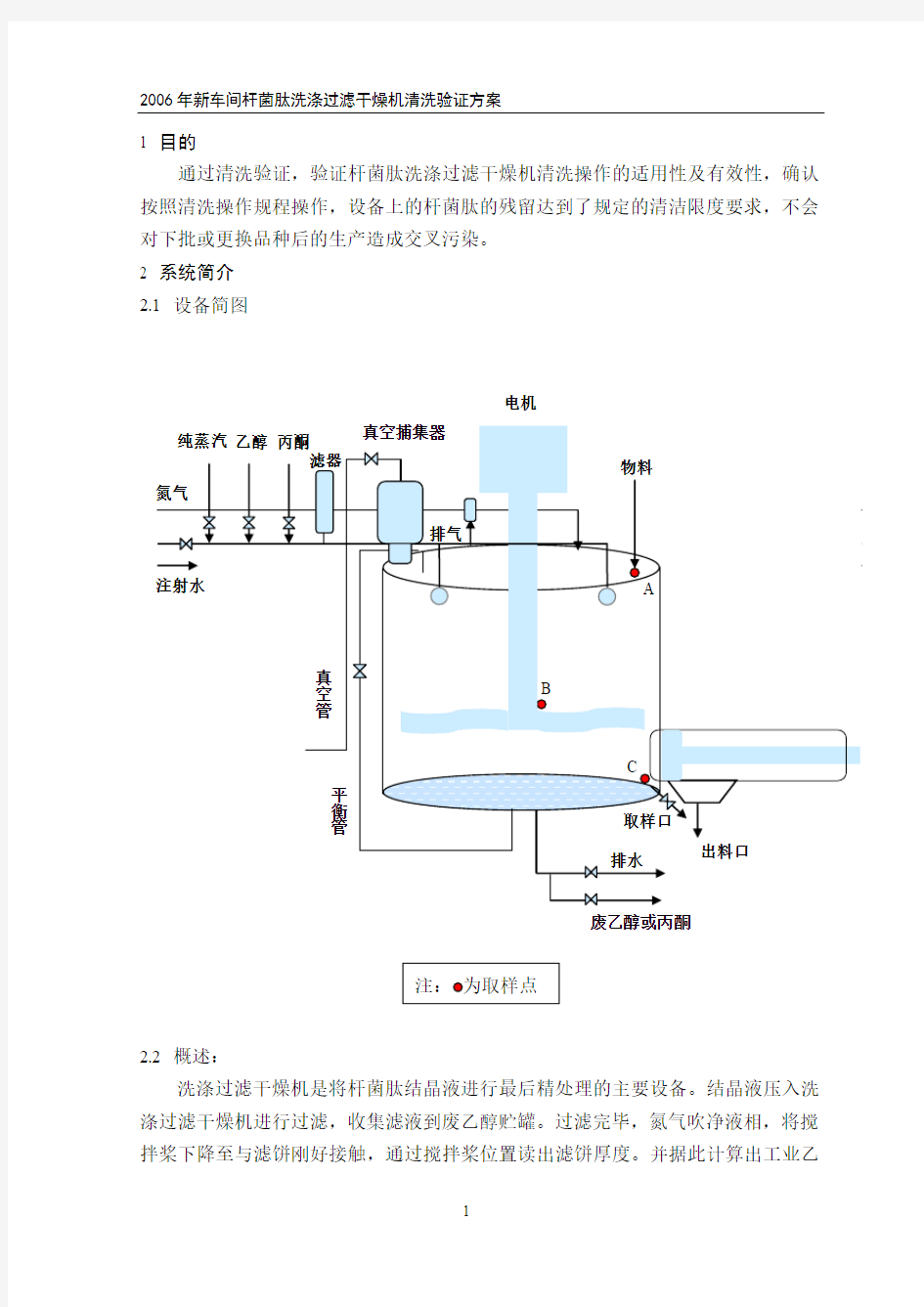
2.1 设备简图
电机
废乙醇或丙酮
2.2 概述:
洗涤过滤干燥机是将杆菌肽结晶液进行最后精处理的主要设备。结晶液压入洗涤过滤干燥机进行过滤,收集滤液到废乙醇贮罐。过滤完毕,氮气吹净液相,将搅拌桨下降至与滤饼刚好接触,通过搅拌桨位置读出滤饼厚度。并据此计算出工业乙
醇、工业丙酮洗涤用量,按每次洗涤加量90~140L工业乙醇、工业丙酮分次洗涤。洗涤液通过精滤器进入洗涤过滤干燥机。先用工业乙醇洗涤,再用工业丙酮洗涤,浸泡一定时间后将液相过滤至废溶媒贮罐,用氮气吹净液相。每次洗涤前将搅拌桨慢下到最低位置,疏松并搅拌滤饼。在加洗涤液的同时,将搅拌桨上升至最高位置。最后用氮气吹干60~120分钟。
开洗涤过滤干燥机搅拌,确认搅拌转速为最小值,将搅拌桨慢下至最低位置。打开平衡阀、真空阀,保持一定的真空度。打开夹套热水阀门,调节水温在所需范围内,待洗涤过滤干燥机温度(下部温度计)升到40℃以上时,开始进行真空干燥。控制洗涤过滤干燥机温度,干燥36~37小时。
本系统为温州亚光机械制造有限公司生产的集过滤、洗涤、干燥为一体的三合一设备。该设备为不锈钢制立式容器,由罐体装置、搅拌装置、轴端密封装置、升降机构、液压装置、罐底座(可拆卸)、加热装置、传动装置、过滤装置、卸料装置、取样装置、洗涤灭菌装置、行程指示装置、触摸屏与手动操作控制装置等部件组成。可在同一容器内完成滤、洗涤、干燥全过程连续操作。设备在真空状态下操作,实现固液分离;其内侧装有两个360°旋转的喷淋球,使清洗液均匀分布于容器内,通过升降桨叶搅拌将滤饼和清洗液混合,使浆状结晶及滤饼得到充分洗涤;滤饼被搅拌桨叶逐层刮松,均匀对滤饼加热,同时用热水对湿物料加热,加速蒸发干燥。排气口安装有过滤精度为0.2μm的呼吸器,进罐溶媒经过0.2μm的过滤器。管路全部使用卫生级不锈钢管道连接,设计和安装时避免了死角、盲端。
杆菌肽在生产过程中容易附着在洗涤过滤干燥机的内壁、搅拌桨等部位上。在批号结束时,需要将这些残留在设备上的杆菌肽清洗干净才能进行下一批的生产,以避免对下一批产品产生污染。
两性霉素B不易溶于水,所以采用人工擦拭与三乙胺的乙醇溶液浸泡的方法,用注射用水进行最后的冲洗。
洗涤过滤干燥机每批清洗一次。
3 范围
本验证是对杆菌肽洗涤过滤干燥机的清洗方法的验证。
4 可接受的标准
4.1注射用水
4.2清洗水样
4.3表面擦拭样
3000g )
cm ()g/cm (B cm 222积洗涤过滤干燥机内表面的含量两性霉素每 <0.1%
其中:3000g 为试生产以来的最小投料量。 5 验证小组
6 验证方案 6.1 仪器仪表检测
a ) H PLC 仪
b ) 气相色谱仪
6.2 清洗方法
批号结束后,打开并卸下洗涤过滤干燥机底部滤板,1小时后操作人进入洗涤过滤干燥机内,用洁净抹布擦拭内壁、搅拌桨、波纹管、出料口等部位,直至目测无可见污物。擦拭完毕后上好滤板。拆洗真空捕集器。
打开洗涤过滤干燥机注射用水阀及两个清洗球阀,冲洗溶媒过滤器及洗涤过滤干燥机5~10分钟。清洗过程中,开搅拌,打开洗涤过滤干燥机排水阀排水。清洗结束后,排干净洗涤过滤干燥机中的水后关闭排水阀。
检查并确认结晶罐可以向洗涤过滤干燥机加三乙胺乙醇溶液。打开洗涤过滤干燥机进料阀,用氮气将结晶罐内三乙胺乙醇溶液压到洗涤过滤干燥机中。溶液压完后浸泡30~60分钟,浸泡时开着搅拌(搅拌桨处于最低位)。浸泡结束后,打开洗涤过滤干燥机排水阀,排干净溶液。
排干净三乙胺乙醇溶液后从结晶罐向洗涤过滤干燥机中压注射用水,冲洗进料管道。
打开洗涤过滤干燥机注射用水阀及两个清洗球阀,冲洗溶媒过滤器及洗涤过滤干燥机15~20分钟。清洗过程中,开搅拌,打开洗涤过滤干燥机排水阀排水。清洗结束后,排干净洗涤过滤干燥机中的水后关闭排水阀。
6.3 取样方法
6.3.1清洗水样
在注射用水清洗过程中,取第5,10分钟的清洗水样各20ml;在清洗结束后,打开注射用水阀2~3分钟,由罐底排水处取最终淋洗水样20ml于洁净取样瓶中,标明名称、编号及取样日期。
6.3.2 表面擦拭样
6.3.2.1清洗结束后,用DMSO润湿棉签,并将其靠在溶剂瓶上挤压除去多余的DMSO,擦拭设备的指定部位A、B、C点(见设备简图)。
6.3.2.2将棉签头按在取样表面上,用力使其稍弯曲,平稳而缓慢地擦拭取样表面。擦拭过程应覆盖整个表面,翻转棉签,将棉签的另一面也在取样表面上擦拭,但与前次擦拭方向垂直(见下图)。擦拭区域约为10cm×10cm,每擦拭一个区域,换一根棉签。
6.3.2.4擦拭完毕后将棉签放入盛有5mlDMSO的试管中,加塞,标识次序。放置10min 钟使两性霉素B溶出,挤干棉签,用5mlDMSO润洗三次,测定DMSO溶液中两性霉素B的含量。
6.3.2.5 制备对照样品:按照6.3.2.2相关内容,湿润棉签,将棉签直接放入试管并加塞。
6.3.2.6 在试管上标明名称、位置、编号及取样日期。
6.3.3 取样方法验证
6.3.3.1 准备一块500×500mm的不锈钢板,平整即可。
6.3.3.2 用钢锥划出400×400mm的区域,每搁100mm划线,形成16个100×100mm 的方块。
6.3.3.3 精密称取0.0080g两性霉素B,DMSO溶解后用乙醇定容至50ml。
6.3.3.4 用微量移液器准确移取0.625ml上述溶液,分五次移至100×100mm的方块中,使其均匀分布。
6.3.3.5 根据实际转移的溶液体积计算单位面积的两性霉素B的含量(约1μg/cm2)。
6.3.3.6 自然干燥或用电吹风温和地吹干表面。
6.3.3.7 按照6.3.2.2擦拭表面,每擦一个方块(100cm2)换一根擦拭棒,共擦6个方块。
6.3.3.8擦拭完成后,将棉签放入盛有5mlDMSO的试管中,加塞。放置10min钟使两性霉素B溶出,挤干棉签,用5mlDMSO润洗三次,测定DMSO溶液中两性霉素B的含量。
6.3.3.9 计算回收率和回收率的RSD。
6.3.3.10 可接受的标准
取样回收率大于70%,体现重现性多次取样回收率的RSD小于20%。
6.4 检查方法
6.4.1 污物目测
人工擦拭后,检查洗涤过滤干燥机罐壁、罐底、搅拌桨、波纹管及出料口处,无明显的目测污物。
6.4.2 HPLC法
表面擦拭样用HPLC法测定。
6.4.2气相色谱法
清洗水样用气相色谱法测定。
6.5 检查频率
全面检查设备的清洗效果3次,连续检查3批。
7 验证记录
8 数据分析
对检验数据进行统计分析,记录偏差及处理情况。
9 偏差分析
对验证实施中出现的偏差及处理情况进行评价,确认偏差级别。
10 验证小结
11 附录
回收率验证记录
年AmB-QX-01
洗涤过滤干燥机清洗检查记录
年AmB-QX-02 上次生产产品批号第次验证
上次生产产品批号第次验证
上次生产产品批号第次验证
洗涤过滤干燥机清洗验证检验记录
年AmB-QX-03
检验分析方法的验证和确认 一、法规要求二、分析方法验证三、分析方法确认四、分析方法验证和确认总结一、法规要求:新版GMP(2010年修订)第二百二十三条物料和不同生产阶段产品的检验应当至少符合以下要求:(一)企业应当确保药品按照注册批准的方法进行全项检验。(二)符合下列情形之一的,应当对检验方法进行验证。1. 采用新的检验方法;2. 检验方法需变更的;3. 采用《中华人民共和国药典》及其他法定标准未收载的检验方法;4. 法规规定的其他需要验证的检验方法。(三)对不需要进行验证的检验方法,企业应当对检验方法进行确认,以确保检验数据准确、可靠。法规要求:中国药典(2010年版)凡例1. 检验方法和限度。2. 二十三、本版药典正文收载的所有品种,均应按规定的方法进行检验。如采用其他方法,应将该方法与规定的方法做比较试验,根据试验结果掌握使用,但在仲裁时仍以本版药典规定的方法为准。法规要求:分析方法确认或验证相关指南二、分析方法验证 1. 分析方法验证的定义 2. 分析方法验证的目的 3. 分析方法验证范围 4. 分析方法验证的时机 5. 需验证的分析方法类型 6. 分析方法验证的具体内容 7. 验证检测项目小结 8. 分析方法验证的方式和步骤 9. 分析方法验证常见问题1. 分析
方法验证的定义是根据检测项目的要求,预先设置一定的验证内容,并通过设计合理的试验来验证所采用的分析方法能否符合检测项目的要求。 2. 分析方法验证的目的(1)证明采用的分析方法是科学、合理。(2)证明分析方法能有效控制药品的内在质量。? 验证过程和结果均应记载在标准起草或修订说明中。 3. 分析方法验证范围(1)适用范围:化学药品的理化分析方法和仪器分析方法的验证与确认;清洁验证方法的验证。(2)不适用:化学药品的微生物方法;生物制品分析方法验证。 4. 分析方法验证的时机(1)建立新的药品质量标准;(2)药品生产工艺变更;(3)制剂的组分变更;(4)对原分析方法进行修订时。方法验证理由、过程和结果均应记载在药品标准起草说明或修订说明中。 5. 需验证的分析方法类型(1)鉴别试验(2)杂质定量或限度检查(仪器或非仪器检测方法)(3)原料药或制剂中活性成分以及制剂中选定组分(如防腐剂等)的定量测定含量测定(4)化学药品/中药制剂中其他需控制成分(如残留物、添加剂等)的测定(5)制剂溶出度、释放度等检查(6)原料药粒度检测 6. 分析方法验证的具体内容(1)专属性(2)线性(3)范围(4)准确性(5)精密度(6)检测限(7)定量限(8)耐用性(9)系统适用性根据检测的类型,采用的技术检测方法,确定具体方法拟订验证的内容。专属性1. 鉴别、杂质和含量测定的方法学
洗瓶机的清洁验证方案 一、概述 二、验证目的 三、验证范围 四、验证人员 五、验证内容 1 验证条件 2 可接受标准 3 清洁过程 4 取样及样品处理 5 偏差分析及处理 六、验证结论及评价 七、再验证周期 八、验证进度计划
一、概述 洗瓶机是用于疫苗的重要辅助设备,对设备进行彻底地清洁,保证设备的清洁卫生影响到疫苗的质量。故需对洗瓶机的清洗效果进行验证,确认清洗后设备的清洁状况满足预定要求。清洁验证共需进行3次,方可证明清洁规程能持续稳定达到要求。 二、验证目的 验证本公司的洗瓶机按清洁规程进行清洁后的清洁效果能达到预定要求,符合制品生产的要求。 三、验证范围 本验证方案主要适用于洗瓶机的清洁验证。 1 验证条件 1.1 设备应为完好设备 1.2 人员:包括设备管理部门、使用部门、QA人员、QC人员及具体岗位操作人员。 1.2.1 在岗人员均GMP知识、药品管理法及其实施细则、生物制品管理办法、产品质量法等法律法规的培训。 1.2.2 在岗人员为经过岗位SOP、岗位安全操作法、工艺规程、卫生清洁规程等岗位专业知识培训,并持有上岗证的熟练工人。 1.3 清洁剂条件:中性或弱碱性,对设备无腐蚀;不含4A沸石等不溶性助剂,洗涤后无溶性残留;对制品、人体无毒害。 2 可接受标准 2.1 目检法:设备表面应无可见的残留物,并无残留物的气体。 2.2 浊度检查:取最终淋洗水50ml与纯化水50ml比色,目测应无可视差异。 2.3 残留物限度:应无上批残留物。 2.4 微生物限度:≤20CFU/ml。 3 清洁过程 3.1 清洁操作:按《盘式过滤器清洁规程》对设备进行清洁至目测合格。 3.2 清洁剂:饮用水、纯化水、注射水。 4 取样及样品处理 4.1 目测法:目视检查设备内外表面,目视检查合格后方可进行取样检查。
方案批准 注:在方案批准部分签字表明签字者同意方案中规定的检测项目检测方法和记录要求。在执行本方案的过程中可能会出现影响严格执行本方案的偏差,对较小的偏差将通过偏差报告的形式来解决,对于关键性偏差,如对方法的调整、对参数或接受标准的调整必须制定出增补方案并按照原方案批准程序得到批准才能进行。所有的偏差报告和增补方案必须在提交验证报告供批准时一同提交。
目录 1.概述 (3) 2.参考资料 (4) 3. 职责 (4) 4. 色谱系统及色谱条件 (4) 5. 器材与试剂 (5) 6. 验证试验 (5) 6.1系统适应性 (5) 6.2专属性 (6) 6.3耐用性 (7) 6.4定量限 (8) 6.5检测限 (8) 6.6线性与围 (8) 6.7准确度 (9) 6.8精密度 (11) 7.再验证周期 (12) 8.偏差及纠正措施 (13) 9.最终审核和批准 (13) 药品残留溶剂顶空分析方法草案 (14)
1.概述 1.1根据ICH对药品中残留溶剂含量的要求及盐酸噻氯匹定生产工艺,必须控制盐酸噻氯匹定生产工艺中使用到的溶剂乙醇、丁酮、甲苯、N,N-二甲基甲酰胺(DMF)的残留量。限度分别为:乙醇≤5000ppm、丁酮≤5000ppm、甲苯≤890ppm、DMF≤880ppm。 1.2分析方法草案见附件。 1.3本分析方法属于杂质定量分析,因此需要验证的项目有:系统适应性、专属性、线性、 准确度、检测限、定量限、精密度、耐用性,具体参数及接受标准要求见下表:
2.参考资料 ICH Q3C (R3), November 2005. ICH Q2 (R1), November 2005. <467> Residual Solvents, United States Pharmacopoeia 31, November 2007. <20424> Residual Solvents, European Pharmacopoeia 6.0, June 2007. 3. 职责 4.1色谱系统
修订记录Revisi ons Recor d 版本号Release 修订描述Description of Revision C01 由KHB-SOP-40067/B00《搪瓷配料罐配制输送操作规程》和KHB-SOP-42067/B00《搪瓷配料罐清洁消毒规程》以及KHB-SOP-30015/B00《配制罐地槽清洁消毒规程》合并升版而来。 姓名 Name 职务 Job Title 签字 Signature 日期 Date 起草人Author(s)陈国良 酶免生产部 配制人员 审核人Reviewer(s)朱玲 酶免生产部 配制经理 批准人Approver(s) 滕浩 酶免生产部 总监 生效日期Valid From
1目的 规范搪瓷配料罐的操作、清洁与消毒的操作,最大限度的降低发生污染与交叉污染的风险,同时确保人员的安全。 2适用范围 本规程适用于搪瓷配料罐的使用、清洁和消毒。 3职责划分 3.1生产部配制人员负责搪瓷配料罐的操作、清洁灭菌的操作,并及时记录; 3.2生产部配制人员负责洗涤剂的配制以及配制记录的填写。 4定义与缩写 4.1定义 NA 4.2缩写 NA 5操作规程 5.1总则 5.1.1清洁消毒程序:为先上后下,先零后整,先里后外,先清洁后消毒; 5.1.2清洁消毒的人员要经过培训合格的配制操作人员; 5.1.3每天使用前应检查一遍各连接管道的密封情况,以防止液体的泄露。 5.2搪瓷配料罐的操作: 5.2.1配制操作: 关闭物料阀,配料罐内注入规定体积纯化水,打开投料口,启动搅拌器进行搅拌,按照溶液配制操作规程向罐内投放原料,同时打开气阀,直至原料完全溶解,关闭气阀; 在搅拌器搅拌的状态下加入Tween-20,然后注入纯化水进行定容; 启动搅拌器搅拌60分钟,关闭搅拌器,打开取样口进行取样测量,测试合格,配制完成,待用。 5.2.2输送操作: 用专用的管道将罐内溶液虹吸至专用桶内。 5.3清洁程序 5.3.1清洁: 5.3.1.1搪瓷配液罐的清洁: 清洁时间: 每次生产结束后 清洁剂 纯化水、饮用水 清洁程序 ●打开搪瓷配料罐的所有下水阀门; ●用饮用水冲洗配料罐外表面,直至外表面冲洗干净; ●打开饮用水,对罐内进行喷淋清洗10分钟; ●用纯化水冲洗10分钟;
TQ-3.6M3提取罐清洁验证方案
TQ-3.6M3提取罐清洁验证方案目录 1.目的 2.适用范围 3.责任者 4.内容 4.1概述 4.2.验证目的 4.3验证小组成员和职责 4.3.1验证小组成员 4.3.2验证小组成员职责 4.4验证方法 4.5执行的清洁程序及相关执行文件 4.6关键部位的确定 4.7物理外观检查 4.8微生物检测 4.9残留量检测 4.10验证结果评定与结论 4.11验证周期
TQ-3.6M3提取罐清洁验证方案 1.目的:制订详细、合理的验证方案,对TQ-3.6M3提取罐清洁全过程进行验证,确保TQ-3.6M3提取罐的清洁达到规定要求,以防止污染和交叉污染,生产出质量稳定、符合质量标准的产品。 2.适用范围:适用于TQ- 3.6M3提取罐的清洁验证。 3.责任者:验证小组成员对本方案的实施负责。 4.内容: 4.1概述 TQ-3.6M3提取罐是提取物料的设备,为了保证产品质量,避免污染和交叉污染,对提取后的TQ-3.6M3提取罐进行清洁是非常必要的,因此我们建立了《TQ-3.6M3提取罐清洁操作规程》,并按此清洁操作规程对设备进行清洁和消毒,以除去设备内外表面可见和不可见物质。清洁该设备后,以该设备内表面最难清洁部位、药品最难溶活性成分残留量及微生物数不超过预定的限量为指标,对《TQ-3.6M3提取罐清洁操作规程》进行验证。 验证以每批清血八味片浓缩结束后,TQ-3.6M3提取罐的清洁作为验证数据收集及评估单位,连续取样3批进行试验,并分析数据,综合整个验证过程,得出验证结论。 4.2验证目的 采用物理、化学分析方法和微生物检测方法来确认按《TQ-3.6M3提取罐清洁操作规程》对TQ-3.6M3提取罐进行清洁后,该设备内表面最难清洁部位的药品最难溶活性成分残留量及微生物数不超过规定的限量,证明《TQ-3.6M3提取罐清洁操作规程》的可行性和可靠性,以表明本清洁方法能够达到防止污染与交叉污染的目的,特制定本验证方案进行验证。 4.3验证小组成员及职责 4.3.1验证小组成员
方法验证试验的一般内容及要求---青岛科标检测 方法验证一般要求: 1.标准编制组应编制方法验证方案,根据影响方法的精密度和准确度的主要因素和数理统计学的要求,选择合适的实验室、样品类型、含量水平、分析人员、分析设备、分析时间等内容。 2.标准编制组除可以使用有证标准物质/标准样品外,还应提供实际样品进行方法验证,实际样品应尽量覆盖方法标准的适用范围。 3.在方法验证前,参加验证的操作人员应熟悉和掌握方法原理、操作步骤及流程,必要时应接受培训。 4.方法验证过程中所用的试剂和材料、仪器和设备及分析步骤应符合方法相关要求。 5.参加验证的操作人员及标准编制组应按照要求如实填写《方法验证报告》中的“原始测试数据表”,若有必要,应附上与该原始测试数据表内容相符的图谱或其他由仪器产生的记录打印条等。 6.标准编制组根据方法验证数据及统计、分析、评估结果,最终形成《方法验证报告》。 具体要求 1.检出限的验证 确定检出限,按方法操作步骤及流程进行分析操作,计算结果的平均值、标准偏差、相对标准偏差、检出限等各项参数。最终的方法检出限为各验证实验室所得数据的最高值。 2.精密度的验证
有证标准物质/标准样品的测定:采用高、中、低3 种不同含量水平(应包括一个在测定下限附近的浓度或含量)的统一样品,每个样品平行测定6 次以上,分别计算不同浓度或含量样品的平均值、标准偏差、相对标准偏差等各项参数。实际样品的测定:各验证实验室应对1 ~3 个含量水平的同类型样品进行分析测试,按每个样品平行测定6 次以上,分别计算不同样品的平均值、标准偏差、相对标准偏差等 各项参数。 对各验证实验室的数据进行汇总统计分析,计算实验室间相对标准偏差、重复性限r 和再现性限R。 3.准确度的验证 若各验证实验室使用有证标准物质/标准样品进行分析测定确定准确度,则需对1 ~3个不同含量水平的有证标准物质/标准样品进行测定,按全程序每个有证标准物质/标准样品平行测定6 次以上,分别计算不同浓度或含量水平有证标准物质/标准样品的平均值、标准偏差、相对误差等各项参数。 若实验室对实际样品进行加标分析测定确定准确度,则需对每个样品类型的1 ~3 个不同含量水平的统一样品中分别加入一定量的有证标准物质/标准样品进行测定, 每个加标样品平行测定6 次以上,分别计算每个统一样品的加标回收率。 对各验证实验室的数据进行汇总统计分析,计算其相对误差或加标回收率的均值及变动范围。
3 定义 3.1 检验方法验证:证明采用的方法适用于相应检测要求。 3.2 检验方法确认:证明使用法定方法在目前实验室条件下是否能获得可靠结果,是否适用于相应的检测工作。在本质上和验证一样,但不一定是验证项目的全部。 3.3 药典方法:经过国家药监部门批准的药典收载的质量标准和检验方法 3.4 法定方法:法定方法包括药典方法、国标方法等。 3.5 准确度:是指用该方法测定的结果与真实值或参考值接近的程度,一般用回收率表示。 3.6 精密度:是指在规定的测试条件下同一个均匀供试品经多次取样测定所得结果之间的接近程度。 3.7 重复性:在相同条件下,由同一个分析人员测定所得结果的精密度称为重复性。3.8 中间精密度:在同一个试验室,不同时间由不同分析人员用不同设备测定结果之间的精密度称为中间精密度。 3.9 重现性:在不同实验室由不同分析人员测定结果之间的精密度称为重现性。 3.10 专属性:是指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的方法能正确测定出被测物质的特性。 3.11 检测限:是指供试品中被测物能被检出的最低量。 3.12 定量限:是指供试品中被测物能被定量测定的最低量。 3.13 线性:是指在设计范围内,测试结果与试样中被测物浓度直接成正比关系的程度。 3.14 范围:是指能达到一定精密度、准确度和线性,测试方法适用的高低浓度或量的区间。 3.15 耐用性:是指在测定条件有小的变动时,测定结果不受影响的承受程度。 4 职责 4.1 标准验证岗 4.1.1 提升现行质量标准工作时,对研究后确定的标准草案进行检验方法验证工作,以确保检验方法的适用性、科学性。 4.1.2 对技术部移交的新品质量标准草案进行确认,以确保检验方法适用性、科学性。 4.1.3 对技术部移交的新品应研究建立设备清洁验证残留物检验方法,并进行方法学验证。
化胶罐清洗验证方案北京鑫航成科技发展有限公司
制定部门:制定人:日期:审核部门:生产部审核人:日期:审核部门:质量管理部审核人:日期:批准人:日期:
化胶罐清洗验证方案 1、目的:防止同品种物料批与批之间的交叉混杂和不同品种物料之间的交 叉污染,特制定切实有效的清洗程序以便把这种交叉混杂或交叉污染的程度控制在一个可以允许的限度之内,从而确保物料的品质符合标准要求。 验证小组名单 2、验证步骤及方法 2.1清洗程序的建立 2.1.1清洗的原则规定 2.1.1.1设备和容器连续使用一周后 2.1.1.2当物料出现质量问题时 2.1.1.3设备检修前后 2.1.1.4设备和容器静置超过一定时间后,在重新使用前 2.1.1.5更换品种时 2.2清洗剂的类型 2.2.1根据物料的化学性质不同,选择适宜的清洗剂。
2.2.2明胶的性质:明胶由18种氨基酸组成,一般含16%左右的水份,脂肪 和灰份0.5%以下,平均分子量10万左右。 明胶的特性:明胶在冷水中吸水膨胀不溶解,水温约在35℃以下即溶解成溶胶,温度降低即成凝胶。 2.2.3化胶罐清洗剂为80℃的纯水。 2.3清洗方法和步骤 2.3.1检查各种阀门是否按要求关闭 2.3.2化胶锅内加入纯水至2/3处 2.3.3打开加热罐的蒸汽阀门 2.3.4打开循环泵前的热水开关,循环泵后的热水开关。 2.3.5打开循环开关 2.3.6待化胶锅内水温开至80℃以上,打开搅拌开关,搅拌1h。 2.3.7在出料口接上皮管,打开出料末关把水放掉。 2.3.8再往罐内加入纯水,按上法搅拌30分钟,停止搅拌,用刷子刷净罐壁 粘附的胶液。 2.3.9将罐内水放完,再用纯水冲洗一遍。 2.3.10挂上已清洗牌 3、清洗后的检查验收
检验方法验证方案 目的:证明所采用的检验方法适于相应的检测要求,具有可靠的准确度、精密度。范围:含量的检定方法的前验证 编定依据:《药品生产质量管理规范》1998年修订版及验证管理办法 职责:验证小组人员 目录 1.概述 2.验证目的 3.职责 3.1验证小组 3.2品质部 3.3化验室 4.验证内容 4.1验证的准备工作 4.2适用性验证 4.2.1准确度试验 4.2.2精密度试验 4.3拟订验证周期 4.4验证结果评定与结论 5.附件
1. 概述 对小容量注射剂的含量测定,本公司采用福林酚测定法,该检验方法具有测量准确、精密度高、专属性强、定量准确可靠、方法简便易行的特点,可满足小容量注射剂含量测定的要求。检验方法标准操作规程。用本方法进行转移因子注射液、胸腺肽注射液的含量测定。 2. 验证目的 为确认对转移因子注射液、胸腺肽注射的含量测定的紫外分光光度法,适合相应的检测要求,特制订本验证方案,进行验证。 验证过程应严格按照本方案规定的内容进行,若因特殊原因确需变更时,应填写验证方案变更申请及批准书,报验证工作小组批准。 验证前,应首先对验证所需的仪器、设备进行验证,对所需仪器、仪表、量具等进行校正。 3. 职责 3.1 验证工作小组 负责验证方案的审批。 负责验证的协调工作,以保证本验证方案规定项目的顺利实施。 负责验证数据及结果的审核。 负责验证报告的审批。 负责发放验证合格证书。 负责再验证周期的确认。 3.2 品质部 负责验证所需仪器、设备的安装、调试,并做好相应的记录。 负责组织验证所需仪器、设备的验证。 负责仪器、仪表、量具等的校正。 负责拟订检验方法的再验证周期 3.3 化验室 负责验证所需的标准品、样品、试剂、试液等的准备。 负责验证方案指定的试验的实施。 负责收集各项验证、试验记录,并对试验结果进行分析后,报验证工作小组。 4. 验证内容 4.1 验证的准备工作 4.1.1 验证所需文件资料 品质部负责提供验证所需的文件资料,包括该检验方法的标准操作规程。以及负责提供验证所需仪器、设备的验证报告以及仪器、仪表、量具等的校正报告。 检查人:日期:
分析方法验证报告 2019QQHJKX01 分析方法:《水质石油类的测定紫外分光光度法(试行)》(HJ970-2018) 验证人员: 验证时间:2019年01月23日—25日 乌拉特前旗环境保护监测站
1参加人员情况 2仪器、标准物质情况 3 工作曲线的测定 3.1 工作曲线的测定条件 分析日期:2019年01月23日 温度:20℃湿度:18% 测定波长:225nm 3.2 工作曲线的测定
3.2 标准曲线的绘制 4 方法检出限的测定 依据《环境监测分析方法标准制修订技术导则》(HJ168-2010)附录A 方法特性指标确定方法。 方法检出限的一般确定方法: 按照样品分析的全部步骤,重复n(≥7)次空白试验,将各测定结果换算为样品中的浓度,计算n次平行测定的标准偏差,按公式A-1计算,方法检出限按公式A-2计算。
标准偏差S= (A-1) 式中:n —— 样品的平行测定次数 X i —— 单次测定值 X —— 测定平均值 MDL=t (n-1,0.99)×S (A-2) 式中:MDL —— 方法检出限 n —— 样品的平行测定次数 t —— 自由度为n -1 ,置信度为99%时的t 分布 S —— n 次平行测定的标准偏差 ( ) 1 1 2 - ? ? ? ? ? - ∑ = n X x n i i
5 精密度、准确度测试 分别对标准浓度为0.4mg/L、0.8mg/L、1.0mg/L和有证标准物质BW022四个浓度进行6次测定,测定结果见表5-1。
6 评价与验证结论 6.1 评价 根据《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)对本实验的检出限、精密度、准确度进行相关评价。 6.1.1 空白值最低检出限评价 根据《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)中的检出限为0.01 mg/L,本实验石油类的检出限为0.004mg/L,符合标准方法要求。 6.1.2 精密度评价 本次实验分别对油浓度为0.4 mg/L、0.8 mg/L、1.0 mg/L和有证标准物质BW022进行测试,相对标准偏差分别为4.6%、2.9%、4.7%、5.0%,符合标准方法要求。 6.1.3 准确度评价 根据方法条件,本次实验测定的加标回收率为96%-104%,平均值为99.75%,对标准浓度为0.4 mg/L、0.8 mg/L、1.0 mg/L和有证标准物质BW022的测定,相对误差分别为 2.5%、-1.2%、0.0%、5.0%,其结果均在标准范围内。 6.2 结论 通过对上述指标的验证,证明本站具备按照《水质石油油类的测定紫外分光光度法(试行)》(HJ970-2018)进行监测的能力,该项目可在本监测站正常开展。 分析者:复核者:审核者: 报告编写时间:2019年01月26日
方法验证报告 检测项目:硅酸盐岩石中锂、铍、钪等44个元素 量测定 方法名称及编号: 《硅酸盐岩石化学分析方法第30部分:44个元素量测定》GB/T 14506.30-2010 二O二O年三月
一、方法依据: 根据GB/T 14506.30-2010电感耦等离子体质谱法测定硅酸盐岩石中锂、铍、钪等44个元素的含量。 二、方法原理 样品用氢氟酸和硝酸在封闭溶样器中溶解,电热板上蒸发赶尽氢氟酸,再用硝酸密封溶解,稀释后用ICP-MS外标法直接测定。 三、仪器、试剂及标准物质 3.1 仪器 电感耦合等离子体质谱仪--安捷伦7700 感量天平--赛多利斯科学仪器有限公司 烘箱—上海一恒科学仪器有限公司 3.2 试剂 3.3 标准物质
四、样品 4.1 样品采集和保存 按照HJ/T166的相关规定进行土壤样品的采样和保存,样品采集和保存应使用塑料或玻璃容器,采样量不少于500g,新鲜样品小于4℃时可保存180天。 4.2 样品的制备 将采集的土壤样品放置于风干盘中自然风干,适时压碎、翻动,检出砂砾、植物残体。 在研磨室将风干的样品倒在有机玻璃板上,用木锤敲打,压碎,过孔径2mm尼龙筛,过筛后的样品全部置于无色聚乙烯薄膜上,充分搅匀,用四分法取两份,一份留样保存,一份用作样品细磨。 用于细磨的样品混匀,再用四分法分成四份,取一份研磨到全部过孔径0.074mm筛,装袋待分析。 4.3 样品前处理 称取约0.025g(精确到0.0001g)样品,置于50ml聚四氟乙烯
(PTFE)消解罐中,加1ml氢氟酸,0.5ml硝酸,密封,将溶样器放入烘箱中,加热24h,温度控制在185℃。冷却后取出内罐,置于电热板上加热蒸至近干,再加入0.5ml硝酸蒸发尽干,重复操作此步骤一次。加入5.0ml硝酸,再次密封,放入烘箱中,130℃3h。冷却后取出消解罐,将溶液定量转移至塑料瓶中,用水稀释,定容至25ml,摇匀。此溶液直接用ICP-MS测定。 4.4 实验室空白试样:随同样品进行双份空白试验,所用试剂取自同一瓶,加入同等的量。 4.5 结果计算与表示 计算固体样品中待测物的量: m V c w0? -=) ρ (ρ ) ( 式中:W C—样品中待测元素的量,μg/g; ρ—试样中元素的质量浓度,μg/L; ρ0 —空白试样中元素的质量浓度,μg/L; V—消解后试样的定容体积,ml; m—被称取样品的质量,g; 五、校准曲线 5.1 锂、铍、钒、锰、钴、镍、铜、锌、镓、砷、锶、镉、钡、铊、铅、铋标准曲线 取 5.00ml 100mg/L 多元素混合标准溶液(GSB 04-1767-2004(196046-1))于100ml容量瓶,以1%硝酸定容,得5.00mg/L 标准中间液。取5ml 5.0mg/L的标准中间液浓度于100ml容量瓶中,用1%
工作场所空气中甲醛分光光度法验证方案 方案编号:
目录
1. 概述 本文件验证了工作场所空气中甲醛的分光光度法本检测方法实施细则参照 GBZ/T 《工作场所空气有毒物质测定脂肪族醛类化合物》中甲醛的酚试剂分光光度法的定量测定为主要依据,以及在此标准的基础上根据本检测中心实际配置的仪器和实验条件的情况下将标准中的技术要素编写了相关的作业指导书。 为了保证此分析方法的可行性、准确性、可操作性和适用性、用科学的方法进行相应的验证程序特别编写了此验证方案,为今后在实际工作中将起到指导和借鉴的作用。 在验证空气中甲醛的测定方法中,做了方法的线性范围实验、检出限实验、方法精密度实验、准确度实验,以证明该方法适用于测定工作场所空气中甲醛的的浓度。 2. 目的 通过验证工作,确保测定工作场所空气中甲醛浓度的分析方法在广德众康职业卫生检测服务有限公司适用。 3. 分光光度计操作条件: 4. 试剂 实验用水为蒸馏水 至刻度。 开机准备 检查仪器,准备10mm石英比色皿。 打开紫外可见分光光度计,同时预热分光光度计10min 预热完毕,仪器自检完毕后,准备实验。 在成套的石英比色皿中加入参比溶液,做基线校准与校准能量。 校准完毕后,输入本方法所需要的波长645nm,确认,实验开始。 5.方法验证 线形范围
在3操作条件下将紫外可见分光光度计调节至最佳测定状态,标准曲线的绘制:取7只 具塞比色管,分别加入、、、、、、甲醛标准溶液,加水至,各加2ml酚试剂溶液,摇匀,于43±1℃水浴中放置10min,期间摇动几次,加入硫酸铁铵溶液,摇匀,再放入水浴中加热10min,取出放冷至室温,在645nm 波长下测量吸光度。每个浓度重复测定3 次, 以吸光度均值对相应的甲醛的含量(g)绘制标准曲线。 见表1: 表1:方法验证线性范围数据表 序号 1 2 3 4 5 6 7 标准含量(μ g) 吸光度(A) 相关系数 线形方程Y=+ 检出限 在3工作条件下将仪器调至检测状态,连续测量10次空白溶液,按公式计算检出限: C L = 3σ/s 式中:C L —检出限,g/mL; σ—测量10次空白溶液的浓度标准偏差; s —方法的灵敏度,即工作曲线斜率,mL/μg。 表2:连续测定10次空白溶液检出线数据 序号 1 2 3 4 5 吸光度值 浓度(μg /mL) 序号 6 7 8 9 10 吸光度值 浓度(μg
3定义 3.1检验方法验证:证明采用的方法适用于相应检测要求。 3.2检验方法确认:证明使用法定方法在目前实验室条件下是否能获得可靠结果,是否适用于相应的检测工作。在本质上和验证一样,但不一定是验证项目的全部。 3.3药典方法:经过国家药监部门批准的药典收载的质量标准和检验方法 3.4法定方法:法定方法包括药典方法、国标方法等。 3.5准确度:是指用该方法测定的结果与真实值或参考值接近的程度,一般用回收率表 示。 3.6精密度:是指在规定的测试条件下同一个均匀供试品经多次取样测定所得结果之间的接近程度。 3.7重复性:在相同条件下,由同一个分析人员测定所得结果的精密度称为重复性。 3.8中间精密度:在同一个试验室,不同时间由不同分析人员用不同设备测定结果之间的精密度称为中间精密度。 3.9重现性:在不同实验室由不同分析人员测定结果之间的精密度称为重现性。 3.10专属性:是指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的方法能正确测定出被测物质的特性。 3.11检测限:是指供试品中被测物能被检出的最低量。 3.12定量限:是指供试品中被测物能被定量测定的最低量。 3.13线性:是指在设计范围内,测试结果与试样中被测物浓度直接成正比关系的程度。 3.14范围:是指能达到一定精密度、准确度和线性,测试方法适用的高低浓度或量的区间。 3.15耐用性:是指在测定条件有小的变动时,测定结果不受影响的承受程度。 4职责 4.1标准验证岗 4.1.1提升现行质量标准工作时,对研究后确定的标准草案进行检验方法验证工作,以确保检验方法的适用性、科学性。 4.1.2对技术部移交的新品质量标准草案进行确认,以确保检验方法适用性、科学性。 4.1.3对技术部移交的新品应研究建立设备清洁验证残留物检验方法,并进行方法学验证。
2000L配料罐(1)清洁再验证方案 湖北御金丹药业有限公司
目录1 再验证组织及各部门职责 2概述 3再验证目的 4再验证计划及进度 5资料文件确认 6验证内容 6.1清洁规程的建立 6.2设备清洁验证中参照物质的选定 6.3 关键部位 6.4 验证方法 6.5 验证的实施 7再验证周期
1 再验证组织及各部门职责 1.1 再验证小组: 组长: 组员: 质量管理部: 设备工程部: 1.2 再验证小组职责 1.2.1 负责组织再验证方案的编写和审定 1.2.2 负责再验证工作的组织、协调和实施,以确保再验证工作的顺利进行。 1.2.3 负责再验证数据及有关资料的收集及审核 1.2.4 负责再验证结果的审核 1.2.5 负责再验证周期的确定 1.2.6 负责再验证报告的审批和发放 1.3 再验证小组职责分工 组长:负责组织再验证方案的编写,并对再验证方案进行审批;负责再验证工作
的组织、协调和实施,对再验证结果进行评价。 设备工程部:负责再验证方案的编写、2000L配料罐清洁再验证运行及性能确认的验证工作。 质量管理部:负责2000L配料罐清洁再验证运行及性能确认的各项检测验证工作,负责再验证合格证书的审批和发放。 1.3.1运行确认: 参与部门:设备工程部、生产车间 1.3.2 性能确认: 参与部门:质量管理部、设备工程部、生产车间 1.3.3 检定周期的确定 参与部门:质量管理部 1.3.4 再验证结果及评价: 参与部门:质量管理部、设备工程部、生产车间 1.3.5 再验证合格证书的审批和发放: 审批和发放部门:质量管理部 2概述 2000L配料罐用于液体制剂车糖浆剂配料,该机的清洁验证同清热解毒口服液的工艺验证同时进行(每批工艺验证后进行清洁验证)为证实所制订清洁SOP能有效防止交叉污染,对该设备的清洁效果具有良好的稳定性和可靠性,在设备按《2000L配料罐清洁SOP》清洁后对关键清洁部位进行取样检测,并对各检测结果进行综合评估。3再验证目的 证明经过清洁程序清洁后,设备上残留物(可见的和不可见的:包括前一产品的残留物或清洗过程中洗涤剂的残留物)达到了规定的清洁限度要求,不会对将生产的
验证内容:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐用性。 一、准确度:是指用该方法测定的结果与真实值或参考值接近的程度,一般以百分回收率表示。至少用9次测定结果进行评价。 二、精密度:是指在规定的条件下,同一个均匀样品,经过多次取样测定所得结果之间的接近程度。用偏差、标准偏差或相对标准偏差表示。 1、重复性:相同条件下,一个分析人员测定所得结果的精密度称为重复性。至少9次。 2、中间精密度:一个实验室,不同时间不同分析人员用不同设备测定结果的精密度。 3、重现性:不同实验室,不同分析人员测定结果的精密度。分析方法被法定标准采用应进行重现性试验。 三、专属性:指在其他成分可能存在的情况下,采用的方法能准确测定出被测物的特性,用于复杂样品分析时相互干扰的程度。鉴别反应、杂质检查、含量测定方法,圴应考察专属性。 四、检测限:指试样中被测物能被检测出的最低量,无须定量。用百分数、ppm或ppb 表示。 五、定量限:指样品中被测物能被定量测定的最低量,测定结果应具一定的精密度和准确度。 六、线性:系指在设计的范围内,测试结果与试样中被测物浓度直接呈正比关系的程度。 七、范围:能达到一定的精密度、准确度和线性的条件下,测试方法适用的高低限浓度或量的区间。 八、耐用性:指在一定的测定条件稍有变动时,测定结果不受影响的承受程度。
方法验证内容如下。 一、准确度 准确度系指用该方法测定的结果与真实值或参考值接近的程度,一般用回收率(%)表示。准确度应在规定的范围内测试。 1.含量测定方法的准确度 原料药可用已知纯度的对照品或样品进行测定,或用本法所得结果与已知准确度的另一个方法测定的结果进行比较。 制剂可用含已知量被测物的各组分混合物进行测定。如不能得到制剂的全部组分,可向制剂中加入已知量的被测物进行测定,或用本法所得结果与已知准确度的另一个方法测定结果进行比较。 如该分析方法已经测试并求出了精密度、线性和专属性,在准确度也可推算出来的情况下,这一项可不必再做。 2.杂质定量测定的准确度 可向原料药或制剂中加入已知量杂质进行测定。如不能得到杂质或降解产物,可用本法测定结果与另一成熟的方法进行比较,如药典标准方法或经过验证的方法。在不能测得杂质或降解产物的响应因子或对原料药的相对响应因子情况下,可用原料药的响应因子。应明确表明单个杂质和杂质总量相当于主成分的重量比(%)或面积比(%)。 3.数据要求 在规定范围内,至少用9个测定结果进行评价,例如,设计3个不同浓度,每个浓度各分别制备3份供试品溶液,进行测定。应报告已知加入量的回收率(%),或测定结果平均值与真实值之差及其相对标准偏差或可信限。 (意见3:是否对所设定的浓度范围作出要求,如:该方法用于药品的含量测定,回收率试验的样品浓度应设定于含量100%的±20%之间;用于溶出(释放)曲线考察时,回收率试验的样品浓度应设定于全曲线范围的上、中、下部位。) 二、精密度 精密度系指在规定的测试条件下,同一个均匀样品,经多次取样测定所得结果之间的接近程度。精密度一般用偏差、标准偏差或相对标准偏差表示。 在相同条件下,由一个分析人员测定所得结果的精密度称为重复性;在同一个实验室,不同时间由不同分析人员用不同设备测定结果之间的精密度,称为中间精密度;在不同实验室由不同分析人员测定结果之间的精密度,称为重现性。 含量测定和杂质的定量测定应考虑方法的精密度。 1.重复性 在规定范围内,至少用9个测定结果进行评价,例如,设计3个不同浓度,每个浓度各分别制备3份供试溶液,进行测定。或100%的浓度水平,用至少测定6次的结果进行评价。 2.中间精密度 为考察随机变动因素对精密度的影响,应设计方案进行中间精密度试验。变动因素为不同日期、不同分析人员、不同设备。 3.重现性 当分析方法将被法定标准采用,应进行重现性试验,例如,建立药典分析方法时通过协同检验得出重现性结果。协同检验的目的、过程和重现性结果均应记载在起草说明中。应注意重现性试验用的样品本身的质量均匀性和贮存运输中的环境影响因素,以免影响重现性结果。 4.数据要求 均应报告标准偏差、相对标准偏差和可信限。
8 ***原料有关物质分析方法验证方案 20**年**月
验证方案的起草与审批 方案实施日期:
目录 1.验证目的 (4) 2.方法简介与确认范围 (4) 3.标准品、供试品 (4) 4.风险评估 (4) 5.验证的可接受标准 (5) 6.验证步骤 (6) 6.1系统适应性 (6) 6.2专属性 (6) 6.3检测限与定量限 (8) 6.4线性 (9) 6.5准确度 (9) 6.6精密度 (10) 6.6范围 (10) 6.7耐用性 (10) 6.8样品测定 (11) 7.偏差 (11) 8.风险的接收与评审 (11) 9.再验证 (11) 10.确认结果评审和结论 (11) 11.更改历史 (12) 12. 附录 (12)
1.验证目的 根据法规的要求,采用非药典或其它法规未收载的分析方法应进行验证,证明采用的方法适合于相应的检测要求。 这个验证方案的目的是为验证提供具体方法参数、可接受标准和研究步骤。 2.方法简介与确认范围 ***原料有关物质检测方法为自行开发的液相室色谱方法。为确保方法的准确性和可行性,为日常检测方法提供依据,现对该方法进行验证。方法验证必须按照验证方案进行,此次验证方案提供***原料含量分析方法验证验证,包括:专属性、精密度、线性、范围、准确度、检测限&定量限、耐用性。 3.标准品、供试品 3.1标准品 3.2供试品 4.风险评估 按照《质量风险管理规程》,质量控制部和质量管理部共同对分析方法进行了风险评估,确定了需进行方法确认的项目。具体见下表:
风险评估人: 评估日期: 5.验证的可接受标准
验证报告Validation Report
目录CONTENTS 报告总结Summary Report 1、目的Purpose 2、验证范围Scope 3、验证依据Validation Basis 4、责任者Person Responsibility 5、接受标准Acceptance Criterion 6、有机挥发性物质的测定Determination of Organic Volatile Impurities 6.1有机挥发性物质的限度Limit of Organic Volatile Impurities 6.2溶液配制Preparation 6.3色谱系统Chromatographic System 6.4系统适应性试验System Suitability Test 6.5测定Procedure 6.6结果计算Calculate 6.7样品测定Sample Determination 7、仪器和试药Instruments and Reagents 7.1仪器Instruments 7.2试药Reagents 8、验证内容Validation Contents 8.1专属性(定位)Specificity 8.2检测限Limit of Detection 8.3精密度Precision 8.4线性范围Linearity and Range 8.5准确度/回收率Accuracy/Recovery 9、变更和偏差调查Change and Deviation Investigation 10、结果分析、结论和评价Comprehensive, Conclusion and Assessment 11、附录A ppendix
配液罐清洁验证方案 质量部[] 生产部[] 计划供应部[] 工程部[] 技术中心[] 人力资源部[] 行政部[] 企划部[] 信息管理中心[] 财务部[] 审计监察部[] 市场部[] QC 室[] 针剂车间[] 提取车间[] 验证方案 1适用范围 本方案适用于配液罐的清洁验证。 2职责验证领导小组 2.1.1 负责验证方案的审批。
2.1.2负责验证的协调工作,以保证本验证方案规定项目的顺利实施。 2.1.3负责验证数据及结果的审核。 2.1.4 负责验证报告的审批。 2.1.5 负责清洁验证周期的确认。 质量部 2.3.1各种理化检验、微生物检验的准备、取样及测试工作。 2.3.2负责根据检验结果出具检验报告单。 2.3.3负责验证工作的现场监督。 生产部 2.4.1负责指定设备的清洁人员。 2.4.2负责按照相关的标准清洁操作程序对清洁验证对象进行清洗。 3概述配液罐用于提取车间生化提取工段中间体的药液混合、分装,为保证生产中不产生交叉污染,使用本生产设备总混分装产品时没有来自上次产品及清洗过程所带来污染的风险,我们将对清洗后的设备,采用不同的方法检测,对最终结果进行评价。 4验证目的确认当设备按已制定的清洁规程进行清洁后,可将设备上残留的污染物的量清除到规定的限度标准要求。即通过设备清洗过程,消除了即将分装产品受前产品遗留物及清洗过程中污染物污染的风险。5验证内容 原理 本验证方案根据我公司生产品种的实际情况,配液罐用于XXX的混合、分装,因 XXX较难清洁,固本次验证采取对生产XXX的配液罐进行清洁验证,我们采用最终清洗水取样检验,将检验结果与可接受限度比较,证实清洁规程的有效性。 执行的清洁程序 参见《配液罐清洁SOP》 验证方法 配液罐按《配液罐清洁SOP进行清洁后,取冲洗液液(纯化水)进行水质检测、无菌检查,同时对设备进行物理外观检查。 检验方法 5.4.1物理外观检查
异烟肼含量测定分析方法验证方案验证原因:验证类型: 新项目验证再验证 其它 预验证 回顾性验证转移验证 方法描述: 本分析方法为中国药典2010版二部方法。为确保其检测结果准确,对该分析方法的专属性、精密度(系统精密度、方法精密度、中间精密度)、线性和范围、准确度、耐用性进行评价。 验证依据: 中国药典2010年版分析方法(295页) 验证时间: 2010年07月09日~2010年07月10日 验证项目组成员及职责:
验证内容:-
a)人员培训: b)仪器设备、标准品和试剂: 仪器设备 标准品和试剂 c)样品
色谱条件 色谱条件 色谱柱:agilent ODS-2 长度:250cm ,内径:4.6mm ,填料 C18 ,填料粒度:5μm 检测波长:262nm,带宽30 柱温:25℃ 进样量:20μl 流速:1.0ml/min 流动相A:0.02mol/l磷酸氢二钠溶液(用磷酸调pH至6.0),流动相B:甲醇 A:B=85:15 停止时间:12min 1.系统精密度 1.1.溶液配制 系统精密度溶液:取异烟肼10mg,置100ml容量瓶中,精密称量,用水溶解并稀释至刻度。 1.2验证过程及结果 系统精密度溶液连续进样6次,记录其异烟肼峰面积、保留时间。 可接受标准:异烟肼峰面积RSD≤2.0%,保留时间RSD≤2.0%。 结论:
2.重现性试验(方法精密度) 2.1.溶液配制 2.1.1.对照溶液:取异烟肼工作标准品10mg,精密称量,置100ml容量瓶中,用水溶解 并稀释至刻度。 2.1.2.方法精密度溶液:取异烟肼样品10mg置100ml容量瓶中,精密称定,用水溶解并 稀释至刻度。用此方法配置同一批号的样品溶液6份。 2.2.验证程序及结果 工作标准品溶液进2针,样品溶液各进2针。记录异烟肼峰面积,计算样品含量。 可接受标准:异烟肼含量的RSD≤2.0%。 结论: