
测量不确定度评定报告1、评定目的识别实验室定量项目检测结果不确定度的来源,明确评定方法,给临床检测结果提供不确定度依据。 、评定依据2CNAS-GL05《测量不确定度要求的实施指南》 JJF 1059-1999《测量不确定度评定和表示》 CNAS— CL01《检测和校准实验室能力认可准则》 、测量不确定度评定流程3 测量不确定度评定总流程见图一。
概述 建立数学模型,确定被测量Y与输入量 测量不确定度来源 标准不确定度分量评 B类评定评类A 计算合成标准不确定 评定扩展不确定 编制不确定度报告 图一测量不确定度评定总流程 测量不确定度评定方法、4建立数学模型 4.1.1 数学模型根据检验工作原理和程序建立,即确定被测量Y(输出量)与影响量(输入量)X,X,…,X间的函数关系f来确定,即:N21 Y=f(X,X,…,X)N12建立数学模型时应说明数学模型中各个量的含义和计量单位。必须注意, 数学模型中不能进入带有正负号(±)的项。另外,数学模型不是唯一的,若采用不同测量方法和不同测量程序,就可能有不同的数学模型。 4.1.2计算灵敏系数 偏导数Y/x=c称为灵敏系数。有时灵敏系数c可由实验测定,iii即通过变化第i个输入量x,而保持其余输入量不变,从而测定Y的变化i量。
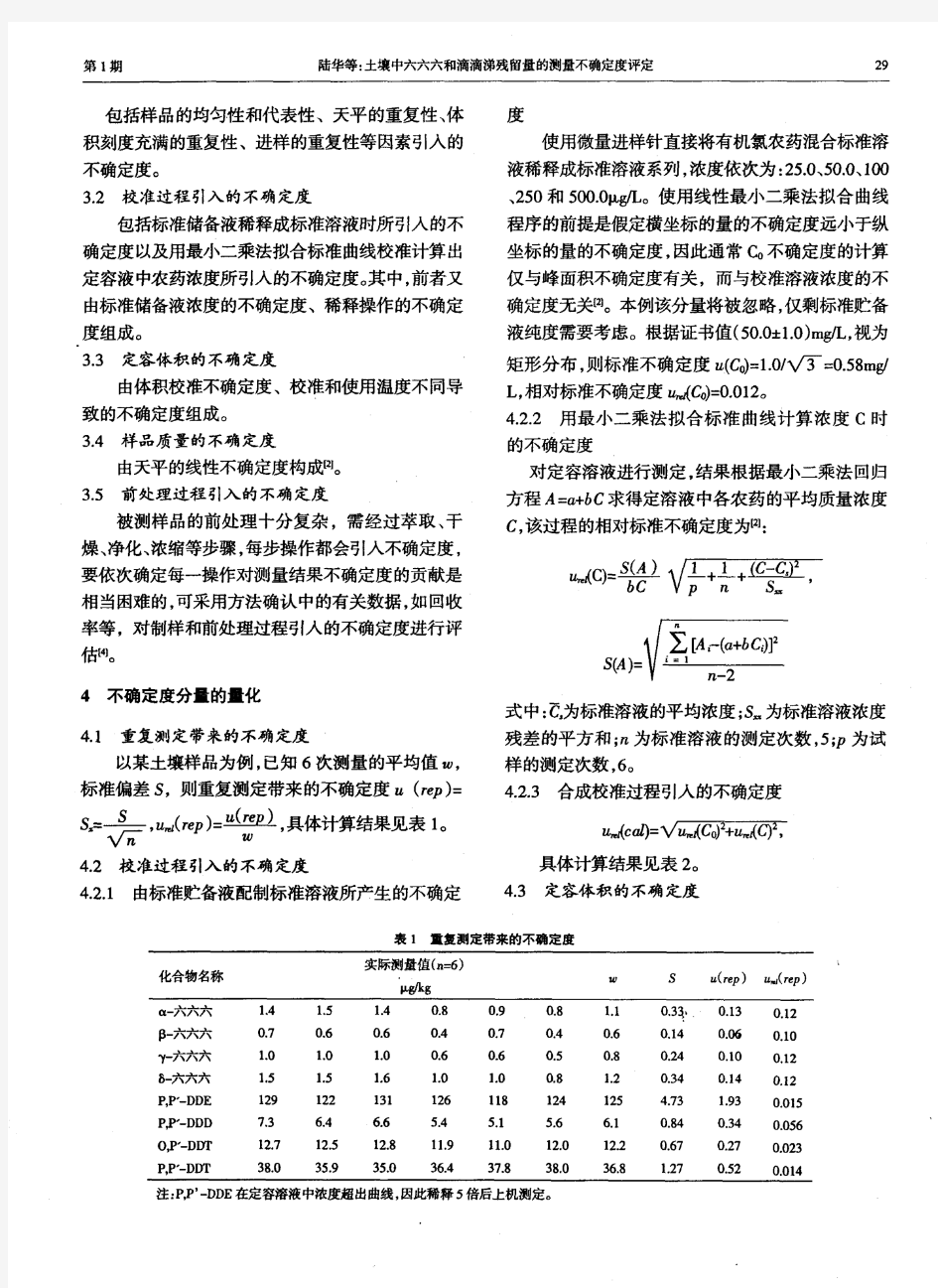
不确定度来源分析 测量过程中引起不确定度来源,可能来自于: a、对被测量的定义不完整; b、复现被测量定义的方法不理想; c、取样的代表性不够,即被测量的样本不能完全代表所定义的被测量; d、对测量过程受环境影响的认识不周全或对环境条件的测量和控制不完善; e、对模拟式仪器的读数存在人为偏差(偏移); 、测量仪器的计量性能(如灵敏度、鉴别力阈、分辨力、死区及稳定性f 等)的局限性; 、赋予计量标准的值或标准物质的值不准确;g 、引入的数据和其它参量的不确定度;h 、与测量方法和测量程序有关的近似性和假定性;i 、在表面上完全相同的条件下被测量在重复观测中的变化。j 标准不确定度分量评定 对观测列进行统计分析所作的评估--4.3.1 A 类评定 , x进行n次独立的等精度测量,得到的测量结果为:a对输入量XI 1为xx,…x。算术平均值n2 n1 ∑xx = in n i=1 由贝塞尔公式计算:s(x单次测量的实验标准差)i 1 n ∑ i—i 2 ( xx )S(x)= n-1 i=1
测量不确定度评定实例 一. 体积测量不确定度计算 1. 测量方法 直接测量圆柱体的直径D 和高度h ,由函数关系是计算出圆柱体的体积 h D V 4 2 π= 由分度值为0.01mm 的测微仪重复6次测量直径D 和高度h ,测得数据见下表。 表: 测量数据 计算: mm 0.1110h mm 80.010==, D 32 mm 8.8064 == h D V π 2. 不确定度评定 分析测量方法可知,体积V 的测量不确定度影响因素主要有直径和高度的重复测量引起的不确定都21u u ,和测微仪示值误差引起的不确定度3u 。分析其特点,可知不确定度21u u ,应采用A 类评定方法,而不确定度3u 采用B 类评定方法。
①.直径D 的重复性测量引起的不确定度分量 直径D 的6次测量平均值的标准差: ()mm 0048.0=D s 直径D 误差传递系数: h D D V 2 π=?? 直径D 的重复性测量引起的不确定度分量: ()3177.0mm D s D V u =??= ②.高度h 的重复性测量引起的不确定度分量 高度h 的6次测量平均值的标准差: ()mm 0026.0=h s 直径D 误差传递系数: 4 2 D h V π=?? 高度h 的重复性测量引起的不确定度分量: ()3221.0mm h s h V u =??= ③测微仪示值误差引起的不确定度分量 由说明书获得测微仪的示值误差范围mm 1.00±,去均匀分布,示值的标准不确定度 mm 0058.0301.0==q u 由示值误差引起的直径测量的不确定度 q D u D V u ??= 3
测量不确定度评定作业指导书 (IATF16949/ISO9001-2015) 1.目的: 规定了测量不确定度的评定方法,保证实验室对测量结果进行不确定度评定和报告出具。 2.适用范围: 适用于各检测项目的不确定度评定与表示。 3.依据的技术文件: JJF1059.1Y2012 测量不确定度的评定与表示。 4. 不确定度的评定方法: 测量不确定度评定依据JJF 1059.1-2012《测量不确定度评定与表示》进行,应对由仪器设备、人员、试验环境、试验方法等各方面可能引入的不确定度分量进行全面分析,然后根据JJF 1059.1-2012的要求合成不确定度,作出正确的分析报告。不确定度愈小,分析测试结果与真值愈靠近,其质量愈高,数据愈可靠。因此,测量不确定度就是对测量结果质量和水平的定量表征。 5.测量不确定度评定的步骤: 5.1一般评定不确定度的流程如下:
5.2建立测量的数学模型 测量的数学模型是指测量结果与其直接测量的量、引用的量以及影响量等有关量之间的数学函数关系。当被测量Y由N个其他量X1、X2、…、XN的函数关系确定时,被测量的数学模型为: Y = f (X1、X2、…、XN) 5.3测量不确定度的来源 一般应从被测量、样本离散性、环境、人员、仪器设备、方法、试剂、用于数据计算的常量及其他参量、测量方法及测量重复性等方面考虑不确定度来源。详细介绍如下: 1、对被测量的定义不完整或不完善 若在定义要求的温度和压力下测量,就可避免由此引起的不确定度。 2、实现被测量定义的方法不理想 如上例,被测量的定义虽然完整,但由于测量时温度和压力实际上达不到定义的要求(包括由于温度和压力的测量本身存在不确定度),使测量结果中引入了不确定度。
测量不确定度评定报告 1、评定目的 识别实验室定量项目检测结果不确定度的来源,明确评定方法,给临床检测结果提供不确定度依据。 2、评定依据 CNAS-GL05《测量不确定度要求的实施指南》 JJF 1059-1999《测量不确定度评定和表示》 CNAS— CL01《检测和校准实验室能力认可准则》 3 、测量不确定度评定流程 测量不确定度评定总流程见图一。 图一测量不确定度评定总流程 4、测量不确定度评定方法 4.1建立数学模型 4.1.1 数学模型根据检验工作原理和程序建立,即确定被测量Y(输出量)与影
响量(输入量)X 1,X 2 ,…,X N 间的函数关系f来确定,即: Y=f(X 1,X 2 ,…,X N ) 建立数学模型时应说明数学模型中各个量的含义和计量单位。必须注意, 数学模型中不能进入带有正负号(±)的项。另外,数学模型不是唯一的,若采用不同测量方法和不同测量程序,就可能有不同的数学模型。 4.1.2计算灵敏系数 偏导数Y/x i =c i 称为灵敏系数。有时灵敏系数c i 可由实验测定,即通 过变化第i个输入量x i ,而保持其余输入量不变,从而测定Y的变化量。 4.2不确定度来源分析 测量过程中引起不确定度来源,可能来自于: a、对被测量的定义不完整; b、复现被测量定义的方法不理想; c、取样的代表性不够,即被测量的样本不能完全代表所定义的被测量; d、对测量过程受环境影响的认识不周全或对环境条件的测量和控制不完善; e、对模拟式仪器的读数存在人为偏差(偏移); f、测量仪器的计量性能(如灵敏度、鉴别力阈、分辨力、死区及稳定性等)的 局限性; g、赋予计量标准的值或标准物质的值不准确; h、引入的数据和其它参量的不确定度; i、与测量方法和测量程序有关的近似性和假定性; j、在表面上完全相同的条件下被测量在重复观测中的变化。 4.3标准不确定度分量评定 4.3.1 A 类评定--对观测列进行统计分析所作的评估 a对输入量X I 进行n次独立的等精度测量,得到的测量结果为: x 1,x 2 , (x) n 。 算术平均值x为 1 n x n= ∑x i n i=1 单次测量的实验标准差s(x i )由贝塞尔公式计算: 1 n S(x i )= ∑ ( x i — x )2 n-1 i=1
水质六六六、滴滴涕的测定气相色谱法 1. 适用范围 本方法适用于地面水、地下水以及部分污水中六六六、滴滴涕的分析。 本方法用石油醚萃取水中六六六、滴滴涕净化后用带电子捕获检测器气相色谱仪测定。 当所用仪器不同时,方法的检出范围不同。γ-六六六通常检测至4ng/L,滴滴涕可检测至200ng/L。样品中的有机磷农药、不饱和烃以及邻苯二甲酸酯等有机化合物在电子捕获鉴定器上也有响应,这些干扰物质可用浓硫酸除掉。 2. 试剂 2.1 载气 氯气:纯度99.9%,氧的含量小于5ppm,用装5A分子筛净化管净化。 2.2 配制标准样品和试样预处理时使用的试剂和材料。 2.2.1 石油醚:沸程30~60℃或60~90℃浓缩20倍后色谱测定无干扰峰,如有干扰需用全玻璃蒸馏器重新蒸馏。 2.2.2 浓硫酸(H2SO4),ρ=1.84g/mL。 2.2.3 无水硫酸钠(Na2SO4):在300℃烘箱中烘烤4h,放入干燥器中冷却至室温,装入玻璃瓶备用。 2.2.4 硫酸钠((Na2SO4·10H2O):20g/L 溶液,使用前用石油醚提取三次,溶液与石油醚之比为10:1。 2.2.5 异辛烷(C8H18)。 2.2.6 苯(C6H6),优级纯。 2.2.7 色谱标准物:α-六六六、γ-六六六、β-六六六、δ-六六六、PP'-DDE、OP'- DDT、PP'-DDD、PP'-DDT,纯度为95~99%。 2.2.8 储备溶液:称取每种标准物(2.2.7)100mg,准确至1mg,溶于异辛烷(2.2.5)( β-六六六,先用少量苯溶解),在容量瓶中定容至100mL。在4℃可储存一年。 2.2.9 中间溶液:用移液管量取八种储备溶液,移至l00mL容量瓶中,用异辛烷(2.2.5)稀释至刻度。八种储备液量取的体积比为Vα-六六六、Vγ-六六六、Vβ-六六六、Vδ-六六、V PP'-DDE、V OP'- DDT、V PP'-DDD、V PP'-DDT=1:1: 3.4:1:3.5:5:3:8。 六
工业热电阻自动测量系统结果不确定度评定实例 用于检定工业热电阻的自动测量系统,根据国家计量检定规程(JJG 229—1998)对不确定度分析时可以在0℃点,100℃点,现在A 级铂热电阻的测量为例. B1 冰点(0℃) B1.1 数学模型,方差与传播系数 根据规定,被检的R(0℃)植计算公式为 R(0℃)=R i 0 =??? ??t dt dR t i = R i 0=??? ??t dt dR * * *0=??? ??-t I dt dR R R ℃)( = R i - 0.00391R * (0℃)×) ℃(0 0.00391R 0* *℃) (R R I - = R i - 0.391×1 .00* *℃) (R R I - = R i - 0.39 [] ℃)( 0* *R R I - 式中: R(0℃)—被检热电阻在0℃的电 阻值,Ω; R i —被检热电阻在0℃附近的测得值,Ω; R *(0℃)—标准器在0℃的电阻值,通常从实测的水三点值计算,Ω; R * i —标准器在0℃附近测的值,Ω。 上式两边除以被检热电阻在0℃的变化率并做全微分变为 dt 0R =d ()391.0R i +d ??? ? ???-2500399.0** 0i R R =dt Ri +dt *0 R +dt *i R 将微小变量用不确定度来代替,合成后可得方差 u 20 R t =u 2i R t +u 2t *0R +u 2t *i R (B-2) 此时灵敏系数C 1=1,C 2=1,C 3=–1。
B1.2 标准不确定分量的分析计算 B1.2.1 u 2i R t 项分量 该项分量是检热电阻在0℃点温度t i 上测量值的不确定度。包括有: a) 冰点器温场均匀性,不应大于0. 01℃,则半区间为0.005℃。均匀分布,故 u 1.1= 3 005.0=0.003℃ 其估计的相对不确定度为20﹪,即自由度1.1ν=12,属B 类分量。 b) 由电测仪表测量被检热电阻所带入的分量。 本系统配用电测仪表多为6位数字表(K2000,HP34401等),在对100Ω左右测量时仍用100Ω挡,此时数字表准确度为 100×106×读数+40×106×量程 对工业铂热电阻Pt100来说,电测仪表带入的误差限(半宽)为 被δ=±(100×100×106-+100×40×106- =±0.014Ω 化为温度:391 .0014 .0±=±0.036℃ 该误差分布从均匀分布,即 u 2.1= 3 036.0=0.021℃ 估计的相对不确定度为10﹪,即1.1ν=50,属B 累类分量。 c) 对被检做多次检定时的重复性 本规范规定在校准自动测量系统时以一稳定的A 级被检铂热电阻作试样检3次,用极差考核其重复性,经实验最大差为4m Ω以内。通道间偏差以阻值计时应不大于2m Ω,故连同通道间差 异同向叠计在内时,重复性为6m Ω,约0.015℃,则 u 3.1= 69 .1015 .0=0.009℃ 3.1ν=1.8,属A 类分量。 d) 被检热电阻自然效应的影响。 以半区间估计为2m Ω计约5mK 。这种影响普遍存在,可视为两点分布,故 u 4.1=1 5=5mK 估计的相对不确定度为30﹪,即4.1ν=5,属B 类分量。
CNAS—CL07 测量不确定度评估和报告通用要求General Requirements for Evaluating and Reporting Measurement Uncertainty 中国合格评定国家认可委员会
测量不确定度评估和报告通用要求 1.前言 1.1中国合格评定国家认可委员会(英文缩写:CNAS)充分考虑目前国际上与合格评定相关的各方对测量不确定度的关注,以及测量不确定度对测量、试验结果的可信性、可比性和可接受性的影响,特别是这种影响和关注可能会造成消费者、工业界、政府和市场对合格评定活动提出更高的要求。因此,CNAS在认可体系的运行中给予测量不确定度评估以足够的重视,以满足客户、消费者和其他各有关方的期望和需求。 1.2CNAS在测量不确定度评估和应用要求方面将始终遵循国际规范的相关要求,与国际相关组织的要求保持一致,并在国际规范和有关行业制定的相关导则框架内制订具体的测量不确定度要求。 2.适用范围 本文件适用于CNAS对校准和检测实验室的认可活动。同时也适用于其它涉及校准和检测活动的申请人和获准认可机构。 3.引用文件 下列文件中的条款通过引用而成为本文件的条款。以下引用的文件,注明日期的,仅引用的版本适用;未注明日期的,引用文件的最新版本(包括任何修订)适用。 3.1Guide to the expression of uncertainty in measurement(GUM).BIPM,IEC, IFCC,ISO,IUPAC,IUPAP,OIML,lst edition,1995.《测量不确定度表示指南》3.2International Vocabulary of Basic and General Terms in Metrology(VIM). BIPM,IEC,IFCC,ISO,IUPAC,IUPAP,OIML,2nd edition,1993.《国际通用计量学基本术语》 3.3JJF1001-1998《通用计量术语和定义》 3.4JJF1059-1999《测量不确定度评定和表示》
色谱分离分析方法测定粮食中的六六六、滴滴涕残留量 一、气相色谱分离分析方法 1、目的:(1)熟悉粮食样品中六六六、滴滴涕提取方法 (2)掌握气相色谱法测定六六六、滴滴涕的原理和方法 2、原理:利用电子捕获检测器对于负电极强的化合物具有较高的灵敏度这一特点,可分别测出微量的六六六和滴滴涕,也可同时分别测定不同异构体和代谢物。出峰顺序为α-666,β-666,γ-666,δ-666,р,р′-DDE,ο,р′DDT,р,р′-DDD,р,р′-DDT。 六六六、滴滴涕及异构体或代谢物含量按下式计算 X=[A1×1000]/[m1(V2/V1)×1000] 式中X—样品中六六六、滴滴涕及其异构体或代谢物的单一含量,㎎/㎏; A1—被测定用样液中六六六或滴滴涕及其异构体或代谢物的单一含量,μg; V1—样品净化液体积,ml; V2—样液进样体积,μL; m1—样品质量,g。 结果的表述报告取平行测定的算术平均值的两位有效数字。 3、仪器及试剂:组织捣碎机、电动振荡器、旋转浓缩蒸发器、吹氮浓缩器、具有电子捕获检测器(ECD)的GC① 丙酮、正己烷、石油醚(沸程30~60℃)、笨、硫酸、无水硫酸钠、20g/L H2SO4(aq)、六六六、滴滴涕标准溶液②,六六六、滴滴涕标准使用液③ 使用的试剂一般为分析纯,有机溶剂需经重蒸馏。 4、步骤及记录 (1)提取:称取约200g具有代表性的样品(适用于生的计烹调加工过的蔬菜、水果或谷类、豆类、肉类、蛋类),加适量水于捣碎机捣碎,混匀。称取匀浆2.00~5.00g于50ml 具塞锥形瓶中,加10~15ml丙酮,在振荡器上振荡30min,过滤于100ml分液漏斗中,残渣用丙酮洗涤4次,每次4ml,用少许丙酮洗涤漏斗和滤纸,合并滤液,加入20ml石油醚,摇动数次,放气。振荡1min,然后加入20ml 20g/L NaSO4(aq),振摇1min静置分层,弃去下层水溶液。用滤纸擦干分液漏斗颈内外的水,然后将石油醚液经盛有约10g无水硫酸钠的漏斗缓缓放出,滤入50ml三角瓶中,再以少量石油醚分三次洗涤原分液漏斗、滤纸和漏斗,洗液并入滤液中,将石油醚浓缩,移入10ml具塞试管中,定容至5.0ml或10.0ml。 (2)、净化:将5.0ml提取液加0.50ml浓硫酸,盖上试管塞振摇数次后,打开塞子放气,然后振摇30s,于1600r/min离心15min,上层清液供气相色谱法分析用。 (3)、测量:电子捕获检测器的线性范围窄,为了便于定量,选择样品进样量使之适合各组分的线性范围。根据样品中六六六、滴滴涕存在形式,相应的绘制各组分的标准曲线,从而计算出样品中的含量。 5、注意事项 ①气相色谱参考条件:色谱柱为玻璃柱,内装涂以OV-17(15g/L)和QF-1(20g/L)的混合固定液的80~100目硅藻土,NL电子捕获检测器内径3~4?长1.2~2m 气化室温度215℃色谱柱温度195℃检测器温度225℃ 载气N2流速90ml/min 纸速0.5?/min ②六六六、滴滴涕标准溶液的配制:准确称取α-666、β-666、γ-666、δ-666、р,р′-DDE、ο,р′DDT、р,р′-DDD、р,р′-DDT各10.0㎎溶于苯,分别移入100ml 容量瓶中,加苯至刻度并摇匀,每ml含农药100.0μg,作为储备溶液存于冰箱中。 ③六六六、滴滴涕标准使用液:将上述标准储备液用己烷稀释至适宜浓度,一般为 0.01μg/ml。
测量不确定度评定举例 A.3.1 量块的校准 通过这个例子说明如何建立数学模型及进行不确定度的评定;并通过此例说明如何将相关的输入量经过适当处理后使输入量间不相关,这样简化了合成标准不确定度的计算。最后说明对于非线性测量函数考虑高阶项后测量不确定度的评定结果。 1).校准方法 标称值为50mm 的被校量块,通过与相同长度的标准量块比较,由比较仪上读出两个量块的长度差d ,被校量块长度的校准值L 为标准量块长度 L s 与长度差d 之和。即: L=L s +d 实测时,d 取5次读数的平均值d ,d =0.000215mm ,标准量块长度L s 由校准证书给出,其校准值L s =50.000623mm 。 2)测量模型 长度差d 在考虑到影响量后为:d =L (1+?? )-L s (1+?s ?s ) 所以被校量的测量模型为: 此模型为非线性函数,可将此式按泰勒级数展开: L =ΛΛ+-++)(θαθαs s s s L d L 忽略高次项后得到近似的线性函数式: )(θαθα-++=s s s s L d L L () 式中:L —被校量块长度; L s —标准量块在20℃时的长度,由标准量块的校准证书给出; ? —被校量块的热膨胀系数; ?s —标准量块的热膨胀系数; ? —被校量块的温度与20℃参考温度的差值; ?s —标准量块的温度与20℃参考温度的差值。
在上述测量模型中,由于被校量块与标准量块处于同一温度环境中,所以?与?s 是相关的量;两个量块采用同样的材料,?与?s 也是相关的量。为避免相关,设被校量块与标准量块的温度差为??,??= ?-?s ;他们的热膨胀系数差为??,??= ?-?s ;将?s = ?-?? 和 ?=??+?s 代入式(),由此,数学模型可改写成: = ][θαδαθδs s s l d l +-+ () 测量模型中输入量??与?s 以及??与?不相关了。 特别要注意:在此式中的??和??是近似为零的,但他们的不确定度不为零,在不确定度评定中要考虑。由于??和??是近似为零,所以被测量的估计值可以由下式得到: L =L s +d () 3).测量不确定度分析 根据测量模型, 即: l = ][θαδαθδs s s l d l +-+ 由于各输入量间不相关,所以合成标准不确定度的计算公式为: )()()()()()()(222222222222θδαδθαδδθαθ αu c u c u c u c d u c l u c l u s d s s c s +++++= () 式中灵敏系数为: 1)(11=+-=??= =θαδαθδs s s l f c c , 由此可见,灵敏系数c 3和c 4为零,也就是说明?s 及? 的不确定度对测量结果的不确定度没有影响。合成标准不确定度公式可写成: )()()()()(22222222θαδαδθu l u l d u l u l u s s s s c +++= () 4).标准不确定度分量的评定 ○ 1标准量块的校准引入的标准不确定度u (l s ) 标准量块的校准证书给出:校准值为l s =50.000623mm ,U = 0.075?m (k =3),
盲样测量不确定度评定报告 1、概述 1.1 测量依据 JJG119-2005《实验室(酸度)计检定规程》 1.2 环境条件: 温度(23±3)℃;相对湿度≤85%RH 1.3 测量标准: pH 标准缓冲溶液,中国计量测试技术研究院提供;酸度计:型号:pHS-3E ; 编号:600709040019;制造厂:上海精密科学仪器有限公司;量程:(0.00~14.00)pH;分辨率:0.01pH;电极编号:05598709J 1.4 被测对象:盲样(新疆维吾尔自治区计量测试研究院提供) 1.5 测量过程: 选用JJG119-2005《实验室(酸度)计检定规程》附录A 表1中规定的一种(或多种)标准溶液,在规定温度的重复性条件下,对pHS-3E 型酸度计进行校准后,测量盲样溶液,重复校准和测量操作6次,6次测量结果的平均值即为盲样的pH 值。 2、数学模型 y=x 3、输入量引入的标准不确定度 3.1测量重复性引入的标准不确定度分量u 1 按照贝塞尔公式计算单次测量的实验标准差: () 1 1 2 --= ∑=n pH pH s n i i (n=6) 平均值的实验标准差: u 1= 6
盲样检测 3.2酸度计引入的不确定度分量u2 用性能已知的pH(酸度)计,对未知pH值的盲样(酸度计溶液标准物质)进行测量。 选用JJG119-2005《实验室(酸度)计检定规程》参照酸度计使用说明书中校准点对传递的酸度计进行校准,用校准过的酸度计对盲样(酸度计溶液标准物质)进行测定6次,得出测量重复性引入的标准不确定度分量u 1 。结合酸度 计引入的不确定度分量u 2和盲样引入的标准不确定度分量u 3 得到合成标准不确 定度,扩展不确定度。
钢卷尺测量不确定度评定报告 1测量方法及数学模型 1.1测量依据:依据JJG4-1999《钢卷尺检定规程》 钢卷尺的示值误差:△L=L a-L s+L a*αa*Δt-L s*αs*Δt 式中:L a——被检钢卷尺的长度; L s——标准钢卷尺的长度; αa——被检钢卷尺的膨胀系数; αs——标准钢卷尺的膨胀系数; Δt——被检钢卷尺和标准钢卷尺对参考温度20℃的偏离值。 由于L a-L s很小,则数学模型: △L= L a-L s +L s*△α*Δt 式中:△α——被检钢卷尺和标准钢卷尺的膨胀系数差 1.2方差及传播系数的确定 对以上数学模型各分量求偏导: 得出:c(L a)=1;c(L s)= -1+△α*Δt≈-1;c(△α)= L s*Δt;c(Δt)= L s*△α≈0 则:u c2 =u2(△L)=u2(L s)+ u2(L a) + (L s*Δt )2u2(△α) 2计算分量标准不确定度 2.1标准钢卷尺给出的不确定度u (L s) (1)由标准钢卷尺的测量不确定度给出的分量u (L s1) 根据规程JJG741—2005《标准钢卷尺》,标准钢卷尺的测量不确定度为: U=0.02mm其为正态分布,覆盖因子k=3,自由度v=∞,故其标准不确定度: u (L s1)= 0.02∕3 =0.007 (2)由年稳定度给出的不确定度分量u (L s2) 根据几年的观测,本钢卷尺年变动量不超过0.05mm,认为是均匀分布,则:L a≤5m:u (L s2)=0.05∕31/2 =0.029mm 估计u (L s2)的不可靠性为10%,则自由度v=1/2×(0.1)-2=50 (3)由拉力偏差给出的不确定度分量u (L s3) 由拉力引起的偏差为:△=L×103×△p/(9.8×E×F)
MMFSCNG0152 食品 六六六 滴滴涕 气相色谱法 薄层色谱法 MM_FS_CNG_0152 食品中六六六、滴滴涕残留量的测定方法 1.适用范围 本标准适用于食品中六六六、滴滴涕残留量的测定。 气相色谱法检测限:α-666、β-666、γ-666、δ-666依次为0.2,0.8,0.3,0.4μg/kg。p,p’-DDE、o,p’-DDT、p,p’-DDD、p,p’-DDT依次为0.5,2.7,1.6,4.0μg/kg。薄层色谱法最低检测量为0.02μg,适宜范围为0.02~0.20μg。 方法一:气相色谱法 2.原理概要 样品中六六六、滴滴涕经提取、净化后用气相色谱法测定,与标准比较定量。电子捕获检测器对于负电极强的化合物具有较高的灵敏度,利用这一特点,可分别测出微量的六六六和滴滴涕。不同异构体和代谢物可同时分别测定。 出峰顺序:α-666、γ-666、β-666、δ-666、p,p’-DDE、o,p’-DDT、p,p’-DDD、p,p’-DDT。 3.主要仪器和试剂 3.1.主要试剂(有机溶剂需经重蒸馏)。 丙酮、正己烷、石油醚(沸程30~60℃)、硫酸钠溶液(20g/L)。 六六六、滴滴涕标准溶液:准确称取甲、乙、丙、丁六六六四种异构体和p,p’-滴滴涕、p,p’-滴滴滴、p,p’-滴滴伊、o,p’-滴滴涕(α-666、β-666、γ-666、δ-666、p,p’-DDT、p,p’-DDD、p,p’-DDE、o,p’-DDT)各10.0mg,溶于苯,分别移入100mL容量瓶中,加苯至刻度,混匀,每毫升含农药100.0μg,作为储备液存于冰箱中。 六六六、滴滴涕标准使用液:将上述标准储备液以己烷稀释至适宜浓度,一般为0.01μg/mL。 3.2.仪器 小型粉碎机、小型绞肉机、组织捣碎机、电动振荡器、旋转浓缩蒸发器、吹氮浓缩器。 气相色谱仪:具有电子捕获检测器(ECD)。 4.过程简述 4.1.提取 称取具有代表性的样品(适用于生的及烹调加工过的蔬菜、水果或谷类、豆类、肉类、蛋类)约200g,加适量水捣碎。称取匀浆2.00~5.00g,于50mL具塞三角瓶中,加10~15mL丙酮,在振荡器上振荡30min,过滤于100mL分液漏斗中,残渣用丙酮洗涤四次,每次4mL,用少许丙酮洗涤漏斗和滤纸,合并滤液30~40mL,加石油醚20mL和加20mL 硫酸钠溶液(20g/L)进行萃取分层,弃水层,然后将石油醚液缓缓放出,经盛有约10g无水硫酸钠的漏斗,滤入50mL三角瓶中。再用石油醚分三次洗涤原分液漏斗、滤纸和漏斗,洗液并入滤液中,将石油醚浓缩,移入10mL具塞试管中,定容至5.0mL或10.0mL。 称取具有代表性的乳样品2.00g,于10mL具塞试管中,加4mL丙酮,振摇1min,加4mL石油醚,振摇1min。静置分层。将上层石油醚溶液移入另一25mL具塞试管中,再加1mL石油醚于原试管中,不摇。取出上层石油醚合并于25mL试管中,重复两次。再加与石油醚等体积的硫酸钠溶液(20g/L),摇混,分层。将上层石油醚溶液取出经无水硫酸钠滤入10mL具塞试管中,再加1mL石油醚于原25mL试管中,不摇。取出上层液合并于10mL
此文档下载后即可编辑 测量不确定度评定报告 1、评定目的 识别实验室定量项目检测结果不确定度的来源,明确评定方法,给临床检测结果提供不确定度依据。 2、评定依据 CNAS-GL05《测量不确定度要求的实施指南》 JJF 1059-1999《测量不确定度评定和表示》 CNAS— CL01《检测和校准实验室能力认可准则》 3 、测量不确定度评定流程 测量不确定度评定总流程见图一。
图一 测量不确定度评定总流程 4、测量不确定度评定方法 4.1建立数学模型 4.1.1 数学模型根据检验工作原理和程序建立,即确定被测量Y (输出量)与影响量(输入量)X 1,X 2,…,X N 间的函数关系f 来确定,即: Y=f (X 1,X 2,…,X N ) 建立数学模型时应说明数学模型中各个量的含义和计量单位。必须注意, 数学模型中不能进入带有正负号(±)的项。另外,数学模型不是唯一的,若采用不同测量方法和不同测量程序,就可能有不同的数学模型。 4.1.2计算灵敏系数 偏导数Y/x i =c i 称为灵敏系数。有时灵敏系数c i 可由 实验测定,即通过变化第i 个输入量x i ,而保持其余输入量不变,从而测定Y 的变化量。
4.2不确定度来源分析 测量过程中引起不确定度来源,可能来自于: a 、对被测量的定义不完整; b 、复现被测量定义的方法不理想; c 、取样的代表性不够,即被测量的样本不能完全代表所定义的被测量; d 、对测量过程受环境影响的认识不周全或对环境条件的测量和控制不完善; e 、对模拟式仪器的读数存在人为偏差(偏移); f 、测量仪器的计量性能(如灵敏度、鉴别力阈、分辨力、死区 及稳定性等)的局限性; g 、赋予计量标准的值或标准物质的值不准确; h 、引入的数据和其它参量的不确定度; i 、与测量方法和测量程序有关的近似性和假定性; j 、在表面上完全相同的条件下被测量在重复观测中的变化。 4.3标准不确定度分量评定 4.3.1 A 类评定--对观测列进行统计分析所作的评估 a 对输入量XI 进行n 次独立的等精度测量,得到的测量结果为: x 1,x 2,…x n 。算术平均值x 为 1 n x n = ∑x i
土壤质量六六六和滴滴涕的测定 气相色谱法 GB/T 14550-93 Soil quality-Determination of BHC and DDT-Gas Chromatography 1 适用范围 1.1 本标准适用于土壤中六六六、滴滴涕的分析。 1.2 本法采用丙酮-石油醚提取,以浓硫酸净化,用带电子捕获检测器的气相色谱仪测定。 1.3 本方法的最低检测浓度为0.00005~0.00487mg/kg。 2 试剂和材料 2.1 载气 氮气,纯度99.99%,经去氧管过滤,氧的含量小于5ppm,氢的含量小于1.0ppm。 2.2 配制标准样品和试样预处理时使用的试剂和材料 使用的试剂系分析纯,有机溶剂经重蒸,浓缩20倍用气谱测定无干扰峰。 2.2.1 色谱标准样品:α-六六六、β-六六六、γ-六六六、δ-六六六、p,p/-DDE、o,p/-DDT、p,p/-DDD、P,P/-DDT,含量98%~99%,色谱纯。 2.2.2 石油醚,沸程60~90℃。 2.2.3 丙酮(CH 3COCH 3 )。 2.2.4 异辛烷(C 8H 18 )。 2.2.5 苯(C 6H 6 ):优级纯。 2.2.6 浓硫酸(H 2SO 4 ):密度为1.84g/mL。 2.2.7 无水硫酸钠(Na 2 SOt):在300℃烘箱中烘烤4h,备用。 2.2.8 硫酸钠溶液:20g/L。 2.2.9 硅藻土:试剂级。
2.2.10 三氯甲烷(CHCl )。 3 2.2.11 脱脂棉(或玻璃棉);用丙酮回流16h,取出晾干后备用。 2.3 制备色谱柱时使用的试剂和材料 2.3.1 色谱柱和填充物(3.6.5)。 2.3.2 涂渍固定液所用溶剂三氯甲烷(2.2.10)。 3 仪器 3.1 主要仪器:带电子捕获检测器的气相色谱仪。 3.2 控制载气的压力表及流量计。 3.3 进样器:全玻璃系统进样器。 3.4 记录仪:与仪器相匹配的记录仪。 3.5 检测器: 3.5.1 类型;电子捕获检测器。 3.5.2 器件的特征:可采用63Ni放射源或高温3H放射源。 3.5.3 检测器极化电压:可采用直流电源或脉冲电源。 3.6 色谱柱: 3.6.1 色谱柱数量:2~3支。 3.6.2 色谱柱特征: 3.6.2.1 材料:硬质玻璃。 3.6.2.2 尺寸:长1.8~2.0m,内径2~3mm。 3.6.3 色谱什类型:螺旋状填充柱。 3.6.4 色谱柱预处理:经水冲洗后,在玻璃柱管内注满热洗液(60~70℃)。浸泡4h,然后用水冲洗至中性,再用蒸馏水冲洗,烘干后进行硅烷化处理,将6%~10%的二氯二甲基硅烷甲醇溶液注满玻璃柱管,浸泡2h,然后用甲醇清洗至中性,烘干备用。 3.6.5 填充物:
第一节有关术语的定义 3.量值value of a quantity 一般由一个数乘以测量单位所表示的特定量的大小。 例:5.34m或534cm,15kg,10s,-40℃。 注:对于不能由一个乘以测量单位所表示的量,可以参照约定参考标尺,或参照测量程序,或两者参照的方式表示。 4.〔量的〕真值rtue value〔of a quantity〕 与给定的特定量定义一致的值。 注: (1) 量的真值只有通过完善的测量才有可能获得。 (2) 真值按其本性是不确定的。 (3) 与给定的特定量定义一致的值不一定只有一个。 5.〔量的〕约定真值conventional true value〔of a quantity〕 对于给定目的具有适当不确定度的、赋予特定量的值,有时该值是约定采用的。 例:a) 在给定地点,取由参考标准复现而赋予该量的值人作为给定真值。 b) 常数委员会(CODATA)1986年推荐的阿伏加得罗常数值6.0221367×1023mol-1。 注: (1) 约定真值有时称为指定值、最佳估计值、约定值或参考值。 (2) 常常用某量的多次测量结果来确定约定真值。 13.影响量influence quantity 不是被测量但对测量结果有影响的量。 例:a) 用来测量长度的千分尺的温度; b) 交流电位差幅值测量中的频率; c) 测量人体血液样品血红蛋浓度时的胆红素的浓度。 14.测量结果 result of a measurement 由测量所得到的赋予被测量的值。 注: (1) 在给出测量结果时,应说明它是示值、示修正测量结果或已修正测量结果,还应表明它是否为几个值的平均。 (2) 在测量结果的完整表述中应包括测量不确定度,必要时还应说明有关影响量的取值范围。 15.〔测量仪器的〕示值 indication〔of a measuring instrument〕 测量仪器所给出的量的值。 注: (1) 由显示器读出的值可称为直接示值,将它乘以仪器常数即为示值。 (2) 这个量可以是被测量、测量信号或用于计算被测量之值的其他量。 (3) 对于实物量具,示值就是它所标出的值。 18.测量准确度 accuracy of measurement 测量结果与被测量真值之间的一致程度。
测量不确定度的评定方法 1适用范围 本方法适用于对产品或参数进行检测时,所得检测结果的测量不 确定度的评 定与表示。 2编制依据 JJF 1059 —1999测量不确定度评定与表示 3评定步骤 3.1概述:对受检测的产品或参数、检测原理及方法、检测用仪器 设备、检测时的环境条件、本测量不确定度评定报告的使用作一简要的描述; 3.2建立用于评定的数学模型; 3.3根据所建立的数学模型,确定各不确定度分量(即数学模型中 的各输入量)的来源; 3.4分析、计算各输入量的标准不确定度及其自由度; 3.5计算合成不确定度及其有效自由度; 3.6计算扩展不确定度; 3.7给出测量不确定度评定报告。 4评定方法 4.1数学模型的建立 数学模型是指被测量(被检测参数)Y 与各输入量 X i之间的函数
关系,若被测量 Y 的测量结果为 y,输入量的估计值为x i,则数学模型为 y f x1 , x2 ,......, x n。 数学模型中应包括对测量结果及其不确定度由影响的所有输入 量,输入量一般有以下二种: ⑴ 当前直接测定的值。它们的值可得自单一观测、重复观测、 依据经验信息的估计,并包含测量仪器读数修正值,以及对周围温度、大气压、湿度等影响的修正值。 ⑵ 外部来源引入的量。如已校准的测量标准、有证标准物质、 由手册所得的参考数据。 4.2测量不确定度来源的确定 根据数学模型,列出对被测量有明显影响的测量不确定度来源,并要做到不遗漏、不重复。如果所给出的测量结果是经过修正后的结果,注意应考虑由修正值所引入的标准不确定度分量。如果某一标准不确定度分量对合成不确定度的贡献较小,则其分量可以忽略不计。 测量中可能导致不确定度的来源一般有: ⑴被测量的定义不完整; ⑵复现被测量的测量方法不理想; ⑶取样的代表性不够,即被测样本不能代表所定义的被测量; ⑷对测量过程受环境影响的认识不恰如其分或对环境的测量 与控制不完善; ⑸对模拟式仪器的读数存在人为偏移;
气相色谱法测定食品中滴滴涕、六六六的残留量 1 原理 样品中六六六、滴滴涕经提取、净化后用气相色谱法测定,与标准比较定量。 电子捕获检测器对于负电性强的化合物具有较高的灵敏度, 利用这一特点, 可分别测出微量的六六六和滴滴涕。不同异构体和代谢物可同时分别测定。 出峰顺序:α-666、γ-666、β-666、δ-666、p,p'-DDE、o,p'-DDT、p,p'-DDD、p,p'-DDT。 2试剂 使用的试剂一般系分析纯, 有机溶剂需经重蒸馏。 2.1 丙酮。 2.2 乙醚。 2.3 95%乙醇。 2.4 石油醚: 沸程30~60℃。 2.5 苯。 2.6 无水硫酸钠。 2.7 草酸钾。 2.8 硫酸。 2.9 2%硫酸钠溶液。 2.10 过氯酸-冰乙酸混合液:1:1。
2.11 六六六、滴滴涕标准溶液:精密称取甲、乙、丙、丁六六六四种异构体和p,p'-滴滴涕、p,p'-滴滴滴、p,p'滴滴伊、o,p'-滴滴涕(α、β、γ、δ-666、p,p'-DDT、p,p'-DDD、p,p'-DDE、o,p'-DDT)各10mg,溶于苯,分别移入100ml容量瓶中,加苯至刻度, 混匀,每毫升含农药100μg,作为贮备液存于冰箱中。 2.12 六六六、滴滴涕标准使用液:临用时各吸取2.0ml,分别移入10ml 容量瓶中, 各加苯至刻度, 混匀。每毫升相当农药20μg, 此浓度适用于薄层色谱法。用气相色谱法时,根据仪器灵敏度还需以己烷稀释至适宜浓度,一般为0.01μg/ml。 2.13 载体: 硅藻土, 80~100目, 气相色谱用。 2.14 固定液:OV-17及QF-1。 3 仪器 GC-2001(ECD) 4 操作方法 4.1提取 4.1.1 粮食:称取20g粉碎后并通过20目筛的样品,置于250ml具塞锥形瓶中,加100ml石油醚,于电动振荡器上振荡30min,滤入150ml 分液漏斗中,以20~30ml石油醚分数次洗涤残渣,洗液并入分液漏斗中, 以石油醚稀释至100ml。 4.1.2 蔬菜、水果: 称取200g样品,置于捣碎机中捣碎1~2min(若样品含水分少,可加一定量的水)。称取相当于原样50g的匀浆,加100ml 丙酮,振荡lmin, 浸泡1h,过滤。残渣用丙酮洗涤三次,每次10ml,洗
6.6 秒表测量误差测量不确定度的评估 6.6.1 概述 6.6.1.1测量依据:JJG237-2010《秒表检定规程》 6.6.1.2 计量标准:主要计量标准为时间检定仪,时间间隔测量范围(1~99999)s 。 表1 实验室的计量标准器和配套设备 6.6.1.3被校对象: 表2 被校准的机械秒表和电子秒表的分类 6.6.1.4 测量方法: 6.6.1.4.1 机械秒表测量误差的测量方法:按被校机械秒表的秒度盘和分度盘的满刻度值两个校准点进行校准,对每一被校准测量点测量3次,按下式(1)计算每次的测量误差,按(2)式取其中误差最大的作为校准结果。 0T T T i i -=? (1) {}Max i T T ?=? (2) 式中: i T —— 每次的测量值; 0T —— 时间检定仪给出的标准值; i T ?—— 每次测量得到的测量误差; T ?—— 校准结果给出的测量误差。 6.6.1.4.2 电子秒表测量误差的测量方法:对电子秒表的测量误差选择10s 、10min 、1h 三个校准点进行校准,对10s 、10min 两个受校点测量3次,1h 受校点测量2次,按下式(1)计算每次的测量误差,按(2)式取其中误差最大的作为校准结果。 6.6.1.5环境条件 1) 环境温度:(20±5)℃,校准过程中温度变化不超过2℃;相对湿度(65±15)%; 2) 周围无影响仪器正常工作的电磁干扰和机械振动; 3) 电源电压在额定电压的±10%,50Hz 。 6.6.2数学模型
{}Max i T T T 0-=? (3) 式中: T ? —— 机械秒表、电子秒表走时示值测量误差; i T —— 被校机械秒表、电子秒表每次走时测量值; 0T —— 时间检定仪给出的标准时间间隔值。 i —— 测量次数, 一般为3次, 当电子秒表测量1h 点时, 为2次。 6.6.3不确定度传播率 )()()(02 222212T u c T u c T u i c +=? 式中,灵敏系数1/1=???=i T T c ,1/02=???=T T c 。 6.6.4机械秒表、电子秒表测量误差标准不确定度的评定 6.6.4.1 输入量T 0的标准不确定度 标准设备时间检定仪标准装置的扩展不确定度为U 0=1.55×10-6×T+0.0092s, k =2 则将校准点3s ,对应的标准时间T 0的扩展不确定度为 U 0=1.55×10-6×3s+0.0092s=0.0092s ,k=2 ;则该标准引起的标准不确定度 分量为:s s k U T u 0046.02 0092.0)(00== =。 6.6.4.2 输入量T i 的标准不确定度 以被校机械秒表、分辨力0.01s 、校准点3s 为例 1)示值重复性引起的不确定度:校准3s 测量点,共进行3次的重复测量,极差为0.005s, 则单次测量的重复性为: s s s d R T s n i 0030.000295.0693 .1005.0)(≈=== 。 因测量误差为取最大的单次测量误差, 则A 类标准不确定度分量为单次测量的重复性为:s T s T u i i 0030.0)()(1==。 2)读数误差引起的不确定度: 由被校准机械秒表的分辨力引起的,采用B 类标准不确定度评定。已知分辨力为0.01s ,则不确定度区间半宽度为0.005s ,按均分布计算, s s T u i 00289.03 005.0)(2== 由于重复性分量包含了人员读数引入的不确定度分量,为避免重复计算,只计算最大影响量)(1i T u ,舍弃)(2i T u 。 6.6.5合成标准不确定度 6.6.5.1主要标准不确定度汇总表3