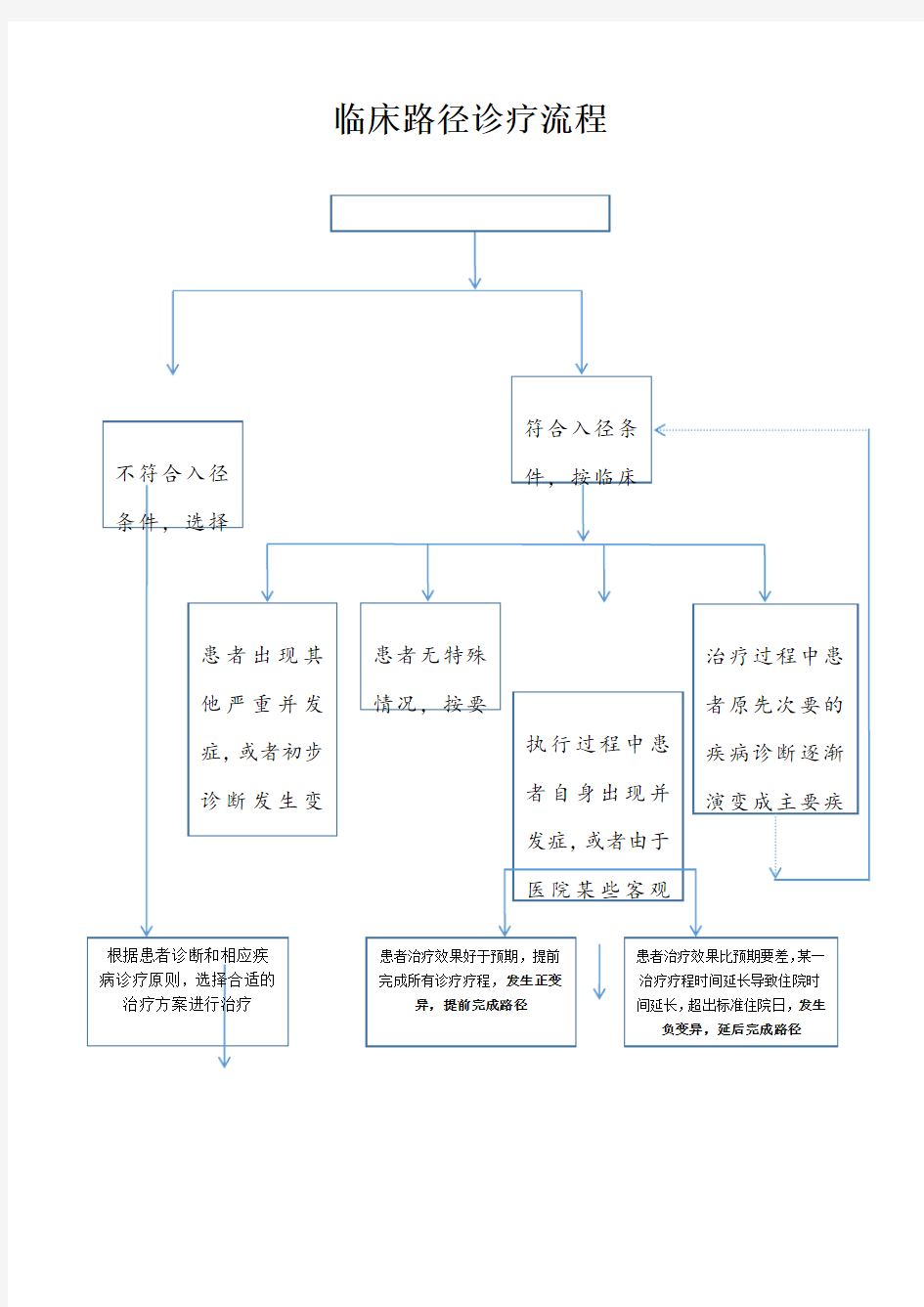

临床路径诊疗流程
1、路径跳转的情况一般发生在病人出现符合临床路径诊断的其他专科性疾病,继续在本科室治疗时使用(本科室有新疾病的路径表单)。如需要转科治疗或者新疾病无临床路径表单,则执行终止路径。
2、三甲医院标准规定:对符合进入临床路径标准的患者,入组率≥50%,入组完成率≥70%。
现在有三种申报方法 一.申报1.1类药进口(进口药) (1)方法:a.等药品在国外上市后,不在国内生产。申请分包进口(类似于代理)。 b.等药品在国外上市后,完善国内厂房技术,在国内生产。 (2)费用:临床实验37.6万,生产59.39万。 (3)难点:a.需要等药物在国外上市后才可以进行,延长周期。 b.如果在国内生产,需要完善国内厂房技术,延长周期、增加费用。 c.如果国外生产,分包进入国内时可以申请免除临床实验,但是不能确定可以申请成 功。 d.需要所有的临床资料,包括所有的实验数据。 二.申报1.1类新药临床试验(国产药) (1)方法:直接在国内申报1类新药,相关药理毒理研究资料由国外进行,提供相关证明,然后工艺研究、质量研究跟稳定性研究在国内进行,等国内药厂达到生产要求后,将药品在国内生产,按照国内药来报。 (2).费用:申请临床实验19.2万,申请生产43.2万。 (3)难点:a.需要等国内工厂具备相关生产条件后才可以进行申报。 b.无法申请避免临床。 三.申报国际多中心临床试验 (1)基本流程+费用+时间:主要临床基地伦理委员会审(费用...,时间...)+临床审批(费用37.6万,时间205天)+药品清关(费用...+时间...)(时间相对较短) (2)方法:申报程序与国内1.1类化学新药临床试验申报大致相同,但是需要更多的临床资料和
证明性文件。 (3)申请地点:国家药监局北京市西城区宣武门西大街28号大成广场3门一层(总局办公大楼 西侧),邮编100053 (4)流程图: 注:斜线前为一般审批时限,斜线后为特殊审批时限,均为工作日。 (5)申报资料列表: (一)概要部分
临床路径管理制度 第一章总则 第一条为提高医疗质量,保障医疗安全,指导各科室更好开展临床路径工作,根据卫生部《临床路径管理指导原则》等文件要求,特制定本管理制度。 第二条各科室临床路径工作依据此制度执行。 第二章临床路径的组织管理 第三条临床路径管理委员会由医院院长和分管医疗工作的副院长分别任正、副主任及各临床及医技科室主任、相关职能科室主任及专家委员会成员任成员。 第四条管理委员会履行以下职责: (一)临床路径开发与实施的规划和相关制度; (二)协调临床路径开发与实施过程中遇到的问题; (三)确定实施临床路径的病种; (四)审核临床路径文本; (五)组织临床路径相关的培训工作; (六)审核临床路径的评价结果与改进措施。 第五条临床路径指导评价小组由分管医疗工作的副院长任组长,相关职能部门(医务部、护理部、财务部)主任任成员。指导评价小组日常工作由医务部负责。 第六条指导评价小组履行以下职责:
(一)对临床路径的开发、实施进行技术指导; (二)制订临床路径的评价指标和评价程序; (三)对临床路径的实施过程和效果进行评价和分析; (四)根据评价分析结果提出临床路径管理的改进措施。 第七条各科室成立实施小组,由实施临床路径的临床科室主任任组长,护理长任副组长,该临床科室医疗、护理人员和相关科室人员任成员。临床路径实施小组履行以下职责: (一)负责临床路径相关资料的收集、记录和整理; (二)负责提出科室临床路径病种选择建议,会同药剂科、检验科、放射科、超声科、财务部等制订临床路径文本; (三)结合临床路径实施情况,提出临床路径文本的修订建议; (四)参与临床路径的实施过程和效果评价与分析,并根据临床路径实施的实际情况对科室医疗资源进行合理调整。 第八条各科室实施小组设立个案管理员,由临床科室科秘书担任。个案管理员履行以下职责: (一)负责实施小组与管理委员会、指导评价小组的日常联络; (二)牵头临床路径文本的起草工作; (三)指导每日临床路径诊疗项目的实施,指导经治医师分析、处理患者变异,加强与患者的沟通; (四)根据临床路径实施情况,定期汇总、分析本科室医护人员对临床路径修订的建议,并向实施小组报告。 第三章临床路径的选择与制订
阿美宁病例报告表 (Case Report Form) 受试者姓名: 家庭地址: 联系电话: 试验中心名称: 申办单位:江苏豪森药业股份有限公司 在正式填表前请认真阅读下列填表说明 病例报告表填写说明: 1.筛选合格者填写病例报告表。 2.病例报告表填写务必准确、清晰,不得任意涂改,错误之处纠正时需用横线居中化出,并签署修改者姓名缩写及修改时间。 3.填写记录一律用钢笔或碳素签字笔。 4.患者姓名拼音缩写四个需填满,两字姓名填写两字拼音前两个字母;三字姓名填写三字首字母及第三字第二字母;四字姓名填写每个字的首字母。
5.表中凡有“□”的项,请在符合的条目上划“√”。表格中所有栏目均应填写相应的文字或数字,不得留空。 6.所有检验项目因故未查或漏查,请填写ND;具体合并用药剂量和时间不明,请先写NK。 7.试验期间应如实填写合并用药记录表、不良事件记录表。记录不良事件的发生时间、严重程度、持续时间、采取的措施和转归。如有严重不良事件发生(包括临床验证过程中发生需住院治疗、延长住院时间、伤残、影响工作能力危及生命或死亡、导致先天畸形等事件),必须立即通知主要研究单位伦理委员会及申办单位。 8.临床试验应严格按照临床试验方案要求进行。试验不同时期需完成的检查和需记录的项目,请对照临床流程图执行。
受试者知情同意书 敬爱的患者: 我们现在正在进行一项临床研究,该项临床研究是经吉林省药品监督管理局备案的,研究的目的是评价物理治疗的高科技产品----温热电位治疗仪的疗效和安全性。 温热磁疗仪是XXX医疗器械有限公司根据韩国专利技术研制生产的医疗器械,本治疗仪是通过XXXXXXXXXXXXXXXXXXXXXXXXXXXXXX机理,主要用于XXXXXXXXXXXXX治疗。具有平衡机体阴阳,增强脏腑机能,促进新陈代谢,调节植物神经,改善心脑血管血液供应,促进血液循环,调节血管张力,降低血液粘稠度,防治动脉粥样硬化,促进组织的再生修复功能,增强机体免疫功能及消炎止痛等作用。其注册标准已经XX省食品药品监督管理局备案,其技术指标经XX省医疗器械检验所检测合格。现拟进行临床验证。
新药Ⅰ期临床试验申报资料的内容及格式要求 1995年11月美国FDA发布 2009年6月药审中心组织翻译 诺华制药有限公司翻译 北核协会审核 药审中心最终核准
目录 I. II. III. 引言 (1) 现行要求与操作规范 (2) 现行新药临床申请法规的解释 (2) A.封面(FDA格式-1571)[21 CFR 312.23(a)(1)]: (2) B.目录[21 CFR 312.23(a)(2)]: (2) C.介绍性声明与整体研究方案[21 CFR 312.23(a)(3)]: (2) D.研究者手册[21 CFR 312.23(a)(5)]: (2) E.方案[21 CFR 312.23(a)(6)]: (2) F.化学、生产和控制信息[21 CFR 312.23(a)(7)]: (3) G.药理学和毒理学信息[21 CFR 312.23(a)(8)]: (6) H.研究药物既往在人体中使用的经验[21 CFR 312.23(a)(9)]: (9) I. 21 CFR 312.23(a)(10)、(11)(b)、(c)、(d)(e): (10)
新药Ⅰ期临床试验申报资料的内容及格式要求 I. 引言 随着近期FDA成功实现《1992年处方药付费法》(PDUFA)审评行动的目标,使得从递交上市注册申请至批准上市的平均时间和中位时间均显著缩短。FDA已将注意力转移至如何提高药品开发过程中其它部分的效率,同时保证这种效率的提高不得以牺牲美国人所期望获得的、具备长期安全性和有效性药品的标准为代价。其中有一个IND法规特别值得关注,即关于在人体中开始进行药物试验的法规(即Ⅰ期试验),自McMahon 行动会议以来,对此课题已经进行了两年多的积极讨论,并且吸纳了各方不同层次的意见。 本指导原则阐述了在美国将研究药品(包括已进行结构确证的治疗性生物工程类产品)开始用于人体研究时,所需要提供的数据和在21 CFR 312.22和312.23中需要报告的数据1。现有法规对IND申报资料中要求提交的各数据数量和程度给予了很大的灵活性,其中大部分取决于所处于的研究阶段和需要进行的特殊人体试验。但是这种灵活性程度在某些状况中却并不是值得赞赏的,因此,FDA认为将申报资料的要求加以分类,在满足向FDA提供评估拟进行Ⅰ期研究所需要的安全性数据的同时,增加透明度、减少模糊性和不一致性、减少递交资料的数量,将有助于加速新药批准进入临床试验的时间。如果遵照本指导原则,那么IND的1期临床研究递交的资料通常应以不超过两至三英寸、三英寸的带3个环的活页夹(“文件夹”)进行装订。 需要澄清的最重要两点是:1)明确愿意接收毒理学检查结果的汇总总结报告,其根据对已完成动物研究,但未经稽查的毒理学报告草案而撰写,旨在对人体研究提供初步支持;2)适合1期临床试验研究用样品的生产质控数据。对于本指导原则未覆盖的产品,应查询其它FDA指导原则。另外,还可联系负责产品的相应中心以得到指导。 因为已进行结构确证的治疗性生物工程类产品和其它生物制剂之间存在生产和毒 理学差异,因此本指导原则仅适用于药物以及已进行结构确证的治疗性生物工程类产品。对于本指导原则未覆盖的产品,应联系负责产品的相应中心以获得指导。 本指导原则同时适用于商业和个体研究者申请IND。 1 在整篇指导原则中,“药物”一词的含义还包括了已进行结构确证的治疗性生物工程类产品。
临床路径工作管理制度 为加强医疗质量管理,保障医疗安全,控制医疗成本,提高病人满意度,根据卫生部文件精神,结合我科室实际情况,制定临床路径管理制度。 一、单病种临床路径是指由医疗、护理及相关人员在疾病诊断明确后,针对某种疾病或者某种手术制定的具有科学性(或合理性)和时间顺序性的患者照顾计划。 二、开展单病种临床路径均需遵守本制度。 三、单病种临床路径开展应当遵循科学、安全、规范、有效、经济、符合伦理的原则,并与科室功能任务相适应,需具备符合资质的专业技术人员、相应的设备、设施和质量控制体系;各级医务人员要严格执行相关病种的诊疗护理规范、常规,优化质控病种的诊断、治
疗环节质量。 四、设立组织,加强督导。在院领导的领导下,科室内成立单病种质量及临床路径实施小组,由科室主任任组长,医疗、护理人员任成员,主管医师主要负责临床路径的实施,临床路径实施过程的效果评价和分析,质控科负责相关材料的收集、记录和整理及信息上报。 五、质量控制,评估改进。 (一)进入路径病历的选择要求: 1.诊断明确; 2.无其他合并症、并发症和伴发病; 3.病人自愿(签署知情同意书); 4.诊疗过程中未出现其他明显并发症、合并症。 (二)实施过程控制与变异分析 (三)单病种质量控制指标 1.诊断质量指标:出入院诊断符合率、手术前后诊断符合率、临床与病理诊断符合率。 2.治疗质量指标:治愈率、好转率、未愈率、并发症发生率、抗生素使用率、
病死率、一周内再住院率。 3.住院日指标:平均住院日、术前平均住院日。 4.费用指标:平均住院费用、每床日住院费用、手术费用、药品费用、检查费用。 (四)单病种质量控制的主要措施 1.按照卫生部制定的临床路径管理要求,严格执行诊疗常规和技术规程; 2.健全落实诊断、治疗、护理各项制度; 3.合理检查,使用适宜技术,提高诊疗水平; 4.合理用药、控制院内感染; 5.加强危重病人和围手术期病人管理; 6.调整医技科室服务流程,控制无效住院日。 六、高度重视单病种质量管理控制工作,细化工作方案,确定具体工作目标和实施步骤,建立信息报送工作制度,完成单病种每例诊疗后要对病例进行登
目的:为了规范临床试验的操作流程,使每个员工都能正确的掌握操作方法,有序进行临床试验工作,特制定本标准操作规程。 范围:适用于临床研究Ⅱ、Ⅲ期试验整体操作过程。 职责:VPS临床部门项目经理起草;临床部门主管和QA经理审核;公司总裁批准;VPS部门及有关管理部门执行。严格按照本SOP的规定进行临床试验。 内容: 1.试验启动阶段 1.1收集该药物已有的各种资料 理化性质、药理、药化、药效、毒理以及已有的临床研究资料,制作研究者手册。 研究者手册是临床试验开始前的资料汇编。研究者手册的内容一般包括:目录、序言、化学和物理性质、临床前研究、药理学研究、药代动力学研究、毒理学研究、已有临床资料、药品使用信息。 1.2筛选主要研究者 临床基地名录中筛选医院,然后选择合适的主任级医生; 联系主任医生,约定拜访时间; 携研究者手册拜访; 初步交谈,请他阅读研究者手册; 再次拜访,与其讨论方案、试验时间、试验费用等问题; 如此拜访2-3位主任医生,从中选择最佳人选; 1.3试验文件准备
会同CRO、申办者、主要研究者,共同制定临床试验方案、病例报告表、知情同意书样本、原始文件;CRO制定临床试验中其他用表; 1.4其他研究者筛选 从临床基地名录中选择其他可能的研究者,可根据首研者的推荐以及 公司曾经合作的情况进行综合判断; 准备资料:方案、研究者手册、知情同意书样本、病例报告表; 与其谈论方案,要求提供其医院所能提供的病例数、时间进度和经费 预算; 选定合适研究者,征得其医院管理部门的同意; 1.5召开首次研究者会议,讨论试验方案、CRF等; 1.6取得伦理委员会批件 按照GCP的要求,所有临床试验必须得到伦理委员会的批准。在实际进行临床试验时,首先必须取得牵头单位伦理委员会的批准,其他参加单位是否要过伦理根据各家单位的具体要求而定。 伦理委员会将对有关批件、药检报告、研究者手册、知情同意书样本、试验方案、病例报告表进行审批; 1.7试验药品准备 督促申办方进行试验用药品的送检; 生物统计师设计随机分组方案; 根据随机分组方案,设计药品标签; 设计应急信件; 药品包装:为每一个受试者准备好一盒药物,药盒上贴好标签,并装 入相应的应急信件; 1.8各方签订协议
临床路径 管理工作手册 科别: 年度: 东郭中心卫生院
工作手册使用说明 1、本记录本严格遵循巨野县人民医院《2013年单病种临床路径管理实施方案》制作。 2、本工作手册分为九部分:第一部分是《单病种临床路径管理制度》;第二部分是全院实施的单病种临床路径病种名称;第三部分是《单病种临床路径管理实施流程图》;第四部分是科室单病种临床路径管理小组名单及职责;第五部分是科室单病种临床路径标准住院流程及表单; 第六部分是单病种临床路径管理工作计划;第七部分是单病种临床路径管理培训与会议记录;第八部分是科室单病种临床路径变异登记表和记录单;第九部分是科室单病种临床路径实施评价记录及管理工作的汇总与分析;第十部分是附件,《单病种临床路径管理督导反馈单》及《科室单病种临床路径登记表》(登记表已经单独装订成册)。 3、科室每实行1例单病种临床路径病例,都要进行登记;对发生变异的病例要进行分析、讨论与总结,并做好相关记录;每月要进行一次单病种临床路径实施效果评价分析,每季度进行一次单病种临床路径管理工作的汇总、分析与评价,同时做好记录。 4、职能科室要严格履行监管职责,每月对各科室实施的单病种临床路径病例都要进行检查,并将检查情况反馈到科室(反馈表一式两份,一份科室存档,一份主管部门存档)。 5、本工作手册内容作为对科室单病种临床路径管理工作的考核依据,相关人员必须按时、如实填写。 6、科室主任指定专门人员负责本工作手册,检查督导其填写情况,要求各种填报字迹清晰。如有人员变更,应及时移交。 7、记录本按年度编制,每年一册,注意保管。已填写的工作手册由本科室妥善保存备查,保存期限为3年。
:算法的设计思想 本算法采用分支定界算法实现。构造解空间树为:第一个城市为根结点,与第一个城市相邻的城市为根节点的第一层子节点,依此类推;每个父节点的子节点均是和它相邻的城市;并且从第一个根节点到当前节点的路径上不能出现重复的城市。 本算法将具有最佳路线下界的节点作为最有希望的节点来展开解空间树,用优先队列实现。算法的流程如下:从第一个城市出发,找出和它相邻的所有城市,计算它们的路线下界和费用,若路线下界或费用不满足要求,将该节点代表的子树剪去,否则将它们保存到优先队列中,并选择具有最短路线下界的节点作为最有希望的节点,并保证路径上没有回路。当找到一个可行解时,就和以前的可行解比较,选择一个较小的解作为当前的较优解,当优先队列为空时,当前的较优解就是最优解。算法中首先用Dijkstra算法算出所有点到代表乙城市的点的最短距离。算法采用的下界一个是关于路径长度的下界,它的值为从甲城市到当前城市的路线的长度与用Dijkstra算法算出的当前城市到乙城市的最短路线长度的和;另一个是总耗费要小于1500。 伪代码 算法AlgBB() 读文件m1和m2中的数据到矩阵length和cost中 Dijkstra(length) Dijkstra(cost) while true do for i←1 to 50 do //选择和node节点相邻的城市节点 if shortestlength>optimal or mincost>1500 pruning else if i=50 optimal=min(optimal,tmpopt)//选当前可行解和最优解的 较小值做最优解 else if looped //如果出现回路 pruning //剪枝 else 将城市i插入到优先队列中 end for while true do if 优先队列为空 输出结果 else 取优先队列中的最小节点 if 这个最小节点node的路径下界大于当前的较优解 continue
临床试验运行管理流程 步骤一:合作意向洽谈 申办者若有意在我院开展药物临床试验,请首先与GCP中心办公室(下称“中心”)就研究科室、PI 等相关问题进行洽谈。 步骤二:立项审查资料准备 申办者提交“立项申请表”,并按照“药物临床试验送审资料列表”准备临床试验相关材料,交中心秘书 步骤三:立项审核 1.申办者与临床科室和机构共同确定项目PI。 2.PI 提出研究小组成员,填写“临床试验项组成员说明”。 3.机构对送审材料及研究小组成员资质进行审核、立项,确定参加研究者会参会人员。 步骤四:组织/参加研究者会议 1. PI 遵照“PI 工作指引”开展临床试验工作。 2. 若本单位为项目组长单位,PI主持召开研究者会议;若为参加单位,项目组研究骨干参加研究者会议,GCP中心派项目协调员参加。 步骤五:伦理委员会审核 1.申办者/PI将伦理申报材料递交给中心办公室秘书,中心办公室秘书审核资料齐全后,附中心“项目审议表”转交伦理委员会秘书。 2. 申办者/PI依据伦理委员会审查指南配合项目审查,审查后“审批批件”分别交申办方代表及GCP中心办公室秘书各一份存档。 步骤六:临床协议及经费审核 1.取得伦理批件后,申办者、PI、GCP中心就临床试验协议和经费预算进行友好协商,最终正式协议签署后交中心办公室秘书存档。 2.只有在正式协议签署后,才能开始临床试验。 步骤七:临床试验材料及药物的交接 申办者将临床试验材料交项目研究小组,按照“药物的接收、保存、分发、回收、退还的SOP”将试验药物交中心药房。 步骤八:临床专业组项目启动会的召开 由申办者代表与项目PI确定专业组项目启动会召开日程安排,PI负责召集、主持项目启动
白盒测试的测试方法有代码检查法、静态结构分析法、静态质量度量法、逻辑覆盖法、基本路径测试法、域测试、符号测试、Z路径覆盖、程序变异。 其中运用最为广泛的是基本路径测试法。 基本路径测试法是在程序控制流图的基础上,通过分析控制构造的环路复杂性,导出基本可执行路径集合,从而设计测试用例的方法。 设计出的测试用例要保证在测试中程序的每个可执行语句至少执行一次。 在程序控制流图的基础上,通过分析控制构造的环路复杂性,导出基本可执行路径集合,从而设计测试用例。包括以下4个步骤和一个工具方法: 1. 程序的控制流图:描述程序控制流的一种图示方法。 2. 程序圈复杂度:McCabe复杂性度量。从程序的环路复杂性可导出程序基本路径集合中的独立路径条数,这是确定程序中每个可执行语句至少执行一次所必须的测试用例数目的上界。 3. 导出测试用例:根据圈复杂度和程序结构设计用例数据输入和预期结果。 4. 准备测试用例:确保基本路径集中的每一条路径的执行。 工具方法: 图形矩阵:是在基本路径测试中起辅助作用的软件工具,利用它可以实现自动地确定一个基本路径集。 程序的控制流图:描述程序控制流的一种图示方法。 圆圈称为控制流图的一个结点,表示一个或多个无分支的语句或源程序语句 流图只有二种图形符号: 图中的每一个圆称为流图的结点,代表一条或多条语句。
流图中的箭头称为边或连接,代表控制流 任何过程设计都要被翻译成控制流图。 如何根据程序流程图画出控制流程图? 在将程序流程图简化成控制流图时,应注意: n 在选择或多分支结构中,分支的汇聚处应有一个汇聚结点。 n 边和结点圈定的区域叫做区域,当对区域计数时,图形外的区域也应记为一个区域。 如下页图所示 n 如果判断中的条件表达式是由一个或多个逻辑运算符(OR, AND, NAND, NOR) 连接的复合条件表达式,则需要改为一系列只有单条件的嵌套的判断。 例如: 1 if a or b 2 x 3 else 4 y 对应的逻辑为:
民权县人民医院 临床路径知情同意制度 临床路径管理是公立医院改革的核心内容,是改善医疗服务管理的有效措施。按照卫生部开展临床路径管理工作和相关要求,结合我院临床路径病种实际情况,制定本制度: 一、患者知情同意即是患者对病情、诊疗(手术)方案、风险益处、费用等真实情况有了解与被告知的权利,患者在知情的情况下有选择、接受与拒绝的权利。 二、履行患者知情同意可根据操作难易程度、可能发生并发症的风险与后果等情况,决定是口头告知或是同时履行书面同意手续。进行特殊检查、特殊治疗、手术、有创操作、使用贵重物品或药品、试验性临床医疗等医疗活动时,应由患者本人、家属或授权委托人签署知情同意书。 三、由患者本人或其监护人、授权委托人行使知情同意权,对不能完全具备自主行为能力的患者,应由符合相关法律规定的人代为行使知情同意权。 1、不具备完全民事行为能力的患者(如未成年人、
精神疾病患者等),由其法定代理人或其监护人签字; 2、患者因病(如昏迷、气管插管、窒息等)无法正确表达自己真实意愿或无法签字时,由其直系亲属或授权委托人签字;无直系亲属者,由其关系人签字。 3、因实施保护性医疗措施不宜向患者直接说明的,须将有关情况通知患者的直系亲属,由直系亲属签署知情同意书,并及时记录。患者无直系亲属或直系亲属无法签字的,由患者的法定代理人或关系人签署知情同意书。直系亲属、关系人和法定代理人须获得患者的授权委托书。 四、病人存有疑虑拒绝接受检查、治疗,主管医师应进一步做出解释,告知由此可能导致的后果并记录;如病人仍拒绝接受,应向上级医师或科主任报告,并在病程记录中记录。如果病人执意不同意,则不可实行,由病人或授权委托人在知情同意书上签字。 五、由主管医师或其上级医师履行告知义务,对患者及家属提出的问题进行详细的解释。
临床路径的开发与制定的管理制度 第八条我院应按以下条件选择实施临床路径的病种: (一)常见病、多发病; (二)治疗方案相对明确,技术相对成熟,诊疗费用相对稳定,疾病诊疗过程中变异相对较少; (三)结合医疗机构实际,优先考虑卫生行政部门已经制定临床路径推荐参考文本的病种。 第九条临床路径诊疗项目包括医嘱类项目和非医嘱类项目。 医嘱类医疗服务项目应遵循循证医学原则,同时参考卫生部发布,以及组织相关学会(协会)和临床标准组织制定的疾病诊疗常规和技术操作规范,包括饮食、护理、检验、检查、处置、用药、手术等。 非医嘱类服务项目包括健康教育指导和心理支持等项目。 第十条临床路径文本包括医师版临床路径表、患者版临床路径告知单和临床路径变异记录单。 (一)医师版临床路径表 医师版临床路径表是以时间为横轴、诊疗项目为纵轴的表格,将临床路径确定的诊疗任务依时间顺序以表格清单的形式罗列出来。 (二)患者版临床路径告知单 患者版临床路径告知单是用于向患者告知其需要接受的诊疗服务过程的表单。 (三)临床路径变异记录单
变异记录单是用于记录和分析临床路径实施过程中的变异情况 的表单。 实用医院制度 加强制度完善,冲刺等级评审 以病人为中心,提高医疗水平 齐心协力、鼓足干劲、全力迎接“三甲”医院复审。 创“三甲”是每一个人的事,重在全院参与。 以“创三甲”为契机,加强医院内涵建设,全面提高医疗水平。 人人都是得分手,“三甲”复审作贡献。 加强医院文化建设,争创“三甲”医院称号。 热烈欢迎三甲评审工作组莅临检查指导 全院参与,共创“三甲”。 加班加点只争朝夕时不我待誓过三甲 以评促建以评促改以评促转以评促管 三甲目标困难大,创建要靠你我他。 争创三甲院,全员齐努力。 树岗位新风,争“三甲”荣誉。 加强细节管理,推进学科建设。 提高医疗质量,构建和谐医院。
临床路径诊疗流程 1、路径跳转的情况一般发生在病人出现符合临床路径诊断的其他专科性疾病,继续在本科室治疗时使用(本科室有新疾病的路径表单)。如需要转科治疗或者新疾病无临床路径表单,则执行终止路径。 三甲医院标准规定:对符合进入临床路径标准的患者,入组率≥50%,入组完成率≥70%。
我最难忘的一位老师 先生姓谢,名然之,是我初二的语文老师。五十多岁,头发却只剩下周边,中间秃了,同学们戏称为“地中海”。灰色旧西装,不扣扣子,露出浅色衬衫,这是他的经典形象。印象中他好像一直是这样的造型,只是夏天把外套脱了,冬天加了毛衣罢了。 第一次和谢老师接触并不光彩,我和几个同学因没写作业而被他用尺子在手心上打了三下。后来在一次语文课上,谢老师宣读学生写的优秀作文,我被点名,他看到我,微张嘴巴,露出惊喜:“是你!”话音未落,两人相视而笑。而后他表扬我行文流畅,情感丰富,说时带着欣赏和欣慰的表情,可见此公作为师者对学生的爱憎分明,奖罚有度。 然而先生用戒尺罚学生早已淘汰,甚至在鲁迅那个年代也已不普遍化。鲁迅的儿子周海婴长大上学,有一次鲁迅和朋友说起:“听说那个先生(指海婴的老师)是要打手心的!怎么现在还有这样的事吗?”小时候被私塾先生打过手心的鲁迅深知戒尺的厉害,对海婴有可能也被打手心感到担忧和心疼。而从鲁迅惊讶的语气可以推断当时用戒尺打手心已开始渐渐摒弃。但是我们的谢老师,竟然时隔近百年之后仍保留着此传统,亦可见其老派的一面,而我估计他小时候也被他的老师用戒尺打过。不过这也本无可厚非,但其传统而严厉的性格也给他招惹过麻烦。 那天谢老师给我们上《送东阳马生序》,他在正式上课前反复强调这篇课文的重要性,说很多人读了《送东阳马生序》之后开始用功读书等等。我是早已听多了这种话,觉得厌倦了,只等着他讲正文,而从他饱含深情,掷地有声的讲话也足见其对这篇课文的重视。可就在谢老师这样苦口婆心的状态下,后排有学生却一直在聊天,完全没有注意到谢老师的警告,于是谢老师就火了,扔下书本,冲下讲台,把吵得最响的学生“教训”了,边“教训”边说着:“你为什么要打扰我上课!你为什么要打扰我上课!”那个学生当时也没还手,但下课后告诉了校长。后来校长就把谢老师叫去了。 下一节语文课,谢老师在全班同学面前说:“上节课我打了××同学,是我的不对,校长也说了,不应该体罚学生,我向××同学道歉。”于是这件事也就随着谢老师说完而结束了。只是我想,也许在谢老师的心里,他会感到郁闷和无奈,觉得自己所持的教育方式是否真的已经不合时代,一个先生用戒尺打学生是天经地义的时代无可挽留的坠落,只留在了他的记忆里,而眼前,是不能体罚,老师需要向学生道歉的新时代。但是在我看来,谢老师严厉,他的方式可能不合时宜,但他的本意还是好的。总比那些虽然不体罚,表面上和学生相处很融洽,实际对学生毫不负责的老师要好。 其实也正是谢老师这种敢爱敢恨,敢作敢当的人格让我对其增了敬意。谢老师还是全校唯一一个出过书的老师。他编了一本类似于歇后语辞典的书,他说为了编好这本书,十几年来每个假期,拒绝了几乎所有游玩的邀请。又因为谢老师并非名人,他出书需要自费出版,他说他把这些年来不抽烟不喝酒省下来的钱全都花在出书上了。其决心之大可见一斑,而我又佩服他的坚持。 清风吹过枫林,叶子发出沙沙的声响,还是那件灰色旧西装,还是那个瘦瘦高高的身影,裤脚出门前也忘了整理而保持了一天的长短不一。一个五十多岁的语文老师夹着备课本,走在学校新铺的石板路上,索寞的存在于这个狂飙突进的时代。 2、
临床路径管理制度 一、成立医院临床路径管理委员会和临床路径管理工作实施小组,工作开展是在医院试点工作领导小组指导下,由科室试点工作实施小组具体实施,院长任领导小组组长,科室主任为实施小组第一责任人。 二、临床路径管理委员会全面负责全院的临床路径管理工作,组织对相关临床与医技人员进行教育培训,对院内各部门统一协调、督导并定期检查各科室临床路径执行情况。 三、临床路径管理委员会定期召开会议,对本院临床路径实施效果的评估与分析并将结果及时反馈给临床路径实施科室。
四、科室临床路径工作实施小组应会议,根据本科室临床路径执行情况及时进行相应调整和改进对试点疾病的质量、费用及成本进行分析评估,总结影响试点疾病质量监控的问题,对领导小组的反馈意见及时落实,采取措施,持续改进。 五、临床路径管理表单的制定应根据卫生部颁发的临床路径管理病种和文本,结合本院实际情况,严格按照卫生部临床路径管理要求,按照临床路径确定的诊疗流程实施诊疗。 六、尊重患者知情同意权,在患者入院时向其详细介绍临床路径的目的、意义、以及相应的诊疗项目等,并将患者评估结果和实施方案通知护理人员按路径流程实施。
七、经治医师应根据当天诊疗项目完成情况以及病情的变化对当日的变异情况进行分析、处理并做好记录。 八、诊治过程中出现变异的,当及时将变异情况记录在临床路径表单中,并对变异情况定期进行分析总结。对于变异病例,科内进行讨论,找出变异的原因,提出处理意见,讨论情况应写入病程记录。 九、对于因各种情况必须退出临床路径管理的病人,应进行告知,根据病人情况,按相关诊疗常规实施后续治疗,病人退出情况应有记录并定期分析总结。
临床路径工作管理制度 一、科室成立临床路径管实施小组。实施小组由科室主任组长,临床科室医疗、护理人员和相关科室人员任成员。 二、临床路径实施小组职责: 负责临床路径相关资料的收集、记录和整理;提出病种选择建议制订临床路径文本并及时进行修订。在实施过程中,进行效果评价与分析 三、实施小组设立个案管理员,由临床科室具有副高级以上技术职称的医师担任。 四、个案管理员职责: 负责与医务科日常联络;牵头临床路径文本的起草工作。指导每日临床路径诊疗项目的实施,指导经治医师分析、处理患者变异,加强与患者的沟通。根据临床路径实施情况,定期汇总。 五、实施小组负责对科室人员进行培训,培训内容包括: 1、临床路径基础理论、管理方法和相关制度; 2、临床路径主要内容、实施方法和评价制度。 六、临床路径实施流程 1、经治医师完成患者的检诊,会同科室个案管理员对住院患者进行临床路径的准入评估; 2、符合准入标准的,按照临床路径确定的诊疗流程实施诊疗,根据医师版临床路径表开具诊疗项目,向患者介绍住院期间为其提供诊疗服务的计划,并将评估结果和实施方案通知相关护理组;
3、相关护理组在为患者作入院介绍时,向其详细介绍其住院期间的诊疗服务计划(含术前注意事项)以及需要给予配合的内容; 4、经治医师会同个案管理员根据当天诊疗项目完成情况及病情的变化,对当日的变异情况进行分析、处理,并做好记录; 5、医师版临床路径表中的诊疗项目完成后,执行(负责)人应当在相应的签名栏签名。 七、进入临床路径的患者应当满足以下条件:诊断明确,没有严重的合并症,能够按临床路径设计流程和预计时间完成诊疗项目。 进入临床路径的患者出现以下情况之一时,应当退出临床路径: 1、在实施临床路径的过程中,患者出现了严重的并发症,需要改变原治疗方案的; 2、在实施临床路径的过程中,患者要求出院、转院或改变治疗方式而需退出临床路径的; 3、发现患者因诊断有误而进入临床路径的; 4、其他严重影响临床路径实施的情况。 八、临床路径的变异是指患者在接受诊疗服务的过程中,出现偏离临床路径程序或在根据临床路径接受诊疗过程中出现偏差的现象。变异的处理步骤: 1、记录。医务人员应及时将变异情况记录在医师版临床路径表中,记录应当真实、准确、简明。 2、分析。经治医师应当与个案管理员交换意见,共同分析变异原因并制订处理措施。
病例报告表(Case Report Form) 受试者姓名: 家庭地址: 联系电话: 试验中心名称: 申办单位:延吉喜来健医疗器械有限公司
在正式填表前请认真阅读下列填表说明 病例报告表填写说明: 1.筛选合格者填写病例报告表。 2.病例报告表填写务必准确、清晰,不得任意涂改,错误之处纠正时需用横线居中化出,并签署修改者姓名缩写及修改时间。 3.填写记录一律用钢笔或碳素签字笔。 4.患者姓名拼音缩写四个需填满,两字姓名填写两字拼音前两个字母;三字姓名填写三字首字母及第三字第二字母;四字姓名填写每个字的首字母。5.表中凡有“□”的项,请在符合的条目上划“√”。表格中所有栏目均应填写相应的文字或数字,不得留空。 6.所有检验项目因故未查或漏查,请填写ND;具体合并用药剂量和时间不明,请先写NK。 7.试验期间应如实填写合并用药记录表、不良事件记录表。记录不良事件的发生时间、严重程度、持续时间、采取的措施和转归。如有严重不良事件发生(包括临床验证过程中发生需住院治疗、延长住院时间、伤残、影响工作能力危及生命或死亡、导致先天畸形等事件),必须立即通知主要研究单位伦理委员会及申办单位。
8.临床试验应严格按照临床试验方案要求进行。试验不同时期需完成的检查和需记录的项目,请对照临床流程图执行。
受试者知情同意书 敬爱的患者: 我们现在正在进行一项临床研究,该项临床研究是经吉林省药品监督管理局备案的,研究的目的是评价物理治疗的高科技产品----温热电位治疗仪的疗效和安全性。 温热磁疗仪是XXX医疗器械有限公司根据韩国专利技术研制生产的医疗器械,本治疗仪是通过XXXXXXXXXXXXXXXXXXXXXXXXXXXXXX机理,主要用于XXXXXXXXXXXXX 治疗。具有平衡机体阴阳,增强脏腑机能,促进新陈代谢,调节植物神经,改善心脑血管血液供应,促进血液循环,调节血管张力,降低血液粘稠度,防治动脉粥样硬化,促进组织的
临床路径及单病种管理工作制度 一、单病种临床路径是指由医疗、护理及相关人员在疾病诊断明确后,针对某种疾病或者某种手术制定的具有科学性(或合理性)和时间顺序性的患者照顾计划。 二、院内各科室开展单病种临床路径均需遵守本制度。 三、各科室单病种临床路径开展应当遵循科学、安全、规范、有效、经济、符合伦理的原则,并与科室功能任务相适应,需具备符合资质的专业技术人员、相应的设备、设施和质量控制体系;各级医务人员要严格执行相关病种的诊疗护理规范、常规,优化质控病种的诊断、治疗环节质量。 四、设立组织,加强督导 在院长、分管院长的领导下,开展单病种质量及临床路径工作,并负责该工作的管理、督导。医院成立临床路径管理领导小组,隶属于医院医疗质量管理委员会,主要负责制定临床路径管理有关规章制度,对我院单病种质量及临床路径管理质量进行指导、监控和评估,协调临床路径实施过程中遇到的问题。 相关科室成立单病种质量及临床路径实施小组,由临床科室主任任组长,医疗、护理人员任成员,主管医师主要负责临床路径的实施,临床路径实施过程的效果评价和分析,医务科负责相关材料的收集、记录和整理及信息上报。 五、质量控制,评估改进 (一)进入路径病历的选择要求:
1.诊断明确; 2.无其他合并症、并发症和伴发病; 3.病人自愿(签署知情同意书) 4.诊疗过程中未出现其他明显并发症、合并症。 (二)实施过程控制与变异分析 (三)单病种质量控制指标 1.诊断质量指标:出入院诊断符合率、手术前后诊断符合率、临床与病理诊断符合率。 2.治疗质量指标:治愈率、好转率、未愈率、并发症发生率、抗生素使用率、病死率、一周内再住院率。 3.住院日指标:平均住院日、术前平均住院日、。 4.费用指标:平均住院费用、每床日住院费用、手术费用、药品费用、检查费用等。 (四)单病种质量控制的主要措施 1.按照卫生部制定的临床路径管理要求,严格执行诊疗常规和技术规程; 2.健全落实诊断、治疗、护理各项制度; 3.合理检查,使用适宜技术,提高诊疗水平; 4.合理用药、控制院内感染; 5.加强危重病人和围手术期病人管理; 6.调整医技科室服务流程,控制无效住院日。 六、做好临床路径统计工作制度。
临床路径实施流程图 r f 医疗计划护理人员下发患者版临床路 径告知单 护理人员执行医疗计划,并协助做好变异监测 提出改进建议 —路径实施流程、实施流程部分退出临床路径 进入临床路径的患者变异分析与记录
1、经治医师完成患者的检诊工作,与上级医师对住院患者进行临床路径的准入评 2、符合准入标准的,按照临床路径确定的诊疗流程实施诊疗,根据医师版临床路径表开具诊疗项目,向患者介绍住院期间为其提供诊疗服务的计划,并将评估结果和实施方案通知相关护理组; 3、相关护理组在为患者及家属作入院介绍时,向他们详细介绍其住院期间的诊疗计划以及需要给予配合的内容; 4、经治医师根据当天诊疗服务完成情况及病情的变化,对当日的变异情况进行分析、处理,并做好记录; 5、医师版临床路径中的服务项目完成后,执行(负责)人应在相应的签名栏签名。 ?进入临床路径的患者应满足以下条件:诊断明确、没有严重的合并症、能够按 临床路径设计流程和预计时间完成诊疗项目的患者。 ?出现以下情况时,患者应当退出临床路径: (1)在实施临床路径的过程中,患者出现了严重的并发症,需要转入其它科室实施治疗的; (2)在实施临床路径的过程中,患者要求出院、转院或改变治疗方式而需退出临 床路径的; (3)发现患者因诊断有误而进入临床路径的; (4)患者出现严重的医疗相关感染等情况不适应继续完成临床路径的。 ?设立紧急情况警告值管理制度:警告值是指患者在临床路径治疗过程中出现严重异常情况,处于危险边缘,应迅速给予患者有效干预措施和治疗。 二、变异的管理:是临床路径管理的重点 1、变异的定义:指病人在接受诊疗服务的过程中,出现偏离临床路径程序或在根据临床路径接受诊疗过程中出现偏差的现象。 2、变异的分类:变异有正负之分,负变异是指计划好的活动没有进行(或 结果没有产生),或推迟完成,如延迟出院、CT 检查延迟;正变异是指计划好的活动或结果提前进行或完成,如提前出院、CT 检查提前等。 3、按照变异产生的原因分为:疾病转归造成的变异,医院系统造成的变异;医务人员造成的变异;病人需求造成的变异,退出五种情况。 4、变异的处理的遵循以下步骤:
附件 新药I期临床试验申请技术指南 一、前言 为帮助新药注册申请人(药品企业、科研机构和科研人员)申请I期临床试验,提高新药研发与审评效率,保护受试者安全与权益,保证临床试验质量,特发布本技术指南。 本指南阐述了新药在我国开展首次临床试验时需要向国家食品药品监督管理总局药品审评中心(以下简称药审中心)提供的信息。 本指南的目的是:明确新药I期临床试验的技术要求,提高I 期临床试验申报资料的质量;通过规范I期临床试验资料的数据要求,缩短新药研发周期,加快新药上市进程。 本指南适用于创新药和改良型新药,包括化学药品和治疗用生物制品(细胞和基因治疗产品除外)。 二、咨询与沟通交流 如果申请人在研发及申请临床试验过程中有疑问,可通过药审中心网站查询相关指导原则,也可以按照相关规范通过药审中心网站“申请人之窗”就相关问题进行咨询。 递交新药临床试验申请前,申请人可按照《药物研发与技术审评沟通交流管理办法》所规定的方法与工作程序,申请与药审中心召开临床试验申请前会议。 申请人与药审中心间的沟通有助于提高临床试验申请的质量,加快后续研究与审评的进程。 三、I期临床试验申请的技术要求
(一)资料格式及内容 I期临床试验申请的申报资料应以纸质资料和电子资料方式提交,电子资料可以CD的形式送交。格式和内容可参照研究人用药品注册技术要求国际协调会(ICH)通用技术文件(CTD)的要求整理提交。 (二)介绍性说明和总体研究计划 介绍性说明应包括新药的名称、所有的活性成分、药理作用类别、结构式(如果已知)、剂型、制剂处方、给药途径、临床试验目的等。如果有所研究药物用于临床的经验,应提供简短概述,包括在其他国家的研究和上市的经验;若没有,标题下写“无”。 总体研究计划应总结申请临床试验方案的设计依据,主要为拟定的适应症、受试者人群、受试者数量、给药方案(剂量、给药间隔、给药持续时间等)、药物安全性评价方法、风险控制计划等,根据已有信息预期的任何安全性(重要的已确定风险、重要的潜在风险、重要的缺失的资料等)的风险论证。 (三)研究者手册 研究者手册是有关试验药物在进行人体研究时已有的药学、非临床与临床研究(如有)资料总结,旨在为临床研究者提供所研究药物的信息,以保证受试者安全。 当有新的重要信息时,申请人应及时更新研究者手册,使其包括对研究药物的所有重要研究信息的总结。更新的研究者手册应及时报送药审中心。研究者手册的格式和内容可参照ICH E6有关章节撰写。 研究者手册应包括如下内容: 1.封面页:包括药物名称,注册申请人名称,完稿或更新日期
第一部分总则 第一条:为了保证新药临床试验过程中遵循科学和伦理道德的原则,使数据的采集、录入和报告做到及时、完整、准确和一致,使受试者的权益和健康得到保护,并保障其安全,保证临床试验遵循己批准的方案、药物临床试验质量管理规范(GCP)和有关法规,使试验结论科学、可靠,根据《中华人民共和国药品管理法》、《药物临床试验质量管理规范》、《药品注册管理办法》、《赫尔辛基宣言》及ICH《人体生物医学研究国际道德指南》等相关法规文件精神,制定本标准操作程序。 第二条:药品临床试验依其流程、内容和进程不同,将其划分为临床试验前的准备、启动临床试验、临床试验过程、中期协调会和结束临床试验等五个阶段。 第三条:本标准操作规程是根据药品Ⅱ期临床试验设计要求确立,临床进行的Ⅲ、Ⅳ期临床试验包括部分生物等效性试验均参照本程序执行。 第二部分临床试验前的准备 第四条:申办者对临床试验中心的遴选。 ⑴申办者在上报药物的临床前研究资料后,根据所申请药物的性质、作用特点、功能主治以及疾病的流行病学、样本量的大小和药品临床试验基地的专业特长等,初步遴选临床试验参加单位和确定参加单位的数量。 ⑵对初选单位的专业特长、研究资质、人员组成结构、任职行医资格、相关临床试验检查和检测设备以及参研人员参加GCP培训等情况进行现场考察,确认其资质、资源、能力和承担任务量的大小。 ⑶根据现场考查结果,首先确定临床试验组长单位,经与之协商确立临床试验参加单位,并据此草拟临床试验的《多中心临床试验协调委员会联络表》和《临床试验参加单位初选报告》。 ⑷国家食品药品监督管理局临床试验批文下达后,申办者根据批文精神,与临床试验组长单位一道最终确定临床试验参加单位。 第五条:申办者起草临床试验文件。 ⑴申办者与研究者共同商定起草并签署试验方案、CRF和知情同意书等临床试验文件。 ⑵申办者起草《研究者手册》,或其替代文件《供临床医师参考的临床前研究药效学、毒理学试验综述》。 ⑶申办者起草《药品临床试验标准操作规程(SOPs)》、《药品临床试验监查工作标准操作规程(SOPs)》、《药品临床试验研究者履历表》、《药品临床试验筹备会议签到表》、《药品临床试验药品发放、回收、清点登记表》、《受试者用药记录卡》和《药品临床试验实验室检查参考正常值范围表》等文件。 ⑷申办者成立数据和安全监查委员会/数据监查委员会和试验项目小组,根据试验方案设计要求和项目标准由项目小组成员共同制定病例报告表(CRF),监查员可参与部分设计工作。