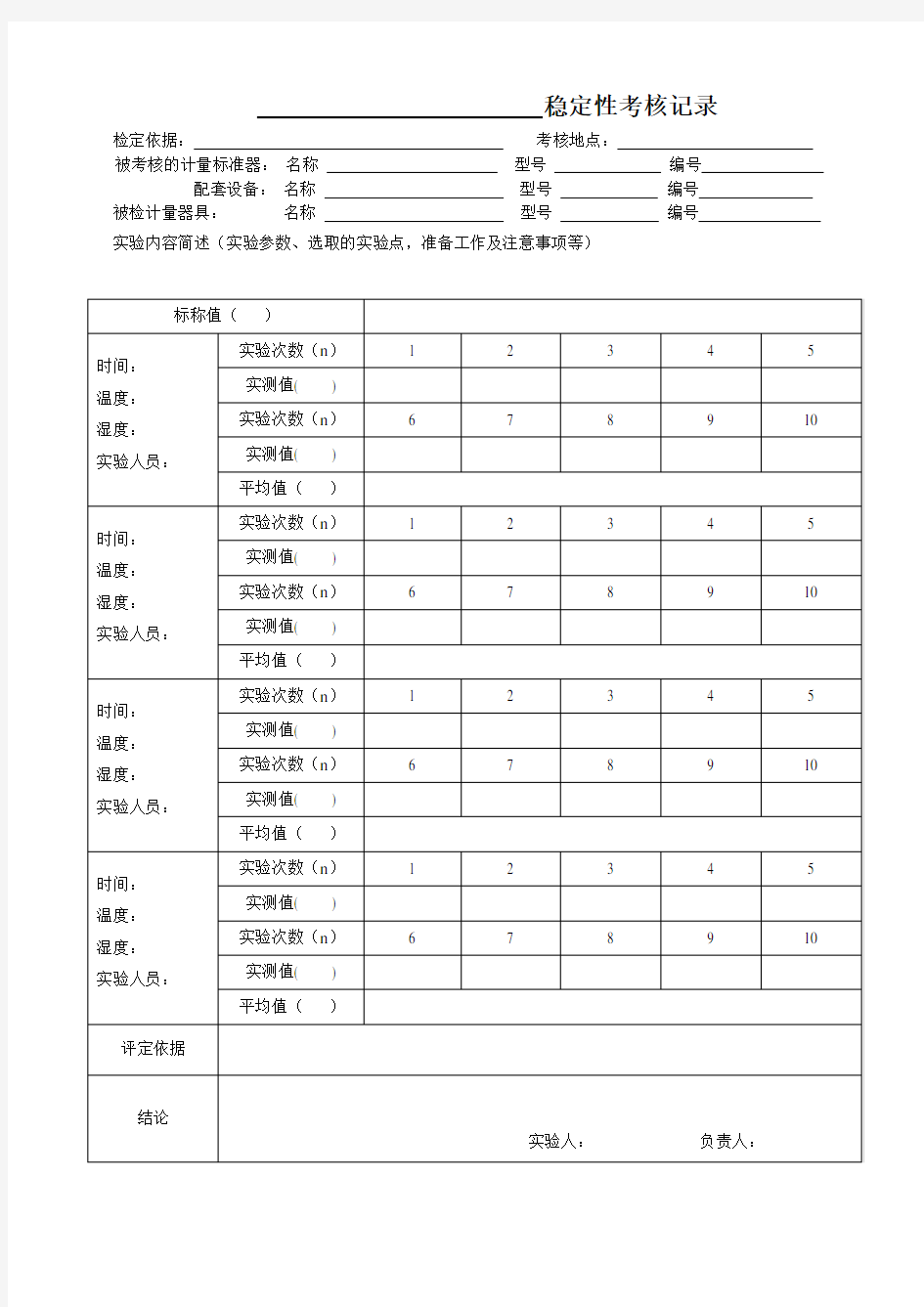
稳定性考核记录检定依据:考核地点:
被考核的计量标准器:名称型号编号配套设备:名称型号编号被检计量器具:名称型号编号实验内容简述(实验参数、选取的实验点,准备工作及注意事项等)
车身尺寸 稳定性控制方法 龚国平(沙济伦博士指导) 2005年11月 奇瑞公司规划设计院
编写本文目的 ?讨论建立车身尺寸稳定性指标的必要性、可行性以及如何实施。 ?介绍车身尺寸稳定性控制方法。 公司目前车身尺寸控制指标 ?目前,公司车身尺寸主要控制指标是IQG值和尺寸符合率(DAR)。 ?这两个指标侧重控制车身尺寸的准确性,也就是精度,但是相对忽视了更重要的一项指标--稳定性。 认识 IQG ?什么是IQG ? 它是法语:Indice Qualide Geometrique 的所写,中文意思是“车身几何质量指数”,它是用来评定钣金零件、分总成及总成重要几何尺寸一致性的一种工具。 ?IQG值是如何计算的? IQG值=所有超差测量特性扣分之和 / 测量特性总数;它的取值范围是0-10之间。 认识尺寸符合率(DAR) ?什么是DAR ? 它是英语:Dimension Accord Rate 的所写,中文意思是“尺寸符合率”,它是用来评定钣金零件、分总成及总成重要几何尺寸符合要求的程度。 ?DAR值是如何计算的? DAR值=未被扣分测量特性之和 / 测量特性总数;它的取值范围是0-1之间。 结论 ?IQG值和尺寸符合率(DAR)都仅仅控制了车身尺寸的准确性或精度,对尺寸的稳定性却没有控制,或仅有很微弱的控制。
?我们迫切地需要一个控制车身尺寸稳定性的指标。 稳定性比准确性更重要 ?为什么这么说? 一个枪手打靶,可能会有如下四种情形: ?很明显,情况1最差,情况4最好。 ?那么情况2和情况3哪一个比较好呢? 2反映了一种准确性或精度,但是它的分散程度很大,3反映了一种稳定性或一致性,但是它偏离目标很大。究竟哪一种情形更好? ?情况3的解决可能仅仅只需要调整一下准心,很容易就解决了问题。 ?情况2呢?必须对打靶所用的枪进行全面检查,详细分析其原因。 ?对于我们的车身尺寸控制(包括调试)也一样。稳定性比准确性更重要。 ?比如说某个测量特性,它的测量结果表明它一直偏离正确位置10mm,怎么办?很容易解决,只需要调整夹具,调过来10mm;就算因特殊原因,不能调整夹具,那改冲压件也可以,会有立竿见影的效果。 ?如果一个测量特性,测量结果表明它在目标值的正负5mm之间波动,这个问题怎么办?通过调夹具能解决吗?通过更改冲压件能解决吗?
稳定性-重复性
压力计计量装置的重复性试验记录 试验时间2015年 2月17日2015年8月16日 被测对象 名称型号编号名称型号编号 压力变 送器 EJA530A S4NC04780 压力变 送器 EJA530A S4NC04780 测量条件符合要求符合要求测量次数测量值(mA)测量值(mA) 1 11.9775 11.9814 2 11.9779 11.9816 3 11.9776 11.9814 4 11.9774 11.9813 5 11.9778 11.9814 6 11.9773 11.9816 7 11.9781 11.9815 8 11.9785 11.9812 9 11.9780 11.9813 10 11.9781 11.9814 y11.9778 11.9814 1) ( ) (1 2 - - =∑ = n y y y s n i i i 0.3702μA0.1291μA 结论符合要求符合要求试验人员孙成霖孙成霖 注:已建计量标准,至少每年进行一次重复性试验,测得的重复性应满足检定或校准结果的测量不确定度的
压力表计量装置的重复性试验记录 试验时间2016年 2月16日2016年8月18日 被测对象 名称型号编号名称型号编号 压力变 送器 EJA530 A S4NC04780 压力变 送器 EJA530 A S4NC04780 测量条件符合要求符合要求测量次数测量值(mA)测量值(mA) 1 11.9814 11.9813 2 11.9812 11.9817 3 11.9815 11.9816 4 11.9814 11.9815 5 11.9813 11.9816 6 11.9815 11.9817 7 11.9813 11.9815 8 11.9818 11.9815
产品稳定性考察管理规程 1.目的 建立一个产品稳定性考察管理规程,使能在产品有效期内监控已上市药品的质量,以发现药品与生产相关的稳定性问题,并确定药品能够在标示的贮存条件下,符合质量的各项要求。 2.范围 已获准上市的市售包装药品。 3.责任 QC部负责执行本规程。 4.内容 4.1 由够资格的专业技术人员制定稳定性计划,报主管部门负责人批准后执行。 4.2 由授权人担任稳定性试验的全面工作。 4.3稳定性分类 4.3.1影响因素试验 4.3.2加速试验 4.3.3长期试验 考察产品分为以下四类: A类:新产品头3批产品做长期稳定性考察;直至转正后。 B类:当影响产品质量的主要因素,如工艺改进、设备变更、改变内包装形 式、主要原辅料供应商变更的头三批产品做长期稳定性考察; C类:生产过程中遇到特殊情况,可能会影响质量稳定性的产品,如返工、或 有回收操作的批次,该批产品做长期稳定性考察。 D类:除上述A、B、C类之外的产品,长期生产的产品每年选择至少1批产品 做长期稳定性考察,除当年未生产 4.4考察原则 4.4.1 正常批量生产的最初一批产品应当列入持续稳定性考察计划,以进一步确认有效 期。 4.4.2 正常情况下,每一品种的每一规格、每一内包装形式的药品每年考察一个批次。 4.4.3 特殊情况下,重大变更或生产和包装有重大偏差的药品以及重新加工、返工或回 收的批次应列入稳定性考察,并增加考察批次,一般应不少于三批。 4.6制定稳定性计划 4.6.1质量保证部QA人员于每年年初依据本年度的生产计划制订年度稳定性试验计划, 确定本年度将进行的和可能要增加进入稳定性试验程序的品种,并于每年年终
稳定性考察方案 制定人:日期: 审核人:日期: 批准人:日期: 北京远策药业有限公司 目录 一、目的 二、范围 三、职责 四、产品介绍 五、药品稳定性考察信息表和检测方法依据 六、具体方案 1.产品批量的选择 2.考察条件、时间 3.样品测试时限的要求 七、稳定性考察报告 1.考察报告内容的要求 2.偏差处理 3.报告的时限要求 4.报告程序 5.记录管理要求 6.档案保存期限 一、目的 药品的稳定性是指原料药及其制剂保持其物理、化学、生物学和微生物学性质的能力。稳定性试验的目的是考察原料药、中间产品或制剂的性质在温度、湿度、光线等条件的影响下随时间变化的规律,为药品的生产、包装、贮存、运输条件和有效期的确定提供科学依据,以保障临床用药的安全有效。并且通过持续稳定性考察可以监测在有效期内药品的质量,并确定药品可以或预期可以在标示的贮存条件下,符合质量标准的各项要求。
二、范围 适用于公司所有成品及原液的考察。药品稳定性考察包括:加速试验和长期(持续)稳定性试验。长期(持续)稳定性考察主要针对市售包装产品,但也需兼顾待包装产品。例如,当待包装产品在完成包装前,还需要长期贮存时,应当在相应环境条件下,评估其对包装后产品稳定性的影响。此外,还应考虑到对贮存时间较长的中间产品进行考察。加速试验主要针对批量放大及上市后变更(如生产设备变更、原辅料变更、工艺调整等)时生产的产品的稳定性试验。 三、职责 质量保证部负责药品稳定性考察方案的起草、审核、实施过程的监督、数据的收集与报告的撰写; 质量控制部:负责药品稳定性考察的检验工作; 质量受权人:负责药品稳定性考察方案及报告的批准。 四、产品介绍: 将本年度需要做稳定性考察产品的信息如:产品名称、批号、规格、包装规格、考察数量、生产日期、有效期等详细信息填入《药品稳定性考察信息表》。
1计量标准的重复性、稳定性考核 一、概述 计量标准是准确度低于计量基准,用于检定或校准其他计量标准或者工作计量器具的计量器具,它处于国家量值传递(溯源)体系的中间环节,起承上启下的作用。因此,计量标准在使用前必须依照JJF1033《计量标准考核规范》的要求,进行各项技术准备,使计量标准符合规范的要求并通过考核。下面主要介绍计量标准的重复性、稳定性考核的内容。 二、计量标准的重复性考核 1.计量标准的重复性 计量标准的重复性即在相同测量条件下,重复测量同一被测量,计量标准提供相近示值的能力。计量标准的重复性通常用测量结果的分散性来定量表示,即用单次测量结果y的实验标准差s(y i)来表示。 计量标准的重复性通常是检定或校准结果的一个不确定度来源。 新建计量标准应当进行重复性试验,并提供试验的数据;已建计量标准,至少每年进行一次重复性 试验,测得的重复性应满足检定或校准结果的测量不确定度的要求。 在计量标准考核中,计量标准的重复性是指在重复性条件(这些条件包括测量程序、人员、仪器、环境等方面)下用该计量标准测量一常规的被测对象时,所得到的测量结果的一致性。为保证在尽量相同的条件下进行测量必须在尽量短的时间内完成重复性测量。 2.重复性的试验方法在重复性条件下,用计量标准对常规的被检定或被校准对象进行 结果为y i(i=1,2,…,n),则其重复性s(y i)为式中:y —n次测量结果的 算术平均值; n—重复测量次数,n应尽可能大,一般应不少于10次。 重复性试验结果也会受被测对象不稳定的影响,所以在进行计量标准的重复性试验时,选择的测量对象应为常规的被检定或被校准计量器具,而不是本身重复性和稳定性都是最佳的被检定或被校准计量器具,这样评定得到的不确定度可以用于大多数的检定或校准结果。 3?计量标准的重复性考核对于新建计量标准,只要按照要求进行重复性试验,并提供试验的重复性数据即可;对于已建计量标准, 至少每年进行一次重复性试验,如果重复性试验结果不大于新建计量标准时的重复性,则重复性符合要求;如果重复性试验结果大于新建计量标准时的重复性时,应按照新的重复性结果重新进行检定或校准结果的测量不确定度评定,并判断检定或校准结果的测量不确定度是否满足被检定或校准对象的需要。 4?《计量标准的重复性试验记录》参考格式及填写说明 (1)《计量标准的重复性试验记录》参考格式申请考核单位原则上应当按照本参考格式填写。如果本参考格式不适用,申请计量标准考核单位可 以自行设计《计量标准的重复性试验记录》格式,但是不应少于参考格式规定的内容。 《计量标准的重复性试验记录》参考格式: 的重复性试验记录 n次独立重复测量,若得到的测量 试验时间、、测量值()月日年年 月日 年 月日 年 月日 年 月日 试验条件 1 2
标题持续稳定性考察管理规程 编制记载分发记载 起草人:年月日文件编码:发布号: 审核人:年月日分发部门:复印号: 批准人:年月日 质量部发布 生效日:年月日 目的:建立持续稳定性考察管理规程,对上市后生产的药品继续进行稳定性考察和研究,监测在有效期内药品的质量,为规范此项工作而建立管理规程。 范围:主要适用于市售包装药品以及待包装产品。 主责:质量部、质量控制实验室。 释义: 关联: 正文: 1 持续稳定性考察的目的: 是在有效期内监控药品质量,以发现药品与生产相关的稳定性问题(如杂质含量或溶出度特性的变化),并确定药品能够在标示的贮存条件下,符合质量标准的各项要求。 2 持续稳定性考察对象: 主要针对市售包装药品。但也需兼顾待包装产品。还应当考虑对贮存时间较长的中间产品进行考察。 3 稳定性考察方案:每个品种需要制定考察方案,考察方案应由质量受权人批准执行。 3.1 考察方案应涵盖药品有效期; 3.2 每种规格、每个生产批量药品的考察批次数;
3.3 制定考察项目、检验方法依据及合格标准; 3.4 容器密封系统的描述; 3.5 考察周期及检验频次; 3.6 贮存条件(采用与药品标示贮存条件相对应的《中华人民共和国药典》规定的 长期稳定性试验标准条件)。 4 稳定性考察留样原则及批次数量: 4.1 产品批准上市后首次生产的前三批验证产品,对不同包装材料及包装规格的销售产品均要分别留样进行长期稳定性考察。 4.2 生产第二年起各年的产品,对生产的每种规格、每种内包装形式的药品,至少每年应当留取一批进行持续稳定性考察。 4.3 有重大变更或生产和包装有重大偏差的药品应当列入稳定性考察。改变原辅料、与药品直接接触的包装材料变更、生产工艺、主要生产设备及其他影响药品质量的主要因素变更时,应当对变更实施后生产的药品留样三批进行加速试验和长期稳定性试验。 4.4 重新加工、返工或回收的批次,也应当留样进行加速试验和长期稳定性试验。 4.5 考察样品留样数量:按照取样频次、考察项目、考察期内所需的全检量。 5 产品稳定性考察试验计划: 5.1 根据每年的生产情况,按要求填写每个品种“产品稳定性考察试验计划表”,按照计划进行稳定性试验管理。 5.2 产品稳定性考察试验计划表内容,产品名称、批号、规格、生产日期、有效期至、取样日期、检验时间、检验用量、考察目的、考察周期、考察期至。 5.3 样品提取要求: 5.3.1 样品必须按照稳定性试验计划从恒温恒湿箱和其他储存条件下按时取出,样品取样时间偏差范围:
持续稳定性考察方案 1.对各品种生产的前三批进行稳定性考察每批拟计划一定量进行 考察其余批次只做一般留样考察留样量为三次复检的全项检验量 2.考察项目:依据《中国药典》 XX版二部中《原料药与药物制剂稳定性试验指导原则》进行确定 3.考察方法: 3.1加速试验: 3.1.1按市售包装在温度40℃± 2℃、相对湿度 75%±5%的条件下放置六个月在试验期间第 1 个月、 2 个月、 3 个月、 6 个月末分别取样一次按稳定性考察要点项目进行检验 3.1.2 在上述条件下如 6 个月内供试品经检测不符合制定的质量标准则应在如下条件温度 30℃± 2℃、相对湿度 65%±5%情况下进行加速试验时间仍为六个月 3.2长期试验: 按市售包装在温度 18~26℃、相对湿度 60%±15%的条件下放置12 个月. 每 3 个月取样一次分别于 0 个月 3 个月 6 个月 9 个月 12个 月末取样按各剂型品种具体的稳定性考察要点项目进行检验12 个月后仍继续考察分别于18 个月 24 个月 36 个月(以此类推)末取样检测将结果与 0 月比较以确定药品有效期 3.3高温试验:
供试品置密封洁净容器中在60℃条件下放置 10 天于第 5 天和第10 天取样检测有关指标如供试品发生显著变化则在40℃下同法进行试验如 60℃无显著变化则不必进行40℃试验 3.4高湿试验: 试品置恒温密闭容器中于25℃相对湿度为 90%±5%条件下放置 10天在第 5 天和第 10 天取样检测检测项目应包括吸湿增重项若吸湿增 重 5%以上则应在 25℃RH75%±5%下同法进行试验;若吸湿增重 5%以 下且其他考察项目符合要求则不再进行此项试验液体可不进行高湿 试验 3.5光照试验:供试品置光照箱或其它适宜的光照容器内于照度 不< 5000Lx 的条件下放置 10 天在第 5 天和第 10 天取样检测 3.6以上为影响因素稳定性研究的一般要求根据药品的性质必要 时可以设计其他试验如酸、碱及氧化降解等 3.7检测时间的规定: 3.7.1取出时间:1个月加速绝不允许提前和推迟;两个月允许±1 天;三个月允许± 1 周;六个月允许± 2 周;一年后允许±四周 3.7.2样品取出先放在常温下一般要求一周内完成检测;温湿度 敏感的产品要及时检测 3.8恒湿条件的获得方式 恒湿条件可以通过在密闭容器如干燥器下部放置饱和盐溶液而获 得根据不同湿度要求可以选择NaCl 饱和溶液 [ 相对湿度为( 75±1)%15.5℃~60℃] 、KNO3饱和溶液 [ 相对湿度为 92.5%25℃] 、NaNO3
关于涡轮叶片尺寸稳定性的实验调查 摘要:本文介绍的是涡轮叶片简易蜡模尺稳定性的实验研究。由于超级合金制作的涡轮叶片,具有严格的尺寸和形位公差。叶片由熔模铸造制作而成,包括压蜡、制壳、脱蜡、浇注及后处理完成。压蜡阶段的尺寸准确性如同后处理工序一样,对最终的叶片尺寸也有很大的影响。此项实验工作的重点是在射蜡阶段,调查过程参数及叶片形位要素对关键尺寸收缩造成的影响。为了降低分析和模具制造的复杂性,按照叶片形状设计了两种模型。一副模具上带有两个穴(形成两个蜡模)。选取射蜡温度和射蜡时间作为可变过程参数。结果会发现,对叶片的弯曲度和不规则的厚度的影响有明显的不同。射蜡时间比射蜡温度起了更加主要的影响。 1.介绍 燃气涡轮的作用是把热能转化为机械能。适用于很多工业领域,如泵,过滤,提纯,发电机及运输。燃气涡轮的一个关键组成部分就是叶片,包括可转动的叶片及静止叶片。叶片在困难运行条件下发挥作用,如高温,高机械压力,高热疲劳或腐蚀性环境等等。涡轮叶片尺寸及形位公差都很小,是由超级合金采用熔模铸造的方式生产出来的。此工序是用于生产高质量、形状复杂的产品。熔模铸造特别是用在,当产品用其他制作方式如锻造或是加工的方法生产时,不划算,不实用,或是不可行的情况。 熔模铸造主要工序包括压蜡、制壳、脱蜡、干燥、浇注及修磨。每一步都对最终产品尺寸有一定的影响,而压蜡和浇注是最主要的影响。 用于做模型的材料,必须有以下特点:底粘度、一定的固体强度,低混合、低收缩率、高稳定性、并且对于制壳用料有化学抗性、有可接合性并且对健康无害。而蜡恰恰具有了以上所有特性,于是被选为做模型的材料。 蜡模的最终尺寸,在射蜡阶段会受到以下因素影响:1)蜡料种类,2)形状,3)过程参数。 从另一方面来说,仅知道所选蜡料的线性(体)收缩率,是不足以预知尺寸的最终结果的。 产品形状和过程参数对最终尺寸具有相当的影响。 蜡与半结晶状热塑聚合物具有类似的性质。它们也有不一样的特点:1)低熔点(100摄氏度以下) 2)低热传导性。3)对高加热速率敏感。 压蜡包括以下几个阶段:1)把固体蜡放入一个用油来加工的容器里溶化。。2)把溶化的蜡传送到射蜡机的桶里。3)用射蜡机把蜡射入模具里。3)冷却蜡模。最后4)取出蜡模。(通常接下来的工序是校正工序)如果校正要求达到既定的尺寸,那或者增加生产周期或者使用更多的工装,无论是哪种方式,都会增加熔模铸造的总成本。图形1展示的是一个典型的射蜡机的简图。 过程参数对最终尺寸影响的程度,会由于叶片的复杂形状受到影响。因此,这就是要进行
文件编号SMP-11-014 稳定性考察标准管理规程 版本号01 黑龙江宝庆隆生物技术有限责任公司 文件名称稳定性考察管理规程 文件编号SMP-11-014 版本号01 拷贝号 起草人起草日期年月日审核人审核日期年月日审核人审核日期年月日批准人批准日期年月日颁发部门质量管理部执行日期年月日分发部门中心化验室 1目的 为产品稳定性考察提供原则及依据,考察药品(包括原料、成品等)在温度、湿度、 光线的影响下随时间变化的规律,为药品的生产、包装、贮存、运输条件和有效期的确定提 供科学依据,同时通过考察建立产品的有效期;并在有效期内监控已上市药品的质量, 以发现药品与生产相关的稳定性问题,确定药品能够在标示的贮存条件下,符合质量标准的各项要求,确保产品质量。 2范围 适用于成品、原料的稳定性考察。 3责任者 中心化验室、质量管理部、质量受权人
文件编号SMP-11-014 稳定性考察标准管理规程 版本号 01 4职责 4.1 中心化验室副主任负责本标准管理规程的起草。 4.2 中心化验室主任负责本标准管理规程的审核。 4.3 质量管理部部长负责本标准管理规程的审核。 4.4 质量受权人负责本标准管理规程的批准。 5依据 《中国药典》2010 年版二部及《药品 GMP 指南》-质量控制实验室与物料系统 6内容 6.1 稳定性考察的类别 6.1.1 上市前阶段:影响因素试验、加速试验、长期试验。 6.1.2 上市后阶段:上市后持续稳定性考察(条件等同于长期稳定性试验)、承诺稳定性试验(条件等同于为加速试验和长期稳定性试验)。 6.1.3 其他稳定性试验:中间产品放置时间稳定性试验、批量放大及上市后变更(如生产设备变更,原辅料变更,工艺调整等 )稳定性试验、特殊目的稳定性试验,例如对偏差调查等的支持性试验。此类稳定性试验的条件均可参考上市前试验的条件,根据不同产品特性和稳定性试验的目的选择。 6.2 已上市阶段稳定性试验的目的 6.2.1 监控已上市药品在有效期内的质量,以发现药品与生产相关的稳定性问题(如含量、有关物质变化),并确定药品可以或预期可以在标示的贮存条件下,符合质量标准的各项 要求。考察产品上市后在生产、包装、质控、使用条件等诸多方面重大的变更对产品稳定 性的影响,考察变更后药品的稳定性趋势,以评价变更的合理性。
标准文件 qqqq药业有限责任公司 文件名称稳定性考察管理规程 起草人审核 人 批准 人 起草日期审核日 期 批准日 期 生效日期文件编 号 09SM1600-003页码1/5 分发部门QA、QC 1. 范围:适用于原料、辅料、内包材、中间产品、成品的稳定性考察试验。 2. 职责 留样管理员:负责原料、辅料、中间产品、成品的稳定性考察,对试验到期品种的稳定性进行评价,并对相关记录归档。 化验员:负责原料、辅料、中间产品、成品的检验。 QC负责人:负责对稳定性试验中出现的异常情况进行处理和总结的审核,监督、检查执行情况。 质量管理部长:对总结的审核和批准。 3. 内容 3.1. 试验前的准备 3.1.1. 计划
由QC化验员起草制订稳定性试验计划表,包括:品名、规格、实验批次、批号、考察条件、考察方式、考察项目及方法、实施部门等。稳定性试验计划表须经QC主任汇审,交质量管理部长审核、批准方可生效。 3.1.2. 包装 成品包装与销售包装一致,原辅料与实际保存包装一致或相似。中间体模拟生产周转包装,与生产保持一致或相似。 3.2. 原辅料稳定性试验 3.2.1. 观察项目:性状、鉴别、含量测定等。 3.2.2. 贮存条件:与该物料规定贮存条件相一致。 3.2.3. 贮存时间:按《留样管理规程》执行。 3.2. 4. 考察方式 编 号09SM1600-003 稳定性考察管理规程页 码 2/5 3.2. 4.1. 影响因素试验 将检品除去包装以后,平放在称量瓶或培养皿中摊成≤5mm厚的薄层,疏松样品摊成≤10mm厚薄层,在以下条件下贮存、观察、检测,考察各项指标变化情况。 高温条件下,温度分别为40℃、6 0℃2个温度水平。将供试品在60℃温度下放置10天,于第5天和第10天取样,按重点考察项目进行检测,若供试品无明显变化则不再进行40℃条件下试验;若供试品有明显变化(如含量下降5%、鉴别不明显、外观色泽变化大等),则须在40℃条件下用同样的方法进行试验。 高湿条件下,相对湿度分别为75%±5%、90%±5%2个湿度水平(温度为25℃)。将供试品置于相对湿度90%±5%湿度下(装有KNO3饱和溶液的干燥器中,用封口胶密封)放置10天,于第5天和第10天取样,按考察项目进行检测,同时准确称定试验前后供试
尺寸稳定性 当要求塑料材料精密尺寸时,即尺寸稳定性能要好,可用热膨胀系数表示,热膨胀系数越小,尺寸稳定性越好。 膨胀系数x10-5k 塑料 10~35 XPE 16~20 EVA 10~22 LDPE 13~17 聚乙烯弹性体 10~20 PU 8~30 有机硅 11~13 HDPE 11.7 聚四甲基戊烯 6~13 ABS 8.3~10.5 四氟乙烯-全氟丙烯共聚物 10 聚四氟乙烯/尼龙12共混物 9 尼龙610 5.8~10.2 均聚PP 8~9 共聚PP 8.5 CPOM 8.3 PA-6 8.1 HPOM
8 PA-66 氯化聚醚 6~9.5 饱和聚酯(PBTP) 6~8 PS 7.2 UHMWPE 6.6 PC 5~8 丙烯酸酯 5.5~10 不饱和聚酯 5.5 聚苯硫醚 5.2~5.6 聚砜 5.2 改性聚苯醚 5.0 热塑性聚酰亚胺 3~6 环氧树脂 3~4.5 酚醛树脂 2~5 醇酸树脂 3.6~3.8 AS 2.2~ 3.6 脲醛 1~3.6 邻苯二甲酸丙酯 1.5 热固性聚酰亚胺 当采用某一塑料品种后,尺寸稳定性仍达不到要求时,可改换别的品种,或是在原塑料品种基础上,玻纤,碳纤,硼纤维,石英纤维,碳芯硼纤维,钨芯碳化纤维,二硼化钛纤维,不锈钢纤维,氧化铝晶
须等超强无机纤维,金属晶须,填料进行增强改性,从此降低膨胀系数。一般添加量为30%,增强改性是以纤维材料或其他材料作为增强材料的。 当采用玻纤时一定要进行表面活性化处理,表面处理以偶联剂和相容剂为主,偶联剂有硅烷类,钛酸酯类;相容剂为树脂对应的马来酸酐接枝聚合物等。其增强塑料一般称为玻璃钢,其力学性能大幅度提高,热性能也提高不少,尺寸稳定性变好。缺点是材料相对密度增加,制品表面平滑性,透明性,光泽性降低,对成型加工设备磨损增大。
5[1].6计量标准的重复性及稳定性考核记录 91、8022 91、8032 91、8242 91、8052 91、8062 91、8272 91、8082 91、8092 91、80102 91、822 91、806其标准偏差:= 2、33μm注:已建计量标准,至少每年进行一次重复性试验,测得的重复性应满足检定或校准结果的测量不确定度的要求。 计量标准的稳定性考核记录参考格式检定游标量具标准组的稳定性考核记录用此套标准装置对300mm游标卡尺1 21、80点测量,编号0061,在规定的间隔时间内,进行4次等精度测量,测量数据如下: 测量时间xx年6月xx年8月xx年10月xx年12月测量次数10次10次10次10次测量值(单位:mm)11
21、801 21、801 21、8021 21、801 21、801 21、801 21、8031 21、801 21、801 21、801 21、8041 21、801 21、801 21、801 21、8051 21、801 21、801 21、801 21、8061 21、801 21、801
21、8071 21、801 21、801 21、801 21、8081 21、801 21、801 21、801 21、8091 21、801 21、801 21、801 21、80101 21、801 21、801 21、801 21、80平均值1 21、801 21、801 21、801 21、80考核人员罗琦郑义罗琦郑义最大值与最小值之差:
0mm根据上述测量数据可知,测量结果的最大值与最小值之差为1 21、80-1 21、80=0mm,其值小于本计量标准的最大允许误差的绝对值0、02mm结论: 该计量标准的稳定性符合要求。注:若计量标准在使用中采用标称值或示值,则稳定性应小于计量标准的最大允许误差的绝对值;如加修正值使用,则应小于修正值的扩展不确定度。计量标准的重复性试验记录参考格式衡器检定装置的重复性试验记录选取一台100kg台秤在装置正常工作的条件下,50kg重量等精度重复测量10次,各次测量值如下:序号 Xi (kg)U=Xi-U21 50、0 30、0 30、00092 50、0 20、0 20、00043 50、00004 50、00005 50、00006 49、97-0、0 30、00097
需要稳定性考察报告怎么打 篇一:稳定性考察范本 一、目的: 为公司新产品以及合同加工产品确定有效期和贮存运输条件提供科学依据;对公司产品以及合同加工产品进行持续稳定性考察,以监控在有效期内药品的质量;由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 二、 适用范围:适用于公司新产品和合同加工产品的投产稳定性考察、公司产品和合同加工产品的持续稳定性考察、 由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 三、责任: 质量保证部、质量检验部。 四、内容: 1.产品稳定性考察的一般规定 1.1产品稳定性考察分类 1.1.1为公司新产品以及合同加工产品确定有效期与贮存运输条件提供科学数据所进行的稳定性考察; 1.1.2为监控公司产品以及合同加工产品在有效期内质量所进行的持续稳定性考察;
1.1.3由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 1.1.3.1重大变更或生产和包装有重大偏差的药品; 1.1.3.2任何采用非常规工艺重新加工、返工、或有回收操作的批次; 1.1.3.3改变主要物料供应商时所作验证的批次。 1.2产品稳定性考察样品批次的规定 1.2.1为公司新产品以及合同加工产品确定有效期与贮存运输条件提供科学数据所进行的稳定性考察,这种情况的稳定性考察需要连续试制的三批样品; 1.2.2公司新产品和合同加工产品正常批量生产的最初三批产品应列入持续稳定性考察计划,以进一步确认有效期。 1.2.3为监控公司产品以及合同加工产品在有效期内质量所进行的持续稳定性考察批次按产品不同规格每年考察一批, 除当年没有生产外。所考察批次采取随机抽取的方式; 1.2.4重大变更或生产和包装有重大偏差的药品批次; 1.2.5任何采用非常规工艺重新加工、返工、或有回收操作的批次; 1.2.6改变主要物料供应商时所作验证的批次。 1.2.7有效期短的原料药,在进行稳定性考察时应适当增加检验频次。
木材属多孔性材料,大的孔隙如细胞腔和纹孔等,小的如微纤丝间隙,而且木材中又含有许多亲水基团,使得水分很容易渗透到木材内部,且与木材中某些成分以化学键或氢键形式结合,引起纤丝的润胀,从而使木材尺寸发生变化,即木材的干绍湿胀性能。 更重要的是,木材为各向异性材料,各方向上随木材含水率变化而不均匀胀绍,易产生翘曲、变形、开裂等缺陷,极大地制约了木材的应用范围。因此,改善木材的尺寸稳定性对木材的高效利用具有重要的意义。 木材的胀缩归根到底是由于水分渗到微纤丝间,增加纤丝间的缝隙,进而影响木材尺寸的变化,而在一定条件下微纤丝间隙的增加是有限度的。如果在其充分润胀的条件下,向这些间隙内境人某种物质,限制其在水分降低时回复到原来状态,即可达到稳定尺寸的目的。但木材中只有吸着水才能引起木材尺寸的变化,这部分水和木材是以化学键和氢键形式结合的,如果将木材中的亲水基团(主要指一OH)除去或减少,由此降低木材的吸水率,也能增加木材尺寸的稳定性。 根据处理方法及效果,美国的Stamm A J把木材尺寸稳定性处理的方法大致分为五类:(1)用交叉层压的方法进行机械抑制,如胶合板生产中的单板按纹理交叉方向组坯就是利用这一原理。(2)防水涂料的内部或外部涂饰,主要有油漆涂刷、石蜡等有机防水剂的浸渍处理。(3)减少木材吸湿性,包括木材中极性物质的抽提或用树脂浸渍处理木材等。(4)对木材细胞组分进行化学交联,现绝大多数化学处理都是应用此原理。(5)用化学药品预先使细胞壁增容,包括树脂浸渍,向木材中浸入不溶性无机盐,将酸、醇等浸入木材后进行酯化反应等。 在实际应用中,经常是同时采用几种方法或是一种方法也能起到多种作用。随着科技的进步,木材的尺寸稳定处理还有更新颖方法,如对木材进行金属化或陶瓷化处理,不但增加了木材的尺寸稳定性,还能增加许多其它优良性能。 1 添加僧水剂向木材中添加一定量的防水剂(憎水剂),如石蜡、干性油、蜂蜡、硅油或亚麻仁油等。这种处理方法操作简单,价格便宜,防水率可达75%一90%,抗胀缩率(A3互)达70%一85%。主要用于刨花板或纤维板生产,将此防水剂以乳液的形式喷人碎料或纤维表面,加入量为1.0%一2.5%。处理后的板材表面活性有所下降,胶合性能和制品的力学性能赂有降低。 2 油漆处理对实木或板材进行油漆处理是使其防湿的行之有效的方法。用油漆将木材表面覆盖,可阻塞水分通往木材内部的通道。通常采用硝基漆、氨基醇酸树脂等合成树脂漆对木材进行多次反复涂刷,这样既达到防湿目的,同时也起到美化木制品的效果。但这种方法是非永久性的,板材表面一旦遭到破坏,防水效果就会下降。在油漆表面用石蜡或烯烃类树脂膜覆盖,可达到更好的效果。 3 树蹭漫演处理将低分子量的酚醛树脂(PF)、腺醛树脂(UF)、三聚氰胺树脂(MF)、聚乙酸酯(PC)等树脂浸入木材内部,加热使其在木材内部缩聚成不溶物,境塞于纤丝间隙、纹孔以及细胞腔内,使纤丝间隙充分胀大,同时阻碍水分进入木材,达到稳定尺寸的目的。处理后木材的A3互随树脂的留存量(尸L)的增加而增加。而且,固化后的树脂沉积于细胞壁内部,能对细胞起到加强作用,因此,经此法处理的木材力学强度会增加很多(可达50%),但韧性有所下降。 例如,以20%浓度的PF浸渍木材,当尸人为46%一49%时A3互为70%。以低分子量MF(摩尔比为1;2.5—3)浸渍木材,处理后A3互达47%,抗吸湿能力(A4五亿)可达36%,且材色无明显变化。用PC 树脂浸渍木材,处理后木材A3互达80%以上,平衡含水率很低(仅3.1%),而且此法处理木材内应力小,力学性能增加,材质结构均匀,表面光洁。采用真空加压的方法,用PVAc(5%)十MF(50%)浸渍木材,不
计量标准的重复性试验记录参考格式 检定游标量具标准组的重复性试验记录 选一把分度值0.02mm ,量程为(0-300)mm 的游标卡尺,在标准器组正常工作的条件下,等精度测量重复5次,各次测量值如下: 其标准偏差: S = =2.33μm 注:已建计量标准,至少每年进行一次重复性试验,测得的重复性应满足检定或校准结果的测量不确定度的要求。
计量标准的稳定性考核记录参考格式 检定游标量具标准组的稳定性考核记录用此套标准装置对300mm游标卡尺121.80点测量,编号0061,在规定的间隔时间内,进行4次等精度测量,测量数据如下: 根据上述测量数据可知,测量结果的最大值与最小值之差为121.80-121.80=0mm,其值小于本计量标准的最大允许误差的绝对值0.02mm 结论: 该计量标准的稳定性符合要求。 注:若计量标准在使用中采用标称值或示值,则稳定性应小于计量标准的最大允许误差的绝对值;如加修正值使用,则应小于修正值的扩展不确定度。
计量标准的重复性试验记录参考格式 衡器检定装置的重复性试验记录 选取一台100kg 台秤在装置正常工作的条件下,50kg 重量等精度重复测量10次,各次测量值如下: X =50kg n i 1 =∑U 2=0.0058 S=±1 ) (1 2 --∑=n X X n i i =± 9 0058.0=±0.025kg=±25g 该装置的最大重复性误差为25g 注:已建计量标准,至少每年进行一次重复性试验,测得的重复性应满足检定或校准结果的测量不确定度的要求。
计量标准的稳定性考核记录参考格式 衡器检定装置的稳定性考核记录 选取一台100kg台秤分度值为0.05kg,在装置正常工作的条件下称量50kg重量,在规定的间隔时间内,进行4次等精度测量,测量数据如下: 根据上述测量数据可知,测量结果的最大值与最小值之差为50.00-50.00=0kg,其值小于本计量标准的最大允许误差的绝对值。 结论: 该计量标准的稳定性符合要求。 注:若计量标准在使用中采用标称值或示值,则稳定性应小于计量标准的最大允许误差的绝对值;如加修正值使用,则应小于修正值的扩展不确定度。
一、目的: 为公司新产品以及合同加工产品确定有效期和贮存运输条件提供科学依据;对公司产品以及合同加工产品进行持续稳定性考察,以监控在有效期内药品的质量;由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 二、 适用范围:适用于公司新产品和合同加工产品的投产稳定性考察、公司产品和合同加工产品的持续稳定性考察、 由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 三、责任: 质量保证部、质量检验部。 四、内容: 1.产品稳定性考察的一般规定 1.1产品稳定性考察分类 1.1.1为公司新产品以及合同加工产品确定有效期与贮存运输条件提供科学数据所进行的稳定性考察;
1.1.2为监控公司产品以及合同加工产品在有效期内质量所进行的持续稳定性考察; 1.1.3由其他原因引起公司产品和合同加工产品需要进行的稳定性考察。 1.1.3.1重大变更或生产和包装有重大偏差的药品; 1.1.3.2任何采用非常规工艺重新加工、返工、或有回收操作的批次; 1.1.3.3改变主要物料供应商时所作验证的批次。 1.2产品稳定性考察样品批次的规定 1.2.1为公司新产品以及合同加工产品确定有效期与贮存运输条件提供科学数据所进行的稳定性考察,这种情况的稳定性考察需要连续试制的三批样品; 1.2.2公司新产品和合同加工产品正常批量生产的最初三批产品 应列入持续稳定性考察计划,以进一步确认有效期。 1.2.3为监控公司产品以及合同加工产品在有效期内质量所进行的持续稳定性考察批次按产品不同规格每年考察一批, 除当年没有生产外。所考察批次采取随机抽取的方式; 1.2.4重大变更或生产和包装有重大偏差的药品批次;
1.目的: 在有效期内监控已上市药品质量,以发现市售包装药品与生产相关的稳定性问题(如杂质含量变化),保证按照固定验证过的生产参数制造的产品质量维持在稳定的趋势,并且经此考察可以在有效期内监控药品质量,并确定药品能够在标示的贮存条件下,符合质量标准的各项要求。 2.范围: 公司的所有市售包装药品。 3.职责: 制订每年的持续性考察计划质量部 每个产品的稳定性试验方案质量部 按计划进行考察,并做好记录,总结考察报告报质量受权人。化验室 4.程序 4.1要求 考察批次数和检验频次应当能够获得足够的数据,以供趋势分析。通常情况下,每种规程、每种内包装形式的药品,至少每年应当考察一个批次,除非当所没有生产。某些情况下,持续稳定性考察中应当额外增加批次数,如重大变更、生产和包装有重大偏差的药品应当列入稳定性考察。此外,重新加工、返工或回收的批次,也应当考虑列入考察,除非已经过验证和稳定性考察。稳定性报告必须定期更新。 4.2稳定性考察计划 质量保证部QA人员于每年年初依据本年度的生产计划制订年度稳定性试验计划,确定本年度将进行的和可能要增加进入稳定性试验程序的品种,并于每年年终对本年度公司的稳定性试验工作做出年度总结报告。 对不符合质量标准的结果或重要的异常趋势进行调查。任何已确认的不符合质量标准的结果或重大不良趋势,都应向当地药品监督管理部门报告;还应考虑是否可能对已上市药品造成影响,必要时应实施召回。 4.3稳定性考察方案 应涵盖药品有效期,至少应包括以下内容: ①每种规程、每种生产批量药品的考察年批次: 考察原因 批次 新产品正常批量生产后前三批产品 3批 为监控公司产品以及合同加工产品在有效期内质量所进行的持续稳定性考察 按产品不同品种、不同规格、不同内包装形式每年考察一批,除当年没有生产外。所考察批次采取随机抽取的方式。 重大变更或生产和包装有重大偏差的药品批次
尺寸稳定性试验记录表 编号: 记录: 审核: 批准: 日期: 日期: 日期: 试验单位 xxx 有限公司 产品规格 xxx 试验 时间 xxxx 管头 螺纹 保护器 样件编号 试验 温度 检测点 序号 A 实际尺寸(mm ) A 平均值尺寸(mm ) B 表读数 B 实际尺 寸(mm ) B 平均 值尺寸 (mm ) C 表读数 C 实际 尺寸 (mm ) C 平均值尺寸(mm ) H 实际尺寸(mm ) H 平均值尺寸(mm ) 齿高(mm ) 平均齿 高(mm ) 小 大 小 大 管 头 xxx 21℃ ① ② ③ ④ 螺纹保护 器 xxx 21℃ ① ② ③ ④ xxx 66℃ ① ② ③ ④ xxx 21℃ ① ② ③ ④ 试验人员签字 Xxx xxx xxx xxx
尺寸稳定性试验记录表 编号: 记录: 审核: 批准: 日期: 日期: 日期: 试验单位 xxxxxx 有限公司 产品规格 xxxxx 试验 时间 xxxxxxxx 管头 螺纹 保护器 样件编号 试验温 度 检测点序号 A 实际尺寸(mm ) A 平均值尺寸(mm ) B 表读数 B 实际尺 寸(mm ) B 平均 值尺寸 (mm ) C 表读数 C 实际 尺寸 (mm ) C 平均值尺寸(mm ) H 实际尺寸(mm ) H 平均值尺寸(mm ) 齿高(mm ) 平均齿 高(mm ) 小 大 小 大 管 头 xxxxx 21℃ ① ② ③ ④ 螺纹保 护 器 xxxxx 21℃ ① ② ③ ④ xxxxx -46℃ ① ② ③ ④ xxxxx 21℃ ① ② ③ ④ 试验人员签字 Xxx xxx xxx xxx
安徽丰汇生物制药有限公司文件 化的规律,为产品生产、包装、贮存、运输条件和有效期的确定提供科学依据,以保障产 品质量。 范围:本标准适用于本公司在研新药的小试、中试,首次商业生产的新产品;经批准进行的有关工艺改进;重要原辅料变更;以及产品包装材料的变更等情况下对产品质量进行的稳定性 考察。 责任:本文件由化验室负责起草,化验室主任审核,生产负责人批准,化验室负责本规程的实施。 正文: 1稳定性考察原则 研发阶段:应进行全面的稳定性试验,以得到注册所需所有数据。此数据用于证明环境因素对产品特性的影响,以确定包装、储存条件、复验周期和有效期。 已上市阶段:产品上市后,应进行适当的持续稳定性考察,监测已上市产品的稳定性。 2稳定性考察分类 a.影响因素试验 b.加速稳定性试验 c.长期稳定性试验 d.持续稳定性试验:公司根据法规的要求,同时开展中间产品放置时间稳定性试验。批量放大及 上市后变更(如生产设备变更,原辅料变更,工艺调整等)稳定性试验、以及特殊目的稳定性试验,例如对偏差调查等的支持性试验。 3. 影响因素试验 影响因素试验目的是考察制剂处方的合理性与生产工艺及包装条件,供试品用1批进行,将供试品脱去外包装,置适宜的开口容器中进行高温试验、高湿度试验、强光照射试验。 3.1. 高温试验 供试品置密封洁净容器中,在60℃条件下放置10天,于第5天和第10天取样,按稳定性重点考察项目进行检测。如供试品发生显著变化,则在40℃下同法进行试验。如60℃无显著变化,则不必进行40℃试验。 3.2.高湿试验 供试品置恒温密闭容器中,于25℃,相对湿度为90%±5%条件下放置10天,在第5天和第10天
13.稳定性实验 在这里你会找到以下问题的答案: 为什么需要进行稳定性试验? 存在什么样的稳定性试验? 运输条件是如何确定的? 怎样的程序和特殊要求是需要特别加以考虑的(指南)? 文件的要求是什么? 如何降低成本,以及在哪些领域能降低成本? 数据应该如何评估(推测)? 如何处理稳定性试验的超标及超出趋势结果? 10.5定义 药品的稳定性是指原料药及其制剂保持其物理、化学、生物学和微生物学性质的能力。稳定型试验的目的是考察原料药、中间产品或制剂的性质在温度、湿度、光线等条件的影响下随时间变化的规律,为药品的生产、包装、贮存、科学依据,以保障临床用药的安全有效。。并且通过持续稳定性考察可以在有效期内监控药品质量,并确定药品可以或预期可以在标示的贮存条件下,符合质量标准的各项要求。 10.6应用范围 稳定性研究是药品质量控制研究的主要内容之一,与药品质量研究和质量标准的建立紧密相关。其具有阶段性特点,贯穿原料药(API),制剂产品及中间产物的药品研究与开发的全过程,一般始于药品的临床前研究,在药品临床研究期间和上市后还应继续进行稳定性研究。 10.7原则 研发阶段:应进行全面的稳定性实验,以得到注册所需所有数据。此数据用于证明环境因素对产品特性的影响,以确定包装、储存条件、复验周期(API而言)和有效期。 已上市阶段:产品上市后,应进行适当的持续稳定性考察,监控已上市药品的稳定性,以发现市售包装药品与生产相关的任何稳定性问题(如杂质含量或溶出度特性的变化);也用于考察产品上市后因变更对产品稳定性的影响。 10.8稳定性分类 按照中国药典2010版及法规要求,我国的稳定性研究可以分为以下几类: 影响因素实验 加速稳定性实验 长期稳定性实验 持续稳定性实验 各公司根据需求及法规规定,还可以进行中间产品放置时间稳定性实验,批量放大及上市后变更(如生产设备变更,原辅料变更,工艺调整等)稳定性实验以及特殊目的稳定性实验,例如对偏差调查等的支持性实验。 10.9要点 10.9.1基本要求(参考中国药典2010) 稳定性试验应遵循具体问题具体分析的基本原则,其设计应根据不同的研究目的,结合原料药的理化性剂型的特点和具体的处方及工艺条件进行。一般性要求如下: a. 影响因素实验用1批原料药或1批制剂进行。加速实验和长期稳定性试验用3批供试品进行。 b. 原料药供试品应是一定规模生产的。供试品量相当于制剂稳定性试验所要求的批量,原料药合成工艺路线、方法、步骤应与大生产一致。药物制剂供试品应是放大试验的产品,