Microwave-assisted rapid synthesis of Ag nanoparticles graphene nanosheet composites
- 格式:pdf
- 大小:1.87 MB
- 文档页数:10

2d-cof的合成方法English Answer:2D-COFs (covalent organic frameworks) are a class of crystalline porous materials with a layered structure. They have attracted considerable attention due to their unique properties, such as high surface area, tunable porosity, and chemical stability. Various methods have been developed for the synthesis of 2D-COFs, including:1. Solution-based synthesis.This method involves the direct reaction of organic building blocks in a solvent. The reaction is typically carried out in a closed vessel at elevated temperature and pressure. The resulting 2D-COF is obtained as a precipitate or powder.2. Interfacial synthesis.This method utilizes the interface between two immiscible solvents to promote the formation of 2D-COFs. The organic building blocks are dissolved in the two solvents, and the reaction takes place at the interface. The resulting 2D-COF forms as a thin film at the interface.3. Template-assisted synthesis.This method uses a template to direct the formation of 2D-COFs. The template can be a metal ion, a metal-organic framework, or a porous support. The organic building blocks are assembled on the surface of the template, and the resulting 2D-COF conforms to the shape of the template.4. Vapor-phase synthesis.This method involves the sublimation of organicbuilding blocks in a vacuum chamber. The vaporized building blocks react in the gas phase to form 2D-COFs, which are deposited on a substrate.5. Microwave-assisted synthesis.This method utilizes microwave irradiation toaccelerate the synthesis of 2D-COFs. The microwaveirradiation provides rapid and efficient heating, which reduces the reaction time and improves the yield of 2D-COFs.The choice of synthesis method depends on the desired properties of the 2D-COF, such as its structure, porosity, and crystallinity. Solution-based synthesis is a versatile method that can be used to prepare a wide variety of 2D-COFs. Interfacial synthesis is suitable for preparing thin films of 2D-COFs. Template-assisted synthesis allows forthe preparation of 2D-COFs with specific shapes and sizes. Vapor-phase synthesis is a relatively new method that canbe used to prepare highly crystalline 2D-COFs. Microwave-assisted synthesis is a rapid and efficient method that can be used to prepare 2D-COFs with high yields.Chinese Answer:2D-COF的合成方法。

铜和对苯二甲酸金属有机骨架材料绿色合成那立艳;张伟;华瑞年;宁桂玲【摘要】采用以水为唯一溶剂的绿色路线合成了金属有机骨架(MOFs)材料。
通过将羧酸类配体转化为水溶性铵盐配体,在水溶液中探索了金属有机骨架材料的绿色合成。
以醋酸铜(Cu(OAc)2)和对苯二甲酸(H2BDC)甲胺盐在水溶液中反应,获得了化合物CuBDC‐H2O 。
同时作为参照,以Cu(OAc)2和H2 BDC在N ,N‐二甲基甲酰胺(DM F)和水的混合溶液中,获得了化合物CuBDC‐DMF 。
通过粉末XRD、IR、SEM、TGA以及N2吸附测试等手段对两种产物进行了表征,结果显示经140℃真空热处理10 h后,化合物CuBDC‐DM F 具有和在水溶液中直接获得的产物CuBDC‐H2 O相同的晶体结构及相似的性质,而且以水作为反应介质的新的合成路线具有便捷、快速、低成本及节约能源等特点。
%A green process for the synthesis of metal‐organic frameworks (MOFs) particles that uses water as the only solvent is presented . The strategy is to convert carboxylic acids ligands into carboxylate ammonium salts ligands which have high solubility in water ,thus allowing the synthesis to be performed in water .CuBDC‐H2O (BDC=1 ,4‐benzenedicarboxylate) is synthesized by mixing aqueous solutions of Cu (OAc)2 and the methylammonium salt of H2 BDC .As a reference ,CuBDC‐DMF is obtained by mixing water solution of Cu (OAc)2 with DMF solution of H2BDC .The two compounds are characterized by powderXRD ,IR ,SEM ,TGA and N2 and adsorption measurements . The experimental results reveal that after evacuation at 140 ℃ for 10 h ,CuBDC‐DMF has the same crystal structure and properties with CuBDC‐H2 O ,andthe new synthetic route performed in water solution has the advantages of being straight forward ,rapid ,cheap and energy‐saving .【期刊名称】《大连理工大学学报》【年(卷),期】2013(000)004【总页数】5页(P474-478)【关键词】金属有机骨架;绿色合成;孔材料【作者】那立艳;张伟;华瑞年;宁桂玲【作者单位】大连理工大学精细化工国家重点实验室,辽宁大连 116024; 大连民族学院生命科学学院,辽宁大连 116600;大连民族学院生命科学学院,辽宁大连116600;大连民族学院生命科学学院,辽宁大连 116600;大连理工大学精细化工国家重点实验室,辽宁大连 116024【正文语种】中文【中图分类】O6410 引言金属有机骨架(MOFs)材料是在微孔晶体材料上发展起来的一种新型功能材料,由于其在气体存储与分离、催化、药物传递和传感器等方面的潜在应用而备受关注[1-5].目前用于合成MOFs材料的方法有溶剂诱导沉淀法[6]、溶剂热合成法[7]、反相微乳液法[8]、微波辅助合成法[9]、超声法[10]等.然而这些合成方法往往存在反应时间长、反应温度高、大量使用有机溶剂或复杂的反应设备等缺点,对其实际应用造成了制约,因而开发快速、简便、节约能源和环境友好的合成路线具有重要意义.DMF由于具有较高的沸点(140℃)和对羧酸类配体良好的溶解能力,是合成MOFs材料经常使用的溶剂.DMF分子中的氧原子通常以配体或客体分子的形式填充到孔道当中,因而,为了获得金属活性中心和开放的骨架结构,通常要对合成的产物进行热处理,以除去配位及孔道中的DMF分子.如果能在反应中以水代替DMF 溶剂,那么不仅能够节约成本,而且在热处理过程中,只需将水分子除去即可,可以明显缩短热处理过程的时间,降低热处理温度,进而有效地节约能源. 本文的合成策略是先将非水溶性的配体对苯二甲酸与甲胺反应,生成水溶性的甲胺羧酸盐;然后利用水溶性配体与金属盐在水溶液中完成聚合反应.由于CuBDC具有独特的磁性、优良的气体分离性能及催化活性[11],本文分别在水溶液及DMF溶液中进行CuBDC 的合成探索,通过对比两种反应体系的合成条件及产物性能,探索羧酸类配体构筑的MOFs材料的快速、高效、低能耗合成.1 实验部分1.1 试剂与仪器原料:试剂均为市售分析纯.仪器:日本岛津XRD-6000 衍射仪;Nicolet FT-IR 740红外光谱仪;日本日立S-4800高分辨电子显微镜;Micromeritics ASAP 2020表面分析仪;美国Perkin-Elmer SDTA 851热失重分析仪.1.2 合成1.2.1 水溶性配体前驱体((NH3CH3)2BDC)的合成取1.667g(10mmol)对苯二甲酸,搅拌下滴加20%~30%的甲胺水溶液至溶液澄清,于真空干燥箱中20℃下减压至溶液黏稠,室温挥发得白色晶体.将上述晶体用水转移至100 mL 容量瓶中,配制成0.1mol/L甲胺盐水溶液,反应方程式如下:1.2.2 CuBDC-H2O的合成称取0.03 g Cu(OAc)2·H2O溶于7 mL 水中,搅拌下滴入0.1mol/L(NH3CH3)2BDC 溶液3 mL,马上产生浅蓝色沉淀,搅拌2h后离心.然后将产物经乙醇洗涤—超声分散—离心两次,自然干燥.1.2.3 CuBDC-DMF的合成称取0.03 g Cu(OAc)2·H2O溶于4mL水中,再称取0.04g H2BDC溶于6 mL DMF 中,两种溶液充分溶解后,搅拌下将配体溶液滴入金属盐水溶液中,马上产生蓝色沉淀,搅拌2h后,静置2h,离心.然后将产物经乙醇洗涤—超声分散—离心两次,自然干燥.2 结果与讨论2.1 XRD 表征两种产物及经140℃真空热处理10h 后CuBDC-DMF的粉末XRD 谱图见图1.由图可知有机溶液中获得的CuBDC-DMF 与水溶液中获得的CuBDC-H2O 峰位明显不同(图1曲线a、c),说明两者结构不同.CuBDC-DMF 是一种已知结构的化合物[11-12](与本文合成方法不同),具有开放的骨架结构,结构中的金属中心铜为五配位,4个氧原子来自于BDC基团,另外一个氧原子来自于DMF分子(图2(a)).当CuBDC-DMF 在140℃真空热处理10h脱除了DMF 基团后,其XRD 谱图(图1曲线b)与CuBDC-H2O(图1 曲线c)相同.这说明CuBDC-DMF高温脱除溶剂分子后,与在水溶液中一步获得的产物CuBDCH2O 具有相同的结构,图2(b)是CuBDC-H2O 的可能结构.图1 粉末XRD 谱图Fig.1 Powder X-ray diffraction(PXRD)patterns图2 晶体结构Fig.2 Crystal structures2.2 红外表征用KBr压片法测定了CuBDC-H2O、CuBDCDMF及经140℃真空热处理10h 后的CuBDCDMF红外光谱,见图3.通过对比吸收峰的位置,发现CuBDC-DMF经热处理后的红外谱图(图3曲线b)与CuBDC-H2O(图3曲线c)基本一致,两个谱图中,1 575cm-1与1 398cm-1处的吸收峰归属为BDC配体中羧酸基团的非对称伸缩振动和对称伸缩振动;1 690~1 730cm-1没有出现羰基的吸收峰,说明配体上的羧酸基团已全部去质子化.而在CuBDC-DMF 的红外谱图(图3曲线a)中,1 688cm-1处的吸收峰为客体DMF中羰基的吸收峰,1 628cm-1处的吸收峰可归属为与金属中心配位的DMF 中羰基的吸收峰,氧原子与金属中心配位作用的存在,使得吸收峰向短波数方向移动.1 292cm-1处的吸收峰为酰胺中C—N 键的伸缩振动.以上3个与DMF相关的特征吸收峰在真空热处理后消失,说明通过加热,已成功去除客体DMF 分子及配位的DMF基团,并且其结构与水溶液中一步获得的产物CuBDC-H2O 相同.图3 红外谱图Fig.3 IR spectra2.3 SEM 表征图4(a)和图4(b)分别是CuBDC-DMF 和CuBDC-H2O 的扫描电镜照片,可以看出,两种合成条件下获得的产物均为方形片状晶体.CuBDCDMF晶体长和宽约为400nm,厚度在50nm 左右,与文献报道该化合物的形貌基本一致[12];CuBDC-H2O 晶体长为10μm,宽为4μm,厚度在1μm 左右.由于在水溶液中进行反应时,配体已经提前完成了去质子化的过程,因而反应速度比较快,所以CuBDC-H2O的尺寸明显大于CuBDC-DMF.图4 扫描电镜照片Fig.4 SEM images2.4 吸附测试采用-196℃下氮气吸附的方法对两种不同条件下获得的化合物进行了氮气吸附等温测试.样品量约为0.2g,测试前均在140℃真空干燥10h,采用BET 方法在p/p0=0.05~0.33获得比表面积.从图5可以看出,两种化合物在相对低的压力范围内(p/p0<0.1),都有一个明显的台阶存在,然后曲线趋于平缓,吸附等温曲线为I型,说明了样品中微孔孔道的存在.CuBDC-DMF和CuBDC-H2O 在低压区(p/p0=0.01)时,氮气的吸附量分别为65cm3/g和68cm3/g,随着压力的升高,吸附量略有增加,在高压区(p/p0=1.0)时,氮气的吸附总量分别为89cm3/g和80cm3/g,BET 比表面积分别为407m2/g和439m2/g.从以上结果可以看出,在CuBDC-DMF脱除客体及配位的DMF基团后,与在水溶液中获得的产物CuBDC-H2O 具有相似的比表面及孔结构信息.图5 氮气吸附等温曲线Fig.5 N2adsorption isotherms采用巨正则蒙特卡罗法(grand canonical Monte Carlo)模拟了CuBDC-DMF 在77K时的氮气吸附曲线[13],见图5.分子模型是通过CuBDC-DMF的实验XRD 晶体学数据建立的.采用通用力场(UFF)描述MOF骨架原子[14],N2力场参数为σ=0.331nm,ε=36 K.利用Music code软件进行模拟[15],并用BET 方法对模拟吸附等温线的线性段(p/p0=0.001~0.015)进行拟合,进而得到BET 方程参数及比表面积,结果如图6所示.模拟吸附曲线及理论比表面积均与实验结果接近.图6 BET 法比表面积计算Fig.6 Specific surface area calculation by the BET method2.5 热稳定性测试对CuBDC-DMF、CuBDC-H2O 及经140℃真空热处理10h后CuBDC-DMF 进行了热失重分析,温度区间为30~500℃,氮气气氛.CuBDCDMF热失重曲线(图7 曲线a)显示该化合物在90~250℃失重值约为27%,100℃左右的失重可归属为水分子的脱除,在140~250℃的失重可归属为配体DMF的失去,温度超过300℃,化合物的骨架开始塌陷.图7中曲线c是CuBDC-H2O的热失重曲线,可以看出化合物在200℃之前基本没有失重,骨架结构可稳定存在至300℃.图7中曲线b 是CuBDC-DMF 经真空热处理后的热失重情况,热失重曲线与CuBDC-H2O 的基本相同,说明热处理过程除去了配位的DMF 分子后,CuBDC-DMF与CuBDC-H2O 拥有相同的骨架热稳定性能.图7 热失重曲线Fig.7 TGA curves3 结论(1)通过水溶性羧酸甲胺盐配体的合成,开发出一种一步绿色合成金属有机骨架材料的新方法.(2)对比有机体系和水体系中CuBDC 合成过程及产物性质,发现有机体系下获得的配合物在真空热处理除去配位的溶剂分子后,与在水溶液中一步获得的产物具有相同的结构和相似的性质.但以水溶液代替有机溶剂使用,不仅降低了合成成本,减少了污染,同时缩短了真空热处理过程的时间,降低了热处理温度.(3)本文开发的水溶液合成路线具有简便、快速、节能和环保的特点,该绿色合成路线具有可推广性,可用于更多以羧酸类配体构筑的金属有机骨架材料的合成.【相关文献】[1]Murray L J,Dinca M,Long J R.Hydrogen storage in metal-organic frameworks [J].Chemical Society Reviews,2009,38(5):1294-1314.[2]Li J R,Kuppler R J,Zhou H C.Selective gas adsorption and separation in metal-organic frameworks[J].Chemical Society Reviews,2009,38(5):1477-1504.[3]WU Chuan-de,HU Ai-guo,ZHANG Lin,et al.A homochiral porous metal-organic framework for highly enantioselective heterogeneous asymmetric catalysis [J].Journal of the American Chemical Society,2005,127(25):8940-8941.[4]Horcajada P,Serre C,Vallet-Regi M,et al.Metalorganic frameworks as efficient materials for drug delivery [J].Angewandte Chemie International Edition,2006,45(36):5974-5978.[5]CHEN Bang-lin,WANG Liang-bo,XIAO Yunqing,et al.A luminescent metal-organic framework with Lewis basic pyridyl sites for the sensing of metal ions[J].Angewandte Chemie International Edition,2009,48(3):500-503.[6]Oh M,Mirkin C A.Chemically tailorable colloidal particles from infinite coordinationpolymers [J].Nature,2005,438(7068):651-654.[7]Jung S,Oh M.Monitoring shape transformation from nanowires to nanocubes and size-controlled formation of coordination polymer particles [J].Angewandte Chemie International Edition,2008,47(11):2049-2051.[8]Rieter W J,Taylor K M,An H,et al.Nanoscale metal-organic frameworks as potential multimodal contrast enhancing agents [J].Journal of the American Chemical Society,2006,128(28):9024-9025.[9]Ni Z,Masel R I.Rapid production of metal-organic frameworks via microwave-assisted solvothermal synthesis[J].Journal of the American Chemical Society,2006,128(38):12394-12395.[10]QIU Ling-guang,LI Zong-qun,WU Yun,et al.Facile synthesis of nanocrystals of a microporous metal-organic framework by an ultrasonic method and selective sensing of organoamines[J].Chemical Communications,2008(31):3642-3644. [11]Carson C G,Hardcastle K,Schwartz J,et al.Synthesis and structure characterization of copper terephthalate metal-organic frameworks [J].European Journal of Inorganic Chemistry,2009(16):2338-2343.[12]XIN Zhi-feng,BAI Jun-feng,SHEN Yong-ming,et al.Hierarchically micro-and mesoporous coordination polymer nanostructures with high adsorption performance [J].Crystal Growth &Design,2010,10(6):2451-2454.[13]Walton K S,Snurr R Q.Applicability of the BET method for determining surface areas of microporous metal-organic frameworks [J].Journal of the American Chemical Society,2007,129(27):8552-8556.[14]Karra J R,Walton K S.Effect of open metal sites on adsorption of polar and nonpolar molecules in metal-organic framework Cu-BTC [J].Langmuir,2008,24(16):8620-8626.[15]Gupta A,Chempath S,Sanborn M J,et al.Objectoriented programming paradigms for molecular modeling[J].Molecular Simulation,2003,29(1):29-46.。

吡咯衍生物的合成研究进展徐虹;白雪峰;吕宏飞;李猛【摘要】吡咯衍生物是杂环化学中化学性质最活泼的一类化合物,广泛应用于医药、农药、感光材料等诸多领域.研究介绍了不同的合成方法:Paal-Knorr反应、Barton-Zard反应、Van Leusen反应.针对这些方法,讨论了它们的反应原理,分析了各自的优缺点.【期刊名称】《黑龙江科学》【年(卷),期】2013(000)003【总页数】3页(P36-38)【关键词】吡咯衍生物;反应原理;合成【作者】徐虹;白雪峰;吕宏飞;李猛【作者单位】黑龙江省科学院石油化学研究院,哈尔滨150040;黑龙江省科学院石油化学研究院,哈尔滨150040;黑龙江省科学院石油化学研究院,哈尔滨150040;黑龙江省科学院石油化学研究院,哈尔滨150040【正文语种】中文【中图分类】TQ251.3吡咯衍生物[1-2]是一类重要的含氮五元杂环化合物,在天然产物化学、有机合成、药物化学及材料化学等领域有着广泛的用途。
近年来,随着天然产物化学的迅猛发展,科研人员[3]发现许多含有吡咯环的化合物具有很强的生物活性或有特殊的用途,如抗菌素藤黄绿脓菌素(Pyoluteorin),从雄性斑蝴蝶中得到的吡咯嗪酮,绿色植物光合作用的催化剂叶绿素,从动物肝脏中提取得到的一种深红色结晶维生素B12等。
这些化合物化学合成的关键问题是如何经济有效地合成吡咯环,因此,吡咯衍生物的合成成为研究的热点。
本研究介绍了几种吡咯衍生物的合成方法及其优缺点。
1 Paal-Knorr反应Paal-Knorr反应是指1,4-二羰基化合物在无水的酸性条件下脱水,生成呋喃及其衍生物,1,4-二羰基化合物与氨或硫化物反应,可得吡咯、噻吩及其衍生物。
该法是合成吡咯中最经典的方法,其可能的生成吡咯反应机理[4]如下:反应第一步生成两份半缩醛胺化合物,再经过亚胺(R =H)或烯胺(R≠H)中间体分步脱水得到吡咯。
1999年,N.Timother等[5]报道了2,5-二羰基正己烷与伯胺在微波作用下生成相应的吡咯,反应在2min之内就能完成,收率最高能达到90%。

过期药品销毁会议纪要范文英文回答:Minutes of Expired Drug Disposal Meeting.Date: March 15, 2023。
Time: 10:00 AM.Location: Conference Room A, Hospital Pharmacy. Attendees:Dr. John Smith, Pharmacy Director.Dr. Jane Doe, Pharmacy Associate Director.Mr. Michael Jones, Pharmacy Technician.Ms. Susan Brown, Pharmacy Technician.Agenda:Review of expired drug disposal procedures.Discussion of new disposal methods.Q&A.Minutes:1. Review of Expired Drug Disposal Procedures.Dr. Smith reviewed the current expired drug disposal procedures with the team. He emphasized the importance of following these procedures to ensure the safe and responsible disposal of expired medications.2. Discussion of New Disposal Methods.The team discussed several new drug disposal methods, including:Incineration: This method involves burning the drugs at high temperatures to destroy them.Microwave-assisted pyrolysis: This method uses microwaves to heat the drugs and break them down into smaller molecules.Chemical degradation: This method uses chemicals to break down the drugs into harmless substances.The team decided to further research these methods to determine the most suitable option for the hospital.3. Q&A.The team had a Q&A session to address any questions or concerns about expired drug disposal. Mr. Jones asked about the safety of incinerating expired drugs. Dr. Doe assured him that it is a safe and effective method when done properly. Ms. Brown asked about the cost of the new disposal methods. Dr. Smith said that he would investigatethe costs and provide an update at the next meeting.Action Items:Dr. Smith will research new drug disposal methods and present the findings at the next meeting.Dr. Doe will revise the expired drug disposal procedures to include the new methods.Mr. Jones will coordinate with the maintenance department to arrange for the installation of equipment for the new disposal method.Adjournment:The meeting was adjourned at 11:30 AM.中文回答:过期药品销毁会议纪要。

1引言社会经济在快速发展,环境恶化也愈加严重。
水环境作为整个环境系统不可或缺的部分,其污染也尤为突出[1]。
污水处理厂面临着原水水质越来越差和出水水质要求越来越高的的矛盾,因此,污水处理厂急需提高污水处理能力及处理效率。
混凝是提高出水水质的强有力手段,但是,中国北方水体具有较高的碱度,而且受工业污染较严重,传统混凝工艺对有机物的去除效率较低。
在水处理过程中,碱度是影响混凝效果的重要因素之一,因此,研究碱度对混凝的影响以及探析碱度调节方式设置对高碱度废水的处理十分有利[2]。
铝系混凝剂作为应用最为广泛的混凝剂之一,本研究以铝盐混凝剂为研究对象,对此问题进行探究。
2碱度对铝盐混凝剂的影响2.1碱度对铝盐混凝剂混凝效果的影响铝是一种被广泛用于制备各种工程材料和化学试剂的元素,同时,铝也是地壳中含量最多的金属元素,铝及其氧化物常被应用于各个方向的基础性研究。
其中,铝溶液化学经历了一个多世纪的发展,已成为一门涵盖范围非常广的学科,包括电镀、催化、环境、农药、微生物、土壤科学等[3]。
铝是一种典型的两性金属,在不同的pH条件下,铝离子会产生不同的水解形态,与污染物发生复杂多变的化学反应,经历典型的水解—聚合—结晶—沉淀过程去除有机物。
铝系混凝剂主要是利用铝盐在水中的水解形成的不同形态的阳离子发生作用,利用阳离子的电中和作用、原位水解形态的架桥吸附作用以及网捕卷扫作用,使水体颗粒物失稳聚集,最终沉降去除[4]。
碱度主要通过影响铝盐的水解而对混凝过程产生影响。
王东升[5]等人通过研究给出了方程式(1)、(2)所示的铝盐水解过程:x Al3+y H2O+z OH-→[Al x(H2O)y(OH)z]3x-z(1)[Al x(H2O)y(OH)z]3x-z→[AlO4Al12(OH)24(H2O)12]7+(2)不难看出,碱度是铝盐水解的重要影响因素。
不同碱度条件下,铝盐水解生成各种羟基铝形态,主要为低聚态铝和不稳定的高聚态铝,这是一个快速反应阶段,受控于碱化的速度和碱液扩散速度[5]。

Microwave assisted synthesis of zeolite A from metakaolinSathy Chandrasekhar *,P.N.PramadaChemical Sciences Division,Regional Research Laboratory,Thiruvananthapuram 695019,Kerala,IndiaReceived 4July 2006;received in revised form 17March 2007;accepted 3April 2007Available online 21April 2007AbstractZeolites are generally synthesized by the hydrothermal reaction of sodium aluminosilicate gels prepared from pure chemicals in alka-line medium using conventional heating systems.In the present study,microwave energy is used as a novel heating tool for the synthesis of zeolite A and metakaolin (Al 2O 3Æ2SiO 2),a calcined product of kaolin (Al 2O 3Æ2SiO 2Æ2H 2O)has been taken as a combined source for silica and alumina.Optimization studies on various reaction parameters have been conducted.Phase pure zeolite A of high crystallinity was synthesized by heating the reaction mixture for 2h at 85°C under microwave hydrothermal system where as conventionally it takes 6–8h.However,initial microwave exposure for 2min at 85°C and 20h ageing at room temperature was found to be a pre-requisite for the crystallization of zeolite A in this system.The products and intermediates at different stages of the reactions were characterized using XRD,IR,solid state NMR and chemical analysis.Morphology was examined using SEM.The crystallization of the zeolite was followed by measuring the water capacities of the product in each experiment.Calcium binding capacity of selected products was also determined.A comparative study of the zeolite A formation from metakaolin under microwave and conventional hydrothermal systems was also carried out.Ó2007Elsevier Inc.All rights reserved.Keywords:Zeolite A;Metakaolin;Microwave;Synthesis;Characterization1.IntroductionZeolites are porous three dimensional aluminosilicates formed by the sharing of oxygen atoms in the frame work aluminium and silicon tetrahedra [1].Out of the various low silica zeolites,Zeolite A is of great commercial impor-tance due to its molecular sieving and ion exchange prop-erties [2].Zeolites are conventionally synthesized from sodium aluminosilicate gel prepared from pure chemicals in air ovens or auto claves [2].Metakaolin (Al 2O 3Æ2SiO 2),a calcined product of kaolin (Al 2O 3Æ2SiO 2Æ2H 2O)with Si/Al ratio $1is a convenient starting material for the syn-thesis of zeolite A [3–5].Microwave energy is a powerful heating source which has not been studied extensively in zeolite synthesis.Thefirst patent is by Chu et al.which reported the synthesis of zeolite A in 12min under microwave heating [6].How-ever,the product was contaminated with hydroxy sodalite and zeolite A was obtained only after prolonged heating.A review on the microwave techniques in zeolite synthesis was reported by Cundy in 1998[7].Slangen et al.reported the effect of ageing on the formation of zeolite A under microwave conditions [8].They reported that ageing of the reactant mixture is a pre-requisite for fast crystalliza-tion of the zeolite.Since the heating by microwaves is rapid,preparation of the reaction mixture has to be more stringent compared to that in the conventional method to get the desired product.Sufficiently aged mixtures can yield zeolite A with crystal sizes ranging from 0.1to 0.3l m,after 1min heating in the microwave.It was also found that zeolite crystallisation from an unaged system can be improved dramatically by adding aged synthesis mixture to it.This suggests that rearrangement of the reaction mix-ture to yield suitable nuclei is the bottle neck in the microwave heating.A two stepped heating at different1387-1811/$-see front matter Ó2007Elsevier Inc.All rights reserved.doi:10.1016/j.micromeso.2007.04.003*Corresponding author.Tel.:+9104712515223;fax:+9104712491712.E-mail address:chandrasekharsathy@rediff (S.Chandrase-khar)./locate/micromesoAvailable online at Microporous and Mesoporous Materials 108(2008)152–161temperatures is reported for the synthesis of zeolite A.i.e. 90s at120°C and then5min at100°C.Microwave assisted crystallization of zeolite A from dense gels was reported by Bonaccorsi et al.[9].Pure zeolite A has been obtained,in a total processing time of1h,by exposing the reaction mixture to a microwave electromagneticfield under atmospheric pressure.A single step with only one output power level was found to be unsuitable for crystal-lizing pure zeolite,even with different exposure times.Fur-ther,it was demonstrated as to how an adequate tailoring of the microwave energy released to a synthesis mixture can lead to pure zeolite A without pre-treatment of the solution.Long ageing is not required and application ofmicrowave radiation is not detrimental even in thefirst stage of the synthesis when a thick gel is formed and nucle-ation has to begin.Finally,it has been established that the strong dissolution effect associated with the oscillating elec-tromagneticfield can be used to reduce water content in zeolite formulation,increasing the yield and production capacity to a point where conventional synthesis leads to impurity formation.Romero synthesized zeolite X by microwave heating and studied its catalytic activity[10].Katsuki et pared microwave and conventional hydrothermal synthesis of zeolite Y.They found that crystallization of zeolite Y in microwave treatment led to increased rate of formation by3–4times than via conventional hydrothermal treatment [11].Somani et al.synthesized ZSM-5at an enhanced crys-tallization rate using microwave energy[12].Inada et al. synthesized zeolite P1from coalfly ash through microwave hydrothermal reaction.They found that the microwave is effective to produce the zeolite from coalfly ash in a short period by control of irradiation schedule in the early stage [13].In all the reported studies on the synthesis of zeolites, sodium aluminosilicate gels prepared from pure chemicals have been taken as the starting material.Even though a number of papers are reported on this,no systematic study has so far been reported on the formation of zeolite A from metakaolin.The present study aims at the optimization of various reaction parameters for the synthesis of zeolite A from metakaolin under microwave(MW)conditions.A comparative study of the zeolite A formed under micro-wave and conventional systems using metakaolin was also carried out.2.Experimental2.1.Raw materialCoating grade kaolin of high purity(Table1)supplied by M/s English Indian Clays Ltd.,Thiruvananthapuram and technical grade sodium hydroxide was used for the synthesis.Metakaolin was prepared by heating the kaolin at700°C for2h at a heating rate of5°C/min in a muffle furnace with a temperature programmer.2.2.SynthesisA fabricated multi mode microwave reactor(MSS05 microwave sintering system)with a thermocouple and pro-grammer for accurate setting and measurement of temper-ature(M/s SV Heat Engineering,Hyderabad)was used for conducting the experiments.Sodium aluminosilicate gel was prepared with the molar ratios SiO2/Al2O3=2;Na2O/SiO2=2.5and H2O/ Na2O=40by mixing calculated amounts of metakaolinite and sodium hydroxide solutions[4].NaOH was dissolved in distilled water and metakaolin was slowly added to the alkali and a thorough mixing was done to get uniformity. The reaction mixture was transferred into100ml teflon vessel and a series of experiments were conducted under different conditions such as ageing time(keeping the sam-ple after mixing for a particular time),heating(reaction) time and temperature(heating the mixture under micro-wave),seeding,etc.Effect of initial microwave exposure of the mixture for a short period before ageing was also studied.The microwave heating was carried out at 2.45GHz and800W.The vessel was not befilled more than50%as a precaution.After each reaction,samples were immediatelyfiltered,washed with alkaline water (pH9–10)and driedfirst over a water bath and then at 110°C for1h.They were then hydrated to equilibrium water content by exposing them to water vapor from satu-rated NaCl solution before characterization.Zeolite A prepared by conventional methods was used as seed material to study its effect on the initiation of the zeolite crystallization.Very small quantities of the zeolite were added in the reaction mixture before the ageing/ hydrothermal treatment.2.3.CharacterizationX-ray diffraction of the sample was carried out in an X’Pert Pro X-ray Diffractometer using Cu K a radiation. The phases formed were identified by comparing the d spacing and relative intensities of the samples with stan-dard reference data in JCPDSfile[14].IR spectra of samples were scanned using a Perkin–Elmer882IR spectrophotometer was used to take the IR Table1Chemical composition of kaolinConstituents%(wt)Theoretical(kaolinite) SiO245.9046.52Al2O338.9039.53Fe2O30.51–TiO20.49–CaO0.05–MgO0.04–Na2O0.08–K2O0.03–LOI13.8013.95Si/Al ratio 1.003 1.0S.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161153spectra of the samples in the range400–4000cmÀ1[15]. Morphology of the samples was examined using a scanning electron microscope,JEOL JSM5600LV.Chemical analysis of the product zeolite was carried out essentially tofind out the Si/Al ratio using standard wet chemical analysis procedure[16].Solid state29Si and27Al NMR spectra of the samples were taken using a DSK 300NMR Spectrometer at59.63and78.21MHz, respectively.The conventional method of heating the samples at 300°C for2h,cooling in a desiccator and rehydrating to the equilibrium water content was adopted for the determi-nation of water adsorption capacity of selected samples[4]. Nearly1g of the zeolite was accurately weighed(a)and heated in a muffle furnace to300°C and a holding time of2h was given.The sample was then cooled in a desicca-tor and again weighed(b).The wt.loss is termed as uncal-cined water capacity(UWC)UWC%¼½ðaÀbÞ=b Â100The sample was kept in a hydrator maintained at35% relative humidity by a saturated solution of NaCl for 24h and then weighed(c).The increase in wt.due to the adsorption of water is termed as the calcined water capac-ity(CWC)[17]CWC c%¼½ðcÀbÞ=b Â100The%crystallinity of the zeolite formed was calculated from the calcined water capacity values.The crystallinity of the products can be directly calculated from the CWC value as%crystallinity¼f½CWCÀCWC a =½CWC cÀCWC a gÂ100 where CWC is the theoretical value of zeolite A(22.19)cal-culated from the molecular formula,CWC a is the value for the amorphous phase(2.93),CWC c is that for the products.The calcium binding capacity(CBC)was determined by exchanging the sodium form of zeolite with calcium ion. An accurately weighed sample was stirred with a standard solution of Ca(300mg of CaO per litreÀ0.786g of CaCl2Æ2H2O dissolved in distilled water and made up to 1l)for10min andfiltered.Thefiltrate was analyzed for its Ca content by complexometry and the difference is attributed to the exchanged amount.The values are expressed as milli equivalence of CaO per gram of zeolite.For the estimation of Ca,10ml of thefiltrate was mixed with25ml2N NaOH and0.2g of P&R indicator and immediately titrated with standard EDTA till the colour of the solution changed from pink to blue(a ml) %CaO=[a·X·100]/w where X=Equivalent CaO in g/ml of EDTA and W=weight of the sample3.Results and discussionThe kaolin under study has a chemical composition as given in Table1and the major constituents are silica and alumina.The concentration of other elements like sodium, potassium,calcium,iron and titanium are very low indicat-ing the high purity of kaolin.The mineralogy studied by XRD analysis confirms that the sample contains high amount of kaolinite and ancillary minerals content is too low to be detected/identified by this technique[5].The loss on ignition corresponds to the water of hydration.A series of experiments were conducted to synthesis zeo-lite A from the metakaolin under microwave(MW)condi-tion.Different sodium aluminosilicate gels were prepared by mixing the reactants at different conditions and the sam-ples are designated as MKAG1-7(Table2).The reaction conditions,values of water capacities and the%crystallin-ity of the products for MW reactions are given in Tables 3a–3e.Details of the products of reactions after mixing and heating for different duration at85°C are given in Table3a.Change in the properties of the products obtained by heating the samples for different timing and at different temperatures is given in Table3b.The reaction mixtures aged for20h at room temperature and heated at 85°C for different timings give products as presented in Table2Formation conditions for gelsSample Initial MW(min)Ageing(h)Seed(%) MKAG1–––MKAG2–20–MKAG32––MKAG4–201 MKAG5220–MKAG6001 MKAG7221Table3aReaction conditions and properties of products from MKAG1(85°C) Sample MW heating(min)UWC(%)CWC(%) MKA11011.66 1.49 MKA22010.12 1.69 MKA33016.22 1.95 MKA46018.71 1.38 MKA59010.51 1.29 MKA612019.25 6.68 MKA715011.4610.67 MKA824021.3721.75Table3bReaction conditions and properties of products from MKAG1at different temperaturesSample MW heating UWC(%)CWC(%) Temperature(°C)Time(min)MKA9906018.17 1.71 MKA10956020.90 1.88 MKA119512016.16 1.8 MKA121101033.40BDL MKA131103021.16 1.83 MKA141106017.3315.58154S.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161Table 3c .Reactions with different seeding conditions were also tried and are given in Table 3d .Reaction conditions such as the time of initial microwave exposure,ageing and heating and the properties of the products obtained are given in Table 3e .The products are designated as MKA1-54.The sample MKA55is the product formed under a combination of microwave and conventional heat-ing.MKA56is the sample after 4h heating (with out initial MW exposure)in a conventional air oven.3.1.Water capacitiesAmorphous products lose their porosity during heating and cannot be further dehydrated.Crystalline products retain their porosity even after heating at 350°C for 1h which shows the stability of zeolite network structure.When the UWC and CWC values come closer to each other and nearer to 22.19(theoretical value of zeolite A),the crystallinity of the sample becomes high provided the zeolite formed is phase pure.The crystallinity of the zeolite is thus directly correlated to its CWC and hence the zeolite crystallization is monitored by determining the same.The reactants were mixed and heated to 85°C for differ-ent duration under MW condition.Tables 3a and 3b show the results obtained using the gel named MKAG1.No crystalline product was formed up to 90min.Zeolite A started crystallizing after 120min and highly crystalline product (MKA8)was obtained after 4h reaction (Fig.1and Table 3a ).Effect of temperature on the hydrothermal reaction was studied (Table 3b ).No zeolite formation is observed by simply mixing the reactants and heating for 60min at 95°C (MKA10).Zeolite A with moderate crystallinity was formed after 60min heating at 110°C (MKA14).Var-iation of %crystallinity with reaction temperature is given in Fig.2.It is seen that above 100°C,the effect of micro-waves on the rate of crystallization is very high.The forma-tion of zeolites takes place under autogeneous pressure.Above 100°C,the water vapour also contributes to the pressure inside the vessel and therefore faster crystalliza-tion takes place.More over,microwaves act strongly on water in vapor phase.Under microwave heating,formation of nuclei is found to be the limiting factor.Ageing is a nucleation aiding stepTable 3cReaction conditions and properties of products from MKAG2(85°C)Sample MW heating (min)UWC (%)CWC (%)MKA15510.82 1.8MKA161013.370.9MKA172018.180.93MKA183015.35 1.23MKA196016.48 1.47MKA2012018.74 2.89MKA2124015.4610.67Table 3dReaction conditions and properties of products (85°C)–different seeding and heating SampleInitial MW (min)Ageing (h)Seed (%)MW heating (min)UWC (%)CWC (%)MKA22––11018.77 1.63MKA23––13013.55 1.62MKA24––16033.18 1.50MKA25––112018.74 2.89MKA26–2011015.75 1.33MKA27–2013019.210.93MKA28–2016018.60 1.49MKA29–20112017.5 6.81MKA302216015.52 2.0MKA312112013.283.56Table 3eReaction conditions and properties of products (85°C)–different initial MW heating and ageing times Sample Initial MW (min)Ageing (h)MW heating (min)UWC (%)CWC (%)MKA320.5212013.86 1.49MKA33512018.618.24MKA342012014.62 2.25MKA351212013.86 1.49MKA36512013.78 1.3MKA372012020.3216.87MKA38226053.07 2.05MKA39212014.56 4.9MKA4059013.35 1.7MKA41512013.93 4.68MKA42159010.76 1.9MKA431512016.512.28MKA44203014.32 1.65MKA45209014.91 1.74MKA462010013.59 4.99MKA472012048.4522.30MKA484412017.8314.25MKA495212020.54 1.79MKA50512015.42 2.8MKA512012015.4010.44MKA5210212010.39 5.52MKA53512014.54 1.55MKA542012014.427.48aMKA552(MW oven)4h a (air oven)aMKA56––4h a (air oven)aConventional heating.S.Chandrasekhar,P.N.Pramada /Microporous and Mesoporous Materials 108(2008)152–161155in the synthesis of zeolites.It was reported that during the ageing period,pre-nuclei are formed.Nuclei can be defined as the smallest viable system capable of forming an identi-fiable crystal phase that can induce crystal growth.For the formation of these nuclei,first the synthesis mixture should be mixed thoroughly to ensure mixing on a molecular scale. It is expected that these nuclei are more abundant in the longer aged synthesis mixture and therefore the reaction time for crystallization gets reduced.Slangen et al reported that for longer aged synthesis mixtures,the heating time does not reduce significantly and ageing has distinct effect only on the crystal size[8].When20h ageing was given to the reaction mixture before MW heating(Table3c),zeolite A with very low crystallinity is formed after120min at85°C(MKA20). The product obtained after4h heating is moderately crys-talline(MKA21).So ageing of the reaction mixture alone does not have much influence in the formation of nuclei of zeolite A from metakaolin under MW conditions.Mixing on a molecular scale is essential for the nucle-ation of zeolite A.Pockets of silica and alumina are formed during the dissolution of metakaolin in alkali which undergo a rearrangement during ageing.In course of time, these pockets combine to form silica-alumina oligomers, with a silicon aluminium ratio of one.Thorough mixing also removes any pockets in which hydroxy sodalite can nucleate.When metakaolin is mixed with alkali,it takes some time to dissolve the metakaolin and form the reactive species.In conventional synthesis the dissolution and the formation of the nuclei takes place during the slow heating. Since microwave heating is very fast,dissolution and orga-nization has to be carried out before heating.Some experiments were carried out by seeding the reac-tion mixture with zeolite A(1%)and applying various reac-tion conditions like,with and with out ageing,2min initial MW exposure and ageing(MKA22-29)(Table3d).It has been observed that seeding does not have much influence in the nucleation/crystallization rate.An initial short microwave exposure is found to favor the dissolution of metakaolin in alkali(samples MKA30-54,Tables3d and3e)When ageing was given after this short MW exposure,the nucleation of zeolite A is very much enhanced and thus crystalline product is formed.Different short initial MW exposure timings from30s to 10min were tried.A series of reactions with different initial MW exposure(0.5,1,2,5and10min)and ageing times(2, 5and10h)was carried out tofind out the optimum expo-sure time to form crystalline zeolite A.Product with max-imum crystallinity is obtained by irradiating the reaction mixture for2min,ageing for20h and heating2h under MW condition(MKA47)(Fig.3).The reaction mixture aged for short durations contains some pockets of inade-quate mixing(along with the zeolite nuclei)which is the reason for the formation of amorphous material.An initial longer heating time than2min has a retarding effect on the formation of zeolite A(Fig.4).This is because there is an optimum MW energy for the effective orientation of the reactive species to form the zeolite nuclei.When the initial heating time is increased,the reactant species do not get organized and the formation of nuclei becomes difficult.The short MW exposure has very significant role on the synthesis of zeolite A from metakaolin.Reduction of syn-thesis time for crystalline zeolite A from metakaolin under156S.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161MW condition is actually achieved through the rate enhancement in the dissolution step(Fig.3).Heating of the reaction mixture for4h in MW condition is result in the formation of highly crystalline zeolite A(MKA8), where as zeolite A with lower crystallinity is formed in the conventional system(MKA56).If an initial2min MW exposure was given to the reaction mixture and heated in an air oven for4h,zeolite A having almost same crystal-linity as MKA8is formed(MKA55).On the contrary6–8h heating is required for the formation of crystalline zeolite A in the conventional synthesis.Efficient synthesis demands carefully controlled initial MW exposure and ageing before heating.When the%crys-tallinity of products is plotted as a function of time,an‘S’shaped curve is obtained.This suggests that crystal growth is preceded by a distinct,slow,nucleation step and a proper understanding of this step is the key to control the synthe-sis conditions[18].3.2.X-ray diffraction analysisXRD patterns of the metakaolin and intermediate gels formed at different stages are shown in Fig.5.Metakaolin has a pattern with a broad hump having maximum at 2h=$22°.The gels formed by mixing the metakaolin with alkali also give amorphous broad bands,but with varia-tions in the height,width and position.These variations are indicative of the differences in the structural environ-ments.The gel formed just after mixing(MKAG1)has the same pattern as metakaolin but with a less hump height.Gel after20h ageing(MKAG2)and the one after 2min MW exposure(MKAG3)show humps slightly broader than that of MKAG1.When1%seeding and 20h ageing was given to the reaction mixture the gel formed is almost like G3but with more broadened band (MKAG4).When2min initial MW exposure and20h age-ing was given,the gel formed is having a hump with a max-imum at2h=$28°(MKAG5).Fig.6gives the XRD patterns of the product after 120min heating(MKA6),after20h ageing and different heating time(MKA18,MKA19and MKA20),the product after20h ageing,with1%seeding and120min heating (MKA29)and the kinetic series after2min initial MW heating and20h ageing(MKA45,MKA46and MKA47).It has been observed that the product with max-imum crystallinity is MKA47formed from the gel MKAG5.Zeolite A with only moderate crystallinity was formed after20h ageing,with1%seeding and120min heating(MKA29)and zeolite A with lower crystallinity by heating the reaction mixture for120min(MKA6), and after20h ageing and120min heating(MKA20).The XRD results are in good agreement with the%crystallinity values and therefore substantiate the values of calcined water capacity.When the reaction mixture after2min ini-tial MW exposure is subjected to conventional heating for 4h,the product obtained is phase pure zeolite(MKA55) with the same crystallinity as obtained after4h heatingS.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161157under MW condition(MKA8).Zeolite A with lower crys-tallinity is formed when the reaction mixture was not exposed to initial MW exposure and was heated4h in air oven(MKA56).The XRD analysis support the fact that when the reac-tion mixture is subjected to a microwave exposure for 2min,ageing for20h and heating for2h,zeolite A with maximum crystallinity is formed.The gel MKAG5is found to have the‘‘ideal precursor gel structure’’for the crystal-lization of zeolite A.3.3.Infra red spectraThe spectra of the gel samples formed after(i)mixing (MKAG1),(ii)mixing and20h ageing(MKAG2),(iii) mixing and2min initial MW(MKAG3)and(iv)2min MW and20h ageing(MKAG5)are taken and given in Fig.7.But no characteristic band of zeolite A is observed in the spectra of the gel samples.The spectra of the selected products(MKA46and MKA47)are also given in Fig.8. The typical bands of zeolite A representing theasymmetric Fig.8.Solid sate27Al and29Si NMR spectra of selected samples.a–MK,b–MKAG1,c–MKAG2,d–MKAG3,e–MKAG4,f–MKAG5and g–MKA47. 158S.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161and symmetric stretches(1050and74cmÀ1),double rings(557cmÀ1)and T–O bend(465cmÀ1)are observed inMKA47and these bands are less intense in MKA46.TheIR spectral analysis is found to substantiate the XRDresults.3.4.29Si and27Al solid state NMR spectraAluminosilicates like clays and zeolites contain SiO4andAlO6/AlO4units and can be studied by the29Si and27Alsolid state NMR spectroscopy.This technique is a power-ful tool for understanding the structure of various reac-tants,intermediates and products in the synthesis ofzeolites,especially those of the intermediates which areamorphous to X-rays.In the29Si spectrum,there are char-acteristic lines for silicon atoms having different environ-ments like Si–O–Al linkages with1,2,3or4Al atoms.The chemical shift values are different for Al atoms with4,5and6coordination in the27Al NMR spectrum.The 29Si and27Al solid state NMR spectra of the metakaolin, gels prepared at various conditions and product zeoliteare given in Fig.8.3.4.1.29Si spectraMetakaolin shows a broad line atÀ97.5ppm which ischaracteristic of silicon atoms in a highly disordered stateas found in amorphous silicates.When kaolinite is dehydr-oxylated,the silicon atoms experience a range of environ-ments of differing distortion and the broadness ofmetakaolinite line is attributed to these variations in theSi–O–Si(Al)bond angles[19].The gel MKAG1has a sim-ilar spectrum(silicon line atÀ99.5ppm)indicating that nochange takes place just after mixing the metakaolin withalkali.After giving an ageing of20h to this,the gelMKAG2gives a small and broad silicon line atÀ89.4ppm.MKAG4is similar to MKAG2.The silicon atoms in this sample appear to have more distorted struc-ture.The distortion in MKAG3and MKAG5is so high that no silicon line is observed in their spectra.The product formed from MKAG5gives sharp single line atÀ89.5ppm which is typical of Si(4Al)units(MKA47).3.4.2.27Al spectraMetakaolin gives a broad line at$6.2and a small line at $24ppm corresponding to the hexa and penta coordinated Al,respectively[4].The gel MKAG1also appears to have a similar structure.As the metakaolin dissolves in alkali to form a gel the environment of Al atoms undergo a change which is clearly indicated in the spectrum of MKAG2giv-ing a sharp line at$59.5(Al IV)and a small one at $6.7ppm(Al VI).The conversion of most of the Al atoms to the Al IV is indicated in the MKAG3also.This obser-vation supports the XRD results of shifting the peak max-ima towards higher2h value.The gel MKG5gives a sharp single line at59.5ppm which can be assigned to the tetra coordinated aluminium(Al IV).The most appropriate pre-nucei for zeoliteA are abundantly formed in this gel which induces fast crystal growth.Hence this gel act as the most suitable precursor for zeolite A crystallization as concluded from other observations.3.4.3.Products from various gelsProperties of products from MKAG1formed at various conditions are given in Tables3a and3b.The product having maximum crystallinity is MKA8which needs4h heating at85°C to form.Heating at higher temperatures does not give any advantage.The gel MKAG2gave a product with low crystallinity after heating for4h (MKA21and Table3c).It is interesting to see that con-ventional heating in an air oven for4h gives a product similar to MKA8.Hence,in these cases,no advantage of MW heating is achieved in the zeolite formation.The NMR studies give an explanation to this behavior.The Si and Al NMR lines of MKG1are almost the same as those of MK.In MKAG2and MKAG3,the Al line at 6.7ppm is retained even though the Si atoms are more dis-torted.These precursors are not responding to the MW heating and zeolite crystallization is not taking place.On the contrary,the MKAG5gel gives Al line at$59.5and highly distorted Si atoms which behaves as the ideal pre-cursor.The NMR spectra of MKAG4are similar to that of MKAG2,but for the more distortion of Si atoms. Hence,a combination of seeding and ageing(2h)followed by MW heating does not give a product zeolite of high crystallinity.The product with maximum crystallinity, MKA47gives silicon and aluminium lines atÀ89.5and 59.5,respectively,in their NMR spectra which are charac-teristic of zeolite A.3.5.Scanning electron microscopic studyMorphology of selected gel and product samples as examined under an electron microscope are given in Fig.9a–c(MKAG1,MKA46and MKA47).The gel sam-ples MKAG1have a morphology showing distorted pseudohexagonal platelets<1l m size as in the metakaolin [4].The material is mostlyfluffier indicating the typical gel morphology.Some cubic crystals of zeolite A along with the gel are observed in the sample MKA46(Fig.9b).The particles in thefinal product(sample MKA47)are beveled cubes and have almost uniform size(1–1.5l m)(Fig.9c).3.6.Chemical assayThe chemical analysis was carried out mainly to get the Si/Al ratio of the intermediates and the products(Table4). The Si/Al ratio of the gel formed just after mixing is0.89 and it increases slightly to1.00during crystallization.Just after mixing the metakaolin does not dissolve in the alkali and therefore the Si/Al ratio remains the same.The gel after2min MW exposure and20h ageing has almost sim-ilar Si/Al ratio of that of zeolite A.During crystallization, a small part of alumina gets leached out resulting in a higher Si/Al ratio.S.Chandrasekhar,P.N.Pramada/Microporous and Mesoporous Materials108(2008)152–161159。
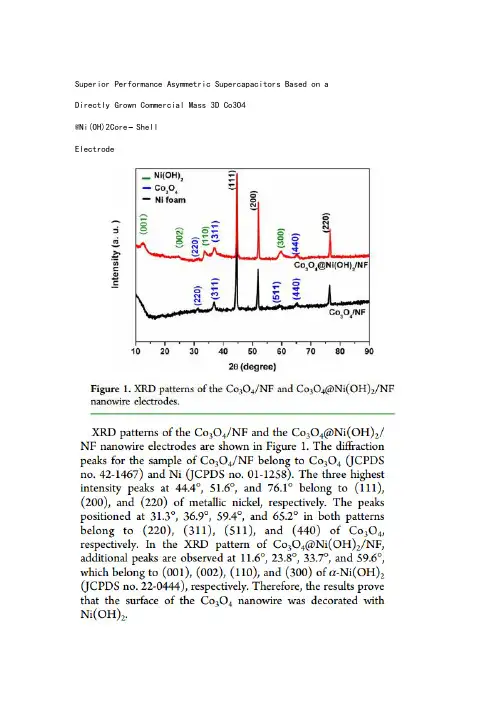
Superior Performance Asymmetric Supercapacitors Based on a Directly Grown Commercial Mass 3D Co3O4@Ni(OH)2Core−ShellElectrodeFacile Synthesis of Hollow MoS2Microspheres/Amorphous Carbon Composites and Their Lithium Storage PropertiesBroad and weak peakIn situ assembly of MnO2nanowires/graphene oxide nanosheets composite with high specific capacitancestrong and clear peakNickelecobalt hydroxide nanosheets arrays on Ni foamfor pseudocapacitor applicationsOne-pot hydrothermal synthesis of reduced graphene oxide/Ni(OH)2 films on nickel foam for high performance supercapacitorsRapid synthesis of graphene/cobalt hydroxide composite with enhanced electrochemical performance for supercapacitorsSynthesis of ultrathin mesoporous NiCo2O4nanosheets on carbon fiber paper as integrated high-performance electrodes for supercapacitorsA sandwich-type three-dimensional layered double hydroxide nanosheet array/graphene composite: fabrication and high supercapacitor performanceUltrathin and highly-ordered CoO nanosheet arrays for lithium-ion batteries with high cycle stability and rate capabilityNi–Co sulfide nanowires on nickel foam withultrahigh capacitance for asymmetricsupercapacitorsOn e-step synthesis of CoNi2S4 nanoparticles for supercapacitor electrodes3D Ni3S2 nanosheet arrays supported on Ni foamfor high-performance supercapacitor and nonenzymatic glucose detectionHigh-performance NiCo2O4@Ni3S2 core/shell mesoporous nanothorn arrays on Ni foam for supercapacitorsNickele Cobalt hydroxide microspheres electrodepositioned on nickelcobaltite nanowires grown on Ni foam for high-performance pseudocapacitorsA novel porous coral-like as an anode material for lithium ion batteries with excellent rate performanceHydrated Cobalt Nickel Molybdate Nanorods as Effectively Supercapacitor Electrode MaterialsMicrowave-assisted synthesis of graphene/CoMoO4 nanocompositeswith enhanced supercapacitor performanceCoMoO4$ nanorods grown on reduced graphene oxide as advanced electrochemical pseudocapacitor materialsFacile fabrication and perfect cycle stability of 3D NiO@CoMoO4 nanocomposite on Ni foam for supercapacitorsFacile hydrothermal synthesis of hierarchical ultrathin mesoporousNiMoO4 nanosheets for high performance supercapacitorsNano a-NiMoO4 as a new electrode for electrochemical supercapacitorsComparison of the Electrochemical Performance of NiMoO4 Nanorods and Hierarchical Nanospheres for Supercapacitor ApplicationsSeaurchin-like hierarchical NiCo2O4@NiMoO4core–shell nanomaterials for high performance supercapacitorsControlled Growth of NiMoO4 Nanosheet andNanorod Arrays on Various Conductive Substrates as Advanced Electrodes for Asymmetric SupercapacitorsDesign and synthesis of 3D Co3O4@MMoO4 (M=Ni, Co) nanocomposites as high-performance supercapacitor electrodesEnhanced performance of supercapacitors with ultrathin mesoporous NiMoO4 nanosheets。

铌酸钾钠压电陶瓷晶体结构分析一、概述铌酸钾钠压电陶瓷是一类具有优良压电性能的陶瓷材料,广泛应用于声表面波滤波器、压电换能器、传感器等领域。
其压电性能与晶体结构密切相关,因此对其晶体结构的研究具有重要意义。
二、铌酸钾钠压电陶瓷的晶体结构1. 基本结构铌酸钾钠压电陶瓷的晶体结构属于钙钛矿结构,空间裙为tetragonal。
其基本单元包括钙钛矿型的四面体ABO3和K+、Na+等离子。
2. 钙钛矿型四面体结构铌酸钾钠压电陶瓷的钙钛矿型四面体结构中,A位离子通常是较大的离子,如钾离子(K+)、钠离子(Na+)等;B位离子为较小的过渡金属离子,如铌离子(Nb5+)等;O位离子为氧离子。
这种结构具有较强的偏压电性能,使得铌酸钾钠压电陶瓷具有优异的压电性能。
3. 离子掺杂结构为了进一步提高铌酸钾钠压电陶瓷的性能,人们常常采用离子掺杂的方法。
通过掺入适量的稀土离子或过渡金属离子,可以有效调控晶格结构,改善压电性能。
三、铌酸钾钠压电陶瓷的制备技术1. 固相法制备固相法制备铌酸钾钠压电陶瓷是最常用的制备方法之一。
该方法主要包括原料粉碎混合、成型、烧结等工艺步骤。
通过精确控制各工艺参数,可以获得均匀致密、晶粒细小的陶瓷制品。
2. 溶胶-凝胶法制备溶胶-凝胶法是一种制备铌酸钾钠压电陶瓷的新型方法。
该方法利用溶胶-凝胶体系的特性,可以制备出颗粒均匀、晶体结构致密的陶瓷制品。
3. 微波一步法制备微波一步法是近年来新兴出现的铌酸钾钠压电陶瓷制备方法。
该方法利用微波加热的特性,可大大缩短制备时间,并且能够得到具有优异性能的铌酸钾钠压电陶瓷制品。
四、铌酸钾钠压电陶瓷的应用前景铌酸钾钠压电陶瓷具有优良的压电性能和稳定的晶体结构,因此在声表面波滤波器、压电换能器、传感器等领域具有广泛的应用前景。
随着技术的不断进步和应用需求的增加,铌酸钾钠压电陶瓷的应用前景将更加广阔。
五、结论铌酸钾钠压电陶瓷具有优良的压电性能和稳定的晶体结构,是一类具有广泛应用前景的陶瓷材料。

V ol.29No.4安徽工业大学学报(自然科学版)第29卷第4期October2012J.of Anhui University of Technology(Natural Science)2012年10月文章编号:1671-7872(2012)04-0338-04微波法合成树枝状纳米硫化锑叶明富1,孔祥荣2,潘正凯1,伊廷锋1,诸荣孙1,任世彪1,雷智平1(1.安徽工业大学化学与化工学院,安徽马鞍山243002;2.北京建筑材料科学研究总院有限公司,北京100141)摘要:以氯化锑、硫脲为原料,采用微波法合成树枝状硫化锑纳米材料。
用X射线粉末衍射(XRD)、扫描电子显微镜(SEM)和X射线能谱(EDS)等对Sb2S3晶体的物相及形貌进行分析表征。
结果表明制备出的是正交相Sb2S3,长度可达数十微米。
并给出树枝状硫化锑纳米材料的可能的形成机理。
关键词:硫化锑;微波合成;纳米材料中图分类号:O614.53文献标识码:A doi:10.3969/j.issn.1671-7278.2012.04.011Microwave-assisted Synthesis of Branch-like Sb2S3NanomaterialsYE Ming-fu1,KONG Xiang-rong2,PAN Zheng-kai1,YI Ting-feng1,ZHU Rong-sun1,REN Shi-biao1,LEI Zhi-ping1(1.School of Chemistry and Chemical Engineering,Anhui University of Technology,Ma'anshan243002,China;2.Beijing Building Materials Academy Co.Ltd.Beijing10004,China)Abstract:Nano-sized branch-like antimony sulfide(Sb2S3)was synthesised at large-scale by microwave heating method and using SbCl3and CS(NH2)2as raw materials.The as-prepared Sb2S3nanomaterials were characterized via X-ray diffraction(XRD),scanning electron microscopy(SEM),energy dispersive X-ray spectrometer(EDS). Results show that the prepared product is the orthogonal Sb2S3csystal phase with length at tens of micrometers. The possible growth mechanism of the Sb2S3nanomaterials was also provided.Key words:antimony sulfide(Sb2S3);microwave synthesis;nanomaterials当粒子的尺寸进入纳米数量级(1~100nm)时,本身和由它构成的纳米固体具有多种传统固体不具备的特殊性质,主要包括量子尺寸效应、小尺寸效应、表面与界面效应、宏观量子隧道效应、介电限域效应等,因而展现出许多特有的性质,在催化、光电、发光、医药、磁介质及新材料等方面有广阔的应用前景,同时推动基础研究的发展[1-3]。

药学领域英文作文Title: Advancements in Pharmaceutical Sciences: A Comprehensive Overview。
Introduction。
The field of pharmaceutical sciences has undergone remarkable advancements in recent years, driven by the convergence of multidisciplinary approaches, technological innovations, and evolving healthcare needs. This essay aims to provide a comprehensive overview of the key developments in pharmaceutical sciences, highlighting their impact on drug discovery, development, and delivery.Drug Discovery and Development。
In drug discovery, the integration of computational modeling, high-throughput screening, and artificial intelligence (AI) has revolutionized the identification of potential drug candidates. Virtual screening techniques,such as molecular docking and pharmacophore modeling, expedite the process of lead compound identification by predicting their interactions with biological targets. Furthermore, AI algorithms analyze vast datasets to uncover novel drug-target associations, accelerating the identification of therapeutic targets and drug repurposing opportunities.Moreover, advancements in synthetic chemistry have facilitated the synthesis of complex molecules with enhanced pharmacological properties and reduced toxicity. Techniques such as microwave-assisted synthesis, flow chemistry, and click chemistry enable the rapid generationof diverse chemical libraries for screening purposes. Furthermore, the development of novel drug delivery systems, including nanoparticles, liposomes, and microneedle patches, enhances the bioavailability and targeting of therapeutic agents, thereby improving their efficacy and minimizingside effects.Clinical Trials and Regulatory Considerations。

http://www.hxtb.org化学通报2005年第12期微波辐射下L.氨基酸的快速消旋方法刘毅郭丽芸吴晓燕焦庆才(南京大学生命科学学院医药生物技术国家重点实验室南京210093)摘要微波辐射作用下的L氨基酸消旋反应是一种新的氨基酸消旋方法,具有对环境友好的优点。
本文报道了在微波辐射下,以1.0mol/L氢氧化钠水溶液替代有机酸作为反应溶剂,水杨醛为催化剂,水杨醛与I厂氨基酸的摩尔比为0.1,L-氨基酸可以快速消旋;消旋反应随微波辐射功率的提高而加快,在600W"时已接近最大反应速率。
同时也讨论了微波作用下L氨基酸的消旋反应机理。
关键词D.氨基酸消旋微波辐射过热效应Microwave--assistedRapidRacemizationofL-AminoAcidsLiuYi,GuoLiyun,WuXiaoyan,JiaoQingcai(StateKeyLaboratoryofPharmaceuticalBiotechnology,NanjingUniversity,Nanjing210093)AbstractApracticalmethodfortheracemizationofL-aminoacidshasbeendevelopedbymicrowaveirradiation.Comparedwiththeconventionalmethodsofracemization,microwavedielectricheatingpromotestheracemizationofL=anlinoacids.1eadingtoslgnmcantincreaseinreactionrates.Theretwomainadvantages:(1)thewholeprocessofracemizationbythismethodbecompletedwithincirca1hinmediumof1.0mol/LNaOHinthepresenceof0.10molarequivalentofsalicylaldehydebyusing600Wmicrowaveirradiation;(2)itiseconomical,convenientandenvironmentallyacceptablewaytoracemizeL-aminoacids.Thefactorsinfluencingtheraeemizationwereinvestigated.Theraeemizationswerefoundtofollowfirst—orderreactionkinetics.ThemechanismoftheracemizationWasdiscussed.KeywordsD.Aminoacids,Racemization,Microwaveirradiation,Superheatingeffects微波辐射促进有机化学反应已成为一个研究热点¨o,并形成一f-i引人注目的全新学科——MORE化学(MicrowaveInducedOrganicReactionEnhancementChemistry)。
噁唑啉化合物合成方法及应用的研究进展摘要噁唑啉是许多天然产物和生物活性分子中经常遇到的结构。
它们也是高分子化学和有机合成的多功能中间体。
噁唑啉环中的N原子含有孤对电子,能与金属离子形成较强的配位键,具有广谱金属配位能力。
此外,手性噁唑啉及其金属配合物已广泛应用于各种类型的不对称催化反应,它们在这些反应中表现出优异的催化活性和立体选择控制能力,因此受到了广泛的关注,特别是对于含有手性噁唑啉环的配体。
近年来,人们开发出了多种噁唑啉配体。
因其有特殊的研究价值,所以,本文主要对其相关研究进展进行了总结梳理。
关键词:噁唑啉;金属配合物;不对称催化Advances in synthesis and application ofoxazoline compoundsABSTRACTOxazolines are frequently encountered structural motifs innumerous natural products and biologically active molecules. They are also versatile intermediates in polymer chemistry andorganic synthesis.The N atom in the oxazoline ring contains lone pair electrons, which can form strongcoordination bonds with metal ions and has broad spectrum metal coordination ability. In addition, chiral oxazolineand its metal complexes have been widely used in varioustypes of asymmetric catalytic reactions, in which they show excellent catalytic activity and stereoselective control ability, so they have received extensive attention,especially for ligands containing chiral oxazoline rings. In recent years, many kinds of oxazoline ligands have been developed.Because of its special research value, this paper mainly summarizes its related research progress.Keywords:Chiral oxazoline; Metal complex; Asymmetric catalysis手性化合物对生命体表现出不同的生理活性,而手性小分子对生命体等生物大分子的功能产生强大的影响,使其成为化学家研究的重要对象,新型手性配体的研究也越来越重要。
紫精-普鲁士蓝凝胶电致变色器件的制备宋伟伟;王跃川【摘要】合成了醛基功能化的紫精,1,1'-二醛己基-4,4'-联吡啶,通过与聚乙烯醇的缩醛反应将紫精固定到凝胶电解质中,对电极用电沉积法沉积普鲁士蓝,制备了新型凝胶型电致变色器件.这种凝胶电致变色器件避免了电解质泄露的风险并拓宽了电致变色器件的应用范围.采用1 HNMR对分子结构进行了表征,通过紫外-可见分光光度计研究了器件的电致变色性能及其影响因素.结果表明:采用将紫精通过共价键并入凝胶的方法,制备的器件在±2V的工作电压下具有47%的对比度,着色时间2.8s,褪色时间3.3s,并且具有良好的循环稳定性.%1,1'-dihexanal-4,4'-bipyridinium chloride was designed and synthesized.For preparing a novel gel eletrochromic device,viologen was immobilized in the PVA gel electrolytes by aldolization,and the counter electrode was deposited Prussian blue via electro-deposition.This device avoids the leakaging risk of liquid electrolytes and extends the appllications of eletrochromic devices.The structure and eletrochromic porperties were studied by1HNMR and UV-Vis spectra.The results indicate that the eletrochromic devices with viologen immobilized in gel electrolytes achieves high-performance in terms of optical contrast (47% at 530 nm),switchingtime(<4 s) and cyclability.【期刊名称】《影像科学与光化学》【年(卷),期】2018(036)001【总页数】6页(P51-56)【关键词】紫精;电致变色;PVA水凝胶【作者】宋伟伟;王跃川【作者单位】四川大学高分子科学与工程学院高分子材料工程国家重点实验室,四川成都610065;四川大学高分子科学与工程学院高分子材料工程国家重点实验室,四川成都610065【正文语种】中文电致变色材料是指在外加电场下光学属性发生可逆变化的材料,在智能窗、显示器、迷彩服等领域有着广阔的应用前景。
南俊民教授简介南俊民,物理化学专业方向骨干教师。
教学方面,近年来讲授的本科课程有《物理化学》、《物理化学实验》、《应用物理化学实验》、《化学与社会》,讲授的研究生课程有《物理化学进展》。
科研方面,光电化学功能材料的制备与应用研究已发表论文50多篇,获专利授权5项,获鉴定成果3项。
研究领域:光电化学功能材料,包括电池材料和功能电极材料、光(电)催化材料等的制备与应用。
工作经历:1. 1994.7-1995.8 国营755厂,技术员2. 1998.10-1999.8 杭州南都电源有限公司,工程师3. 1999.9-2001.7 天津大学应用化学与技术博士后流动站,博士后4. 2001.7-至今华南师范大学化学与环境学院,副教授、教授教育背景:1. 1987.9-1994.6 郑州大学化学系学习,获学士学位2.1991.9-1994.6 郑州大学化学系学习,获硕士学位2. 1995.9-1998.7 厦门大学化学系学习,获博士学位主要论文:29.Xin Xiao, Chao Liu, Ruiping Hu, Xiaoxi Zuo, Junmin Nan*, Laisheng Lia, LishiWang*, Oxygen-rich bismuth oxyhalides: Generalized one-pot synthesis, band structures and visible-light photocatalytic properties, J. Mater. Chem., 2012, 22(43), 22840 – 22843.28.Xin Xiao, Rong Hao, Xiaoxi Zuo, Junmin Nan*, Laisheng Li, Weide Zhang*,Microwave- assisted synthesis of hierarchical Bi7O9I3 microsheets for efficient photocatalytic degradation of bisphenol-A under visible light irradiation, Chem.Eng. J., 2012, 209: 293-300.27.Xiaohua Zhu, Lixuan Zeng, Maotian Xu, Yong Liang*, Junmin Nan*, A glassycarbon electrode modified with electrochemically reduced graphene forsimultaneous determination of guanine and adenine, Anal. Methods, 2012, 4,2935–2939.26.Xiaoxi Zuo*, Chengjie Fan, Xin Xiao, Jiansheng Liu, Junmin Nan*, High-voltageperformance of LiCoO2/graphite batteries with methylene methanedisulfonate as electrolyte additive, J. Power Sources, 2012, 219: 94-99.25.Xiaohui Wu, Xiaoting Hong*, Junmin Nan, Zhiping Luo, Qiuyun Zhang, LaishengLi, Hongyu Chen, K.S. Hui, Electrochemical double-layer capacitor performance of novel carbons derived from SAPO zeolite templates, Micropor. Mesopor.Mater., 2012, 160: 25-31.24.Xin Xiao, Rong Hao, Min Liang, Xiaoxi Zuo, Junmin Nan*, Laisheng Li, WeideZhang*, One-pot solvothermal synthesis of three-dimensional (3D) BiOI/BiOCl composites with enhanced visible-light photocatalytic activities for thedegradation of bisphenol-A, J. Hazard. Mater., 2012, 233-234: 122-130.23.Cong Liu, Junmin Nan*, Xiaoxi Zuo, Xin Xiao, Dong Shu, Synthesis andelectrochemical characteristics of an orthorhombic LiMnO2 cathode materialmodified with poly(vinyl- pyrrolidone) for lithium ion batteries, Int. J.Electrochem. Sci., 2012, 7: 7152-7164.22.Xiaoxi Zuo*, Chengjie Fan, Xin Xiao, Jiansheng Liu, Junmin Nan*, Methylenemethanedisulfonate as an electrolyte additive for improving the cyclingperformance of LiNi0.5Co0.2Mn0.3O2/graphite batteries at 4.4V charge cutoffvoltage, ECS Electrochem. Letters, 2012, 1 (3): A50-A53.21.Rong Hao, Xin Xiao, Xiaoxi Zuo, Junmin Nan*, Weide Zhang*, Efficientadsorption and visible-light photocatalytic degradation of tetracyclinehydrochloride using mesoporous BiOI microspheres, J. Hazard. Mater., 2012, 209– 210: 137– 145.20.Xiaohua Zhu, Qifang Jiao, Chuyi Zhang, Xiaoxi Zuo, Xin Xiao, Yong Liang,Junmin Nan*, Electropolymerization of graphene and 5-[o-(4-bromine amyloxy) phenyl] -10,15,20- triphenylporphrin (o-BrPETPP) composite film electrode for the electrocatalytic oxidation of indirubin, J. Electroanal. Chem., 2012, 681:133-138.19.曹秀芳, 肖信, 郝荣, 左晓希, 南俊民*, 热氧化法制备TiO2薄膜电极及其光电流性能, 上海师范大学学报(自然科学版), 2012, 41(3): 285-291.18.左晓希, 李奇, 刘建生, 肖信, 范成杰, 南俊民*, LiPF6/三氟乙酰胺室温熔盐的制备及在碳-碳电容器中的性能, 化学学报, 2012, 70(4): 367-371.17.易静, 吴绒, 南俊民*, 刘玉英, 叶翠, 胡建强*, Pt-Pd/UCNTs无酶生物传感器的研制与葡萄糖检测的应用, 食品与生物技术学报, 2012, 31(4): 385-390. 16.Qihua Huang, Lishi Wang*, Min Wang, Junmin Nan*, Preparation,characterization and the electrogenerated chemiluminescence behavior of WO3 nanocrystals, J. Alloys Comp., 2011, 509: 9901–9905.15.Keshui Hu, Xin Xiao, Xiufang Cao, Rong Hao, Xiaoxi Zuo, Xiaojing Zhang,Junmin Nan*, Adsorptive separation and photocatalytic degradation of methylene blue dye on titanate nanotube powders prepared by hydrothermal process using metal Ti particles as a precursor, J. Hazard. Mater., 2011, 192: 514–520.14.Qi Li, Xiaoxi Zuo, Jiansheng Liu, Xin Xiao, Dong Shu, Junmin Nan*, Thepreparation and properties of a novel electrolyte of electrochemical double layer capacitors based on LiPF6 and acetamide, Electrochim. Acta, 2011, 58: 330-335. 13.Jinghuan Zhang, Xin Xiao, Junmin Nan*, Hydrothermal-hydrolysis synthesis andphotocatalytic properties of nano-TiO2 with an adjustable crystalline structure, J.Hazard. Mater., 2010, 176(1-3): 617-622.12.Lishi Wang*, Huilian Chen, Xinjian Huang, Junmin Nan*, An experimentalinvestigation of quasireversible maximum of azobenzene on mercury electrode by fourier transformed square-wave voltammetry, Electroanalysis, 2009, 21(6):755–761.11.X.P. Zhou, H.Y. Chen, D. Shu*, C. He, J.M. Nan, Study on the electrochemicalbehavior of vanadium nitride as a promising supercapacitor material, J. Phy. Chem.Solids, 2009, 70(2): 495-500.10.Fang Ye, Junmin Nan*, Lishi Wang, Yan Song, Kuang-Bum Kim, The ultrasonicelectropolymerization of an 5-[o-(4-bromine amyloxy)phenyl]-10,15,20-triphenylporphrin (o-BrPETPP) film electrode and its electrocatalytic properties to dopamine oxidation in aqueous solution, Electrochim. Acta, 2008,53:4156–4160.9. Minjie Yang, Junmin Nan*, Xianlu Hou, Weishan Li, Preparation andelectrochemical performances of nickel metal hydride batteries with specificvolume capacity, Chin. J. Chem. Eng., 2008,16(6):944-948.8. 杨敏杰,南俊民*,侯宪鲁,薛建军,崔燕,崔明,表面包覆CoOOH的球形Ni(OH)2的制备及快充性能,电化学,2006,12(2): 25-29.7. 崔明, 南俊民*, 杨敏杰, 左晓希, 薛建军, 扣式锂离子蓄电池的制备及性能,电源技术, 2006,30(8): 637-640.6. Junmin Nan, Dongmei Han, Minjie Yang, Ming Cui, Xianlu Hou, Recovery ofmetal values from a mixture of spent lithium-ion batteries and nickel-metalhydride batteries, Hydrometallurgy, 2006, 84:75-80.5. Junmin Nan, Dongmei Han, Minjie Yang, Ming Cui, Dismantling, recovery andreuse of spent Ni-MH batteries. J. Electrochem. Soc., 2006,153(1): A101-A105.4. Junmin Nan, Dongmei Han, Ming Cui, Minjie Yang, Linmao Pan,Recycling spentzinc manganese dioxide batteries through synthesizing Zn–Mn ferrite magnetic materials,J. Hazard. Mater., 2006, 133(1-3): 257-261.3. Junmin Nan, Xianlu Hou, Minjie Yang, Dongmei Han, Weishan Li, Effect ofrare-earth elements on the temperature performances of power nickel-metalhydride batteries for electric tools, J. Electrochem. Soc., 2006, 153(6):A1159-1164.2. Junmin Nan, Yong Yang, Zugeng Lin, In situ photoelectrochemistry and Ramanspectroscopic characterization on the surface oxide film of nickel electrode in 30 wt.% KOH solution, Electrochim. Acta, 2006, 51(23): 4873-4879.1. Junmin Nan, Dongmei Han, Xiaoxi Zuo, Recovery of metal values from spentlithium-ion batteries with chemical deposition and solvent extraction,J. Power Sources, 2005, 152: 278-284.。
镍表面阳极氧化膜质量的测试方法易超;熊信柏;曾燮榕;马俊【摘要】Nickel oxide or nickel hydroxide is one of supercapacitor electrode materials with excellent performance .Among different preparing methods ,anodized oxidtion is a promising technology in the field of spercapcitor ,becasuse it can provide nickel oxide or nickel hydroxide with higher electrochemical properties . However ,formation process of anodization film involves two steps of anodizaton weight gain and acid dissolution .Therefore ,the method by directly weighting the sample before and after anodic film can not be adopted .To solve this problem ,a noval test method is proposed ,namely ,a cathodic electrochemical method in85wt% H3 PO4 is used to dissolve oxide film ,which can not harm the substrate .By this ,the mass of the anodic film can be successfully obtained .%氧化镍/氢氧化镍是一种性能优异的超级电容器的电极材料,借助阳极氧化工艺,可发挥其优异的超级电容特性,然而阳极氧化成膜过程涉及增重和酸溶解过程,因此,不能采用成膜前后称量的方法得出膜的质量。
RESEARCH PAPERMicrowave-assisted rapid synthesis of Ag nanoparticles/graphene nanosheet composites and their application for hydrogen peroxide detectionSen Liu •Jingqi Tian •Lei Wang •Xuping SunReceived:15December 2010/Accepted:4May 2011/Published online:15May 2011ÓSpringer Science+Business Media B.V.2011Abstract Ag nanoparticles/graphene nanosheet (AgNPs/GN)composites have been rapidly prepared by a one-pot microwave-assisted reduction method,carried out by microwave irradiation of a N ,N -dimeth-ylformamide (DMF)solution of graphene oxide (GO)and AgNO 3.Several analytical techniques including UV–vis spectroscopy,FT-IR spectroscopy,Raman spectroscopy,X-ray diffraction (XRD),X-ray photo-electron spectroscopy (XPS),and transmission elec-tron microscopy (TEM)have been used to characterize the resulting AgNPs/GN composites.It suggests that such composites exhibit good catalytic activity toward reduction of hydrogen peroxide (H 2O 2),leading to a H 2O 2sensor with a fast amperometric response time of less than 2s.The linear detection range is estimated to be from 0.1to 100mM (r =0.999),and the detection limit is estimated to be 0.5l M at a signal-to-noise ratio of 3.Keywords Graphene nanosheet ÁMicrowave ÁAg nanoparticle ÁHydrogen peroxide detection ÁSensorsIntroductionGraphene,a flat monolayer of sp 2-bonded carbon atoms tightly packed into a two-dimensional (2D)honeycomb lattice,and characterized as ‘‘the thinnest material in our universe’’,has recently received considerable attention and due to its high surface area (*2600m 2/g),high chemical stability,and unique electronic,mechanical properties,which ensure its potential applications in nanoelectronics,nanocom-posites,Li ion batteries,sensors,etc.(Abdelsayed et al.2010;Allen et al.2010;Chen et al.2010a ,b ;Geim and Novoselov 2007;Novoselov et al.2004;Rao et al.2009;Stankovich et al.2006;Zhang et al.2005).In particular,metal nanoparticles/graphene nanosheet (GN)composites have attracted consider-able interest because of the combination remarkable unusual properties of metal nanoparticles with graph-ene and hence their synthesis has become a hot topic of scientific and technological importance (Kamat 2010;Liu et al.2010,2011;Muszynski et al.2008;Sundaram et al.2008;Vinodgopal et al.2010).Among them,Ag nanoparticles/GN (AgNPs/GN)composites have been proved to be a most promising material because AgNPs-containing materials are good candidates for optics,electronics,catalysis,andElectronic supplementary material The online version of this article (doi:10.1007/s11051-011-0410-3)containssupplementary material,which is available to authorized users.S.Liu ÁJ.Tian ÁL.Wang ÁX.Sun (&)State Key Lab of Electroanalytical Chemistry,Changchun Institute of Applied Chemistry,Chinese Academy of Sciences,Changchun 130022,Jilin,China e-mail:sunxp@J.TianGraduate School of the Chinese Academy of Sciences,Beijing 100039,ChinaJ Nanopart Res (2011)13:4539–4548DOI 10.1007/s11051-011-0410-3electrochemistry(Hranisavljevic et al.2002;Sun et al. 2004;Zhang et al.2008).As a result,considerable attention has been paid on the synthesis of AgNPs/GN and AgNPs/graphene oxide(GO)composites(Hassan et al.2009;Li and Liu2010;Lightcap et al.2010;Lu et al.2009;Pasricha et al.2009;Shen et al.2010;Xu and Wang2009;Zhou et al.2009).For example, Pasricha et al.and Li et al.have prepared AgNPs/GN composites though a multiple-step method where AgNPs/GO composites were synthesizedfirst,fol-lowed by the reduction of the composites by an extra reducing agent(Li and Liu2010;Pasricha et al.2009). Zhou et al.and Lightcap et al.have prepared AgNPs/ GN composites by in situ reduction of Ag precursor on GN pre-synthesized from GO(Lightcap et al.2010; Zhou et al.2009).However,all these methods are time-consuming and require complex manipulation process.Recently,a one-pot route has been developed to prepare AgNPs/GN composites where NaHB4or ethylene glycol was used as a reducing agent to reduce GO and AgNO3simultaneously(Hassan et al.2009; Shen et al.2010).On the other hand,microwave irradiation techniques(MIT)has been demonstrated as a rapid and high effective strategy for preparation of graphene-based materials.For example,MIT has been used for exfoliation of graphite(Li et al.2010;Wei et al.2008),reduction of GO(Chen et al.2010a,b; Janowska et al.2010;Zhu et al.2010a,b),and preparation of graphene-based hybrid materials(Guo et al.2010;Jasuja et al.2010;Murugan et al.2009; Zhang et al.2010).Hassan et al.have prepared AgNPs/ GN composites by one-step microwave-assisted heat-ing process in the presence of an extra reducing agent such as hydrazine hydrate,ethylenediamine,or ammo-nium hydroxide(Hassan et al.2009).However,the use of extra reducing agents may introduce heterogeneous impurities,and at the same time,complicate the synthetic procedure,limiting their further practical applications.Herein,we report on one-pot,microwave-assisted preparation of AgNPs/GN composites by microwave irradiation of a N,N-dimethylformamide(DMF)solu-tion of GO and AgNO3for thefirst time,with the use of DMF acting as solvent and reducing agent.We further demonstrate that the resulting composites exhibit good catalytic activity toward the reduction of hydrogen peroxide(H2O2),leading to a H2O2sensor with a fast amperometric response time of less than 2s.The linear detection range is estimated to be from 0.1to100mM(r=0.999),and the detection limit is estimated to be0.5l M at a signal-to-noise ratio of3. ExperimentalChemicals and materialsGraphite powder,AgNO3,H2O2(30wt%)were purchased from Aladin Ltd.NaH2PO4,Na2HPO4, NaNO3,H2SO4(98wt%),KMnO4,and DMF were purchased from Beijing Chemical Corp.All chemi-cals were used as received without further purifica-tion.The water used throughout all experiments was purified through a Millipore system.Phosphate buffer saline(PBS)was prepared by mixing stock solutions of NaH2PO4and Na2HPO4and a fresh solution of H2O2was prepared daily.ApparatusRaman spectra were obtained on J-Y T64000Raman spectrometer with514.5nm wavelength incident laser light.Powder X-ray diffraction(XRD)data were recorded on a Rigaku D/MAX2550diffractometer with Cu K a radiation(k=1.5418A˚).Diffraction patterns were collected under ambient conditions in the2h range of4°–70°at a scanning rate of12°min-1. Transmission infrared spectra were collected in the transmission mode on a Nicolet560Fourier transform infrared(FTIR)spectrometer.X-ray photoelectron spectroscopy(XPS)analysis was measured on an ESCALABMK II X-ray photoelectron spectrometer. Transmission electron microscopy(TEM)measure-ments were made on a HITACHI H-8100electron microscopy(Hitachi,Tokyo,Japan)with an acceler-ating applied potential of200kV.The sample for TEM characterization was prepared by placing a drop of colloidal solution on carbon-coated copper grid and dried at room temperature.Electrochemical measure-ments were performed with a CHI660D electrochem-ical analyzer(CH Instruments,Inc.,Shanghai).A conventional three-electrode cell was used,includinga glassy carbon electrode(GCE,geometric area=0.07cm2)as the working electrode,a Ag/AgCl(3M KCl)electrode as the reference electrode,and platinum foil as the counter electrode.All potentials given in this work are referred to the Ag/AgCl electrode.All the experiments were carried out at ambient temperature.Synthesis of GOGO was prepared from natural graphite powder using modified Hummers method(Hummers and Offeman 1958)as follows:In a typical synthesis,1g of graphite was added into23mL of98%H2SO4, followed by stirring at room temperature over a24h period.After that,100mg of NaNO3was introduced into the mixture and stirred for30min.Subsequently, the mixture was kept below5°C by ice bath,and3g of KMnO4was slowly added into the mixture.After being heated to35–40°C,the mixture was stirred for another30min.After that,46mL of water was added into above mixture during a period of25min. Finally,140mL of water and10mL of30%H2O2 were added into the mixture to stop the reaction. After the unexploited graphite in the resulting mixture was removed by centrifugation,as-synthe-sized GO was dispersed into individual sheets in distilled water at a concentration of0.5mg/mL with the aid of ultrasound for further use.Synthesis of AgNPs/GNAgNPs/GN was prepared by microwave-assisted reduction of GO and AgNO3with the use of DMF as a reducing agent and solvent.In a typical synthesis, 1.0mL of GO aqueous was added into5mL of DMF, followed by addition of40l L of AgNO3aqueous (0.5mol/L).After being immediately sonicated for about2min,the mixture was placed in microwave oven for2min(powder:750W).The AgNPs/GN dispersion was obtained after being sonicated for 10min.The AgNPs/GN nanocomposites were collected by centrifugation and washed by DMF. The resulting precipitates were redispersed in water for characterization and further use.Results and discussionFigure1shows the Raman spectra of GO and AgNPs/ GN composites thus formed.It is established that graphene exhibits two characteristic main peaks:one D band at*1350cm-1arising from a breathing mode of j-point photons of A1g symmetry and one G band at1575cm-1,arising from thefirst order scattering of the E2g phonon of sp2C atoms(Tung et al.2009).In our present study,it is seen that both GO and AgNPs/GN exhibit a D band at1350cm-1 and a G band at1603cm-1and AgNPs/GN has a higher relative intensity of D to G band(0.94)than that of GO(0.80),demonstrating the formation of new graphitic domains after the microwave irradiation(Li et al.2010).Figure1inset shows the photographs of GO–AgNO3mixture before(left)and after(right)the microwave irradiation,revealing a distinct color change from pale-yellow to black.Such observation provides another piece of evidence to support the reduction of GO during the microwave irradiation.Figure2shows the XRD patterns of GO and as-formed AgNPs/GN.Graphite powder shows a sharp (002)peak at26.4°with a typical inter-spacing of 3.34A˚(data not shown)(McAllister et al.2007); however,it is found that GO exhibits a strong peak at 10.02°corresponding to the(002)inter-planar spac-ing of8.2A˚(Fig.2a),indicating graphite has been successfully oxidized by Hummers method(ZhuFig.1Raman spectra of GO(a)and AgNPs/GN(b). Inset photographs of GO–AgNO3mixture before(left) and after(right)microwave irradiationet al.2010a,b).After the microwave irradiation,the peak at10.02°disappears(Fig.2b),confirming the successful reduction of GO(Nethravathi and Raja-mathi2008).Note that another new peaks corre-sponding to(111),(200),and(220)facet of Ag crystal is also observed at38.3°,44.5°,and64.6°, indicating the formation of metallic Ag(Luo et al. 2005).Note that another two new peaks centered at 24.8°and28.1°are observed,which are not corre-sponding to either AgNO3or Ag2O(Fang et al. 2009).Because there are many–COO-groups at the edge of GN(Kim et al.2010),some Ag(I)species are coordinated by–COO-to form coordinated species which may be responsible for these two peaks.Figure S1shows the FT-IR spectra of GO and AgNPs/GN.It is seen that GO exhibits two peaks at1198and 1722cm-1associated with C–O–C and C=O groups. In contrast,such two peaks of AgNPs/GN is observed at1101and1660cm-1,which are shifted97and 66cm-1compared to GO,because the carbonyl of the AgNPs/GN probably catch part electrons cloud of the Ag?,indicating that the Ag?coordinate to COO-(Xu et al.2009).These observations suggest that there are both metallic Ag and Ag?adsorbed on GN.To further confirm the formation of composites structure,the samples were also investigated by XPS technique which has been proven to be a significant measure for observation the inner structure of C for carbon-based materials.Figure3shows the C1s XPS spectra of GO and AgNPs/GN,respectively.The C1s spectra of GO and AgNPs/GN could be deconvoluted into four peaks at284.5,285.6,286.6,and288.4eV, which are associated with C–C,C–OH,C(epoxy/ alkoxy),and C=O,respectively(Park et al.2008).The intensity of all the peaks of carbon to oxygen decreased dramatically after the microwave irradia-tion,suggesting about65%of oxygen-containing functional groups are successfully removed from GO.Figure4shows the Ag3d XPS spectrum of AgNPs/GN.It was reported that the metallic Ag3d peaks are centered at373.9and367.9eV,respec-tively(Pol et al.2002).In contrast,Ag?containing materials show3d peaks at369.6and375.8eV(Cai et al.1998).It is obviously seen the composites thus formed exhibit two strong peaks centered at374.3 and368.2eV,respectively(Tian et al.2010).This observation suggests that there are both metallic Ag and Ag?in AgNPs/GN,which is agreement with the result of XRD.The formation of AgNPs/GN was also evidenced by UV–vis spectrum of the DMF dispersion of the nanocomposites as shown in pared to the spectra of GO and GN,a strong adsorption peak at 452nm assigned to AgNPs is observed,suggesting the formation of AgNPs(Luo et al.2005).Further-more,the absorbance of GN in the spectral region between400and800nm is stronger than that of GO, further indicating the reduction of GO by microwave irradiation.The formation of AgNPs/GN composites was further evidenced by TEM observation as shown in Fig.6.It is clearly seen that the GN has been decorated with small AgNPs about several nanome-ters in diameter,and almost all the AgNPs were distributed uniformly on the GN substrate.Scheme1 represents a scheme(not to scale)to illustrate the formation of AgNPs/GN composites from GO and AgNO3with the use of DMF as reducing and dispersing agent.It should be noted that theformationFig.2XRD patterns of GO(a)and AgNPs/GN(b)of AgNPs and graphene from AgNO3and GO can be attributed to the direct redox between DMF and AgNO3and GO,because there is no other reducing agent in this system.Indeed,DMF has been used as a good reducing agent for preparation of AgNPs (Pastoriza-Santos and Liz-Marza´n2002a)and graph-ene(Murugan et al.2009),respectively.It is expected that DMF can decompose into CO and dimethylamine due to the high temperature([156°C)formed under microwave irradiation,where CO caneffectivelyFig.4Ag3d XPS spectrum of AgNPs/GNreduce GO and AgNO3into AgNPs/GN nanocom-posites(Ai et al.2011).The formation of AgNPs may be also proposed as that Ag?was reduced to Ag0 along with formation of Me2NCOOH from DMF (Pastoriza-Santos and Liz-Marza´n2002b).It also should be mentioned that the resulting dispersion of AgNPs/GN is very stable,suggesting DMF serves as a good dispersing agent for GN and GN-based materials (Park et al.2009;Wang et al.2009).It is well known that the AgNPs exhibit high catalytic activity for reduction of H2O2(Lu et al.2011; Tian et al.2011;Song et al.2009;Welch et al.2005).As a demonstration of application of such AgNPs/GN nanocomposites,they were deposited on a bare GCE surface to test their catalytical performance for H2O2 reduction.Figure7shows cyclic voltammograms (CVs)of a bare GCE,and a AgNPs/GN modified GCE(designated as AgNPs/GN/GCE)in N2saturated 0.2M PBS at pH6.5in the presence of1mM H2O2 (scan rate:0.02V/s).It is obviously seen that the response of the bare GCE toward the reduction of H2O2is pretty weak.In contrast,the AgNPs/GN/GCE exhibits a remarkable catalytic current peak about 40l A in intensity at-0.36V vs.Ag/AgCl.TheseFig.6TEM image of AgNPs/GNScheme1A scheme(not to scale)to illustrate the formation of AgNPs/GN from GO and AgNO3with the use of DMF as a reducing agent and solventFig.7Cyclicvoltammetrys(CVs)of abare GCE(a),and a AgNPs/GN/GCE(b)in N2saturated0.2M PBS at pH6.5in thepresence of1mM H2O2(scan rate:0.02V/s)Fig.8Typical steady-stateresponse of the AgNPs/GN/GCE to successive injectionof H2O2into the stirred N2saturated0.2M PBS at pH6.5.Inset calibration curve(applied potential:-0.3V)observations indicate that the AgNPs/GN nanocom-posites exhibit notable catalytic ability for H2O2 reduction.Figure8shows the typical current–time plot of the AgNPs/GN/GCE in N2-saturated0.2M PBS buffer (pH6.5)on successive step change of H2O2concen-trations under optimized conditions.When an aliquot of H2O2was added into the stirring PBS solution, AgNPs/GN/GCE responded rapidly to the substrate and the current rose steeply to reach a stable value.At the applied potential of-0.30V,the cathode current of the sensor increased dramatically and achieved 95%of the steady-state current within2s,revealing a fast amperometric response behavior.The inset shows the calibration curve of the sensor.The linear detection range is estimated to be from0.1to 100mM(r=0.999),and the detection limit is estimated to be0.5l M at a signal-to-noise ratio of3. ConclusionsMicrowave irradiation has been employed to prepare AgNPs/GN composites by microwave heating of a DMF solution of GO and Ag salts in the absence of an extra reducing agent.The detection of H2O2 without using enzyme in the electrode modified by AgNPs/GN nanocomposites has also been demon-strated and it revealed that the AgNPs contained therein exhibit notable catalytic activity toward the reduction of H2O2.Our presentfindings provide us a low-cost approach to the facile production of nano-particles/GNs composites on a large scale for applications.Acknowledgment This work was supported by National Basic Research Program of China(No.2011CB935800). ReferencesAbdelsayed V,Moussa S,Hassan HM,Aluri HS,Collinson MM,El-Shall MS(2010)Photothermal deoxygenation of graphite oxide with laser excitation in solution and graphene-aided increase in water temperature.J Phys Chem Lett1:2804–2809.doi:10.1021/jz1011143Ai K,Liu Y,Lu L,Cheng X,Huo L(2011)A novel strategy for making soluble reduced graphene oxide sheets cheaply by adopting an endogenous reducing agent.J Mater Chem 21:3365–3370.doi:10.1039/c0jm02865Allen MJ,Tung VC,Kaner RB(2010)Honeycomb carbon:a review of graphene.Chem Rev110:132–145.doi:10.1021/cr900070dCai W,Zhong H,Zhang L(1998)Optical measurements of oxidation behavior of silver nanometer particle within pores of silica host.J Appl Phys83:1705–1711.doi:10.1063/1.366888Chen D,Tang L,Li J(2010a)Graphene-based materials in electrochemistry.Chem Soc Rev39:3157–3180.doi:10.1039/b923596eChen W,Yan L,Bangal PR(2010b)Preparation of graphene by the rapid and mild thermal reduction of graphene oxide induced by microwaves.Carbon48:1146.doi:10.1016/ j.carbon.2009.11.037Fang B,Gu A,Wang G,Wang W,Feng Y,Zhang C,Zhang X (2009)Silver oxide nanowalls grown on Cu substrate as an enzymeless glucose sensor.ACS Appl Mater Interfaces 1:2829–2834.doi:10.1021/am900576zGeim AK,Novoselov KS(2007)The rise of graphene.Nat Mater6:183–191.doi:10.1038/nmat1849Guo S,Wen D,Zhai Y,Dong S,Wang E(2010)Platinum nanoparticle ensemble-on-graphene hybrid nanosheet: one-pot,rapid synthesis,and used as new electrode material for electrochemical sensing.ACS Nano 4:3959–3968.doi:10.1021/nn100852hHassan HMA,Abdelsayed V,Khder AERS,AbouZeid KM, Terner J,El-Shall MS,Al-Resayes SI,El-Azhary AA (2009)Microwave synthesis of graphene sheets support-ing metal nanocrystals in aqueous and organic media.J Mater Chem19:3832–3837.doi:10.1039/b906253j Hranisavljevic J,Dimitrijevic NM,Wurtz GA,Wiederrecht GP (2002)Photoinduced charge separation reactions of J-aggregates coated on silver nanoparticles.J Am Chem Soc124:4536–4537.doi:10.1021/ja012263eHummers WS Jr,Offeman R(1958)Preparation of graphitic oxide.J Am Chem Soc80:1339.doi:10.1021/ja01539a017 Janowska I,Chizari K,Ersen O,Zafeiratos S,Soubane D,Costa VD,Speisser V,Boeglin C,Houlle´M,Be´gin D,Plee D, Ledoux MJ,Pham-Huu C(2010)Microwave synthesis of large few-layer graphene sheets in aqueous solution of ammonia.Nano Res3:126–137.doi:10.1007/s12274-010-1017-1Jasuja K,Linn J,Meltion S,Berry V(2010)Microwave-reduced uncapped metal nanoparticles on graphene:tun-ing catalytic,electrical,and raman properties.J Phys Chem Lett1:1853–1860.doi:10.1021/jz100580xKamat PV(2010)Graphene-based nanoarchitectures.Anchoring semiconductor and metal nanoparticles on a two-dimensional carbon support.J Phys Chem Lett1: 520–527.doi:10.1021/jz900265jKim J,Cote LJ,Kim F,Yuan W,Shull KR,Huang J(2010) Graphene oxide sheets at interfaces.J Am Chem Soc 132:8180–8186.doi:10.1021/ja102777pLi J,Liu CY(2010)Ag/graphene heterostructures:synthesis, characterization and optical properties.Eur J Inorg Chem 1244–1248.doi:10.1002/ejic.200901048Li Z,Yao Y,Lin Z,Moon KS,Lin W,Wong C(2010) Ultrafast,dry microwave synthesis of graphene sheets.J Mater Chem20:4781–4783.doi:10.1039/c0jm00168f Lightcap IV,Kosel TH,Kamat PV(2010)Anchoring semicon-ductor and metal nanoparticles on a two-dimensional catalystmat.Storing and shuttling electrons with reduced graphene oxide.Nano Lett10:577–583.doi:10.1021/nl9035109Liu S,Tian J,Wang L,Li H,Zhang Y,Sun X(2010)Stable aqueous dispersion of graphene nanosheets:noncovalent functionalization by a polymeric reducing agent and their subsequent decoration with Ag nanoparticles for en-zymeless hydrogen peroxide detection.Macromolecules 43:10078–10083.doi:10.1021/ma102230mLiu S,Tian J,Wang L,Sun X(2011)A method for the production of reduced graphene oxide using benzylamine as a reducing and stabilizing agent and its subsequent decoration with Ag nanoparticles for enzymeless hydrogen peroxide detection.Carbon.doi:10.1016/j.carbon.2011.03.036Lu G,Mao S,Park S,Ruoff RS,Chen J(2009)Facile,non-covalent decoration of graphene oxide sheets with nano-crystals.Nano Res2:192–200.doi:10.1007/s12274-009-9017-8Lu W,Liao F,Luo Y,Chang G,Sun X(2011)Hydrothermal synthesis of well-stable silver nanoparticles and their application for enzymeless hydrogen peroxide detection.Electrochim Acta56:2295–2298.doi:10.1016/j.electacta.2010.11.053Luo L,Yu S,Qian H,Zhou T(2005)Large-scale fabrication of flexible silver/cross-linked poly(vinyl alcohol)coaxial nanocables by a facile solution approach.J Am Chem Soc 127:2822–2823.doi:10.1021/ja0428154McAllister MJ,Li JL,Adamson DH,Schniepp HC,Abdala AA, Liu J,Herrera-Alonso M,Milius DL,Car R,Prudˇhomme RK,Aksay IA(2007)Single sheet functionalized graphene by oxidation and thermal expansion of graphite.Chem Mater19:4396–4404.doi:10.1021/cm0630800 Murugan AV,Muraliganth T,Manthiram A(2009)Rapid, facile microwave-solvothermal synthesis of graphene nanosheets and their polyaniline nanocomposites for energy storage.Chem Mater21:5004–5006.doi:10.1021/ cm902413cMuszynski R,Seger B,Kamat PV(2008)Decorating graphene sheets with gold nanoparticles.J Phys Chem C112: 5263–5266.doi:10.1021/jp800977bNethravathi C,Rajamathi M(2008)Chemically modified graphene sheets produced by the solvothermal reduction of colloidal dispersions of graphite oxide.Carbon 46:1994–1998.doi:10.1016/j.carbon.2008.08.013 Novoselov KS,Geim SV,Morozov SV,Jiang D,Zhang Y, Dubonos SV,Grigorieva IV,Firsov AA(2004)Electric field effect in atomically thin carbonfilms.Science 306:666–669.doi:10.1126/science.1102896Park S,An J,Piner RD,Jung I,Yang D,Velamakanni A, Nguyen ST,Ruoff RS(2008)Aqueous suspension and characterization of chemically modified graphene sheets.Chem Mater20:6592–6594.doi:10.1021/cm801932u Park S,An J,Jung I,Piner RD,An SJ,Li X,Velamakanni A, Ruoff RS(2009)Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents.Nano Lett9:1593–1597.doi:10.1021/nl803798y Pasricha R,Gupta S,Srivastava AK(2009)A facile and novel synthesis of Ag–graphene-based nanocomposites.Small 5:2253–2259.doi:10.1002/smll.200900726Pastoriza-Santos I,Liz-Marza´n LM(2002a)Formation of PVP-protected metal nanoparticles in ngmuir 18:2888–2894.doi:10.1021/la015578g Pastoriza-Santos I,Liz-Marza´n LM(2002b)Synthesis of silver nanoprisms in DMF.Nano Lett2:903–905.doi:10.1021/ nl025638iPol VG,Srivastava DN,Palchik O,Palchik V,Slifkin MA, Weiss AM,Gedanken A(2002)Sonochemical deposition of silver nanoparticles on silica ngmuir18: 3352–3357.doi:10.1021/la0155552Rao CNR,Sood AK,Subrahmanyam KS,Govindaraj A(2009) Graphene:the new two-dimensional nanomaterial.Angew Chem Int Ed48:7752–7777.doi:10.1002/anie.200901678 Shen J,Shi M,Li N,Yan B,Ma H,Hu Y,Ye M(2010)Facile synthesis and application of Ag-chemically converted graphene nanocomposite.Nano Res3:339–349.doi:10.1007/s12274-010-1037-xSong Y,Cui K,Wang L,Chen S(2009)The electrodeposition of Ag nanoparticles on a type I collagen-modified glassy carbon electrode and their applications as a hydrogen peroxide sensor.Nanotechnology20:105501.doi:10.1088/ 0957-4484/20/10/105501Stankovich S,Dikin DA,Dommett GHB,Kohlhaas KM, Zimney EJ,Stach EA,Piner RD,Nguyen ST,Ruoff RS (2006)Graphene-based composite materials.Nature 442:282–286.doi:10.1038/nature04969Sun X,Dong S,Kang E(2004)One-step preparation and characterization of poly(propyleneimine)dendrimer-pro-tected silver nanoclusters.Macromolecules37:7105–7108.doi:10.1021/ma048847tSundaram RS,Go´mez-Navarro C,Balasubramanian K,Burg-hard M,Kern K(2008)Electrochemical modification of graphene.Adv Mater20:3050–3053.doi:10.1002/adma.200800198Tian J,Liu S,Sun X(2010)Supramolecular microfibrils of o-phenylenediamine dimers:oxidation-induced morphol-ogy change and the spontaneous formation of Ag nano-particle decorated nanofingmuir26:15112–15116.doi:10.1021/la103038mTian J,Li H,Lu W,Luo Y,Wang L,Sun X(2011)One-step preparation of Ag nanoparticle-decorated coordination polymer nanobelts and their application for enzymeless H2O2detection.Analyst136:1806–1809.doi:10.1039/ c0an00929fTung VC,Allen MJ,Yang Y,Kaner RB(2009)High-throughput solution processing of large-scale graphene.Nat Nanotechnol4:25–29.doi:10.1038/NNANO.2008.329 Vinodgopal K,Neppolian B,Lightcap IV,Grieser F,Ash-okkumar M,Kamat PV(2010)Sonolytic design of graphene–Au nanocomposites.Simultaneous and sequential reduction of graphene oxide and Au(III).J Phys Chem Lett1:1987–1993.doi:10.1021/jz1006093Wang H,Robinson JT,Li X,Dai H(2009)Solvothermal reduction of chemically exfoliated graphene sheets.J Am Chem Soc131:9910–9911.doi:10.1021/ja904251pWei T,Fan Z,Luo G,Zheng C,Xie D(2008)A rapid and efficient method to prepare exfoliated graphite by micro-wave irradiation.Carbon47:337–339.doi:10.1016/ j.carbon.2008.10.013Welch C,Banks C,Simm A,Compton R(2005)Silver nano-particle assemblies supported on glassy-carbon electrodes for the electro-analytical detection of hydrogen peroxide.Anal Bioanal Chem382:12–21.doi:10.1007/s00216-005-3205-5Xu C,Wang X(2009)Fabrication offlexible metal-nanopar-ticlefilms using graphene oxide sheets as substrates.Small5:2212–2217.doi:10.1002/smll.200900548Xu GC,Shi JJ,Li DJ,Xing HL(2009)On Interaction between nano-Ag and P(AMPS-co-MMA)copolymer synthesized by ultrasonic.J Polym Res16:295–299.doi:10.1007/ s10965-008-9229-8Zhang Y,Tan YW,Stormer HL,Kim P(2005)Experimental observation of the quantum Hall effect and Berry’s phase in graphene.Nature438:201–204.doi:10.1038/nature04235 Zhang H,Wang G,Chen D,Lv X,Li J(2008)Tuning pho-toelectrochemical performances of Ag–TiO2nanocom-posites via reduction/oxidation of Ag.Chem Mater 20:6543–6549.doi:10.1021/cm801796qZhang M,Lei D,Yin X,Chen L,Li Q,Wang Y,Wang T (2010)Magnetite/graphene composites:microwaveirradiation synthesis and enhanced cycling and rate per-formances for lithium ion batteries.J Mater Chem 20:5538–5543.doi:10.1039/c0jm00638fZhou X,Huang X,Qi X,Wu S,Xue C,Boey FYC,Yan Q, Chen P,Zhang H(2009)In situ synthesis of metal nanoparticles on single-layer graphene oxide and reduced graphene oxide surfaces.J Phys Chem C113:10842–10846.doi:10.1021/jp903821nZhu C,Guo S,Fang Y,Dong S(2010a)Reducing sugar:new functional molecules for the green synthesis of graphene nanosheets.ACS Nano4:2429–2437.doi:10.1021/ nn1002387Zhu Y,Murali S,Stoller MD,Velamakanni A,Piner RD,Ruoff RS(2010b)Microwave assisted exfoliation and reduction of graphite oxide for ultracapacitors.Carbon48:2106–2122.doi:10.1016/j.carbon.2010.02.001。