
6种方法测定蛋白质含量 一、微量凯氏(kjeldahl)定氮法 样品与浓硫酸共热。含氮有机物即分解产生氨(消化),氨又与硫酸作用,变成硫酸氨。经强碱碱化使之分解放出氨,借蒸汽将氨蒸至酸液中,根据此酸液被中和的程度可计算得样品之氮含量。若以甘氨酸为例,其反应式如下: NH2CH2COOH+3H2SO4――2CO2+3SO2+4H2O+NH3(1) 2NH3+H2SO4――(NH4)2 SO4(2) (NH4)2 SO4+2NaOH――2H2O+Na2SO4+2NH3(3) 反应(1)、(2)在凯氏瓶内完成,反应(3)在凯氏蒸馏装置中进行。 为了加速消化,可以加入CuSO4作催化剂,K2SO4以提高溶液的沸点。收集氨可用硼酸溶液,滴定则用强酸。实验和计算方法这里从略。 计算所得结果为样品总氮量,如欲求得样品中蛋白含量,应将总氮量减去非蛋白 氮即得。如欲进一步求得样品中蛋白质的含量,即用样品中蛋白氮乘以6.25即得。 二、双缩脲法(biuret法) (一)实验原理 双缩脲(NH3CONHCONH3)是两个分子脲经180℃左右加热,放出一个分子氨后得到的产物。在强碱性溶液中,双缩脲与CuSO4形成紫色络合物,称为双缩脲反应。凡具有两个酰胺基或两个直接连接的肽键,或能过一个中间碳原子相连的肽键,这类化合物都有双缩脲反应。
紫色络合物颜色的深浅与蛋白质浓度成正比,而与蛋白质分子量及氨基酸成分无关,故可用来测定蛋白质含量。测定范围为1-10mg蛋白质。干扰这一测定的物质主要有:硫酸铵、tris缓冲液和某些氨基酸等。 此法的优点是较快速,不同的蛋白质产生颜色的深浅相近,以及干扰物质少。主要的缺点是灵敏度差。因此双缩脲法常用于需要快速,但并不需要十分精确的蛋白质测定。 (二)试剂与器材 1.试剂: (1)标准蛋白质溶液:用标准的结晶牛血清清蛋白(bsa)或标准酪蛋白,配制成10mg/ml的标准蛋白溶液,可用bsa浓度1mg/ml的a280为0.66来校正其纯度。如有需要,标准蛋白质还可预先用微量凯氏定氮法测定蛋白氮含量,计算出其纯度,再根据其纯度,称量配制成标准蛋白质溶液。牛血清清蛋白用H2O 或0.9%NaCl配制,酪蛋白用0.05NaOH配制。 (2)双缩脲试剂:称以1.50克硫酸铜(CuSO4?5H2O)和6.0克酒石酸钾钠(KNaC4H4O6?4H2O),用500毫升水溶解,在搅拌下加入300毫升10% NaOH溶液,用水稀释到1升,贮存于塑料瓶中(或内壁涂以石蜡的瓶中)。此试剂可长期保存。若贮存瓶中有黑色沉淀出现,则需要重新配制。 2.器材: 可见光分光光度计、大试管15支、旋涡混合器等。 (三)操作方法 1.标准曲线的测定:取12支试管分两组,分别加入0,0.2,0.4,0.6,0.8,1.0毫升的标准蛋白质溶液,用水补足到1毫升,然后加入4毫升双缩脲试剂。充分摇匀后,在室温(20~25℃)下放置30分
海藻酸钠的提取 实验目的: 1、了解海藻酸钠的基本化学性质 2、掌握从海藻中提取、分离有效成分的一般方法 实验原理: 海藻酸钠(Sodium Alginate ,简称ALG):白色或淡黄色粉末,几乎无臭无味。又称为褐藻酸钠,是从褐藻类的海带或马尾藻中提取的一种由1,4 -聚-β-D-甘露糖醛酸和α-L-古罗糖醛酸组成的线型多糖碳水聚合物,是海藻酸衍生物中的一种,所以有时也称褐藻酸钠、海带胶或海藻胶。ALG易溶于水,不溶于乙醇、乙醚、氯仿和酸。其稳定性以pH值在6—11之间较好,低于6时析出海藻酸,不溶于水;高于11时又要凝聚。黏度在pH值为7时最大,但随温度升高而显著下降。海藻酸钠不耐强酸、强碱及某些重金属离子,因为他们会使海藻酸凝成块状,但钠、钾除外。海藻酸钠水溶液遇酸会析出海藻酸凝胶,遇钙、铁、铅等二价以上的金属离子会立即凝固成这些金属的盐类,不溶于水而析出。 海藻酸钠结构式
试剂与仪器: 海带,15%NaCl溶液,3%Na2CO3溶液,10%CaCl2溶液,稀硫酸,95%乙醇,5%HCl溶液。烧杯若干,纱布,抽滤装置,水浴装置, 实验步骤: 采用钙凝—离子交换法提取海藻酸钠,其工艺流程如下:原料→清洗→干燥→粉碎→浸泡→消化→过滤→钙析→离子交换脱钙→过滤→干燥→粉碎→产品。 1、浸泡:称取10克切碎的海带,放入500mL烧杯中,再往烧杯中加入100mL水在常温下浸泡3个小时。浸泡结束后,用滤布过滤,用水洗涤至洗涤液为无色。 2、消化:放入250mL的烧杯中。然后往烧杯中加入3%的Na2CO3溶液50mL,在 50℃下消化4个小时。2M(ALG)n + nNa2CO3→2nALG+M2(CO3)n。式中, M 为Ca、Fe 等金属离子,ALG为海藻胶 3、过滤:消化后,海带变成了糊状,比较粘稠。要先加入一定体积的水将糊状液体稀释,再过滤。由于直接抽滤这种糊状的液体速度太慢,因此首先用纱布初滤一次,再将滤液用真空泵抽滤。 4、钙析:将滤液用5%盐酸调节至pH值为6—7,取50ml 滤液加入10ml 10 %的氯化钙溶液,使水溶性海藻酸钠转化为非水溶性的海藻酸钙析出: 2NaALG+ CaCl2→Ca(ALG)2 + 2NaCl 。该过程可以使海藻酸钠与大量的水分离,同时将大量的无机盐、色素等水溶性杂质随水排除。 5、离子交换脱钙:由于盐析作用,交换生成的海藻酸钠不溶于交换液中,仍然为凝胶状。所以采用15 % NaCl 溶液间歇多次脱钙,并在洗脱液中滴加适量的稀硫酸直到不生成CaSO4浑液为止。在此过程中,海藻酸钙凝胶中的Ca2 + 被Na+交
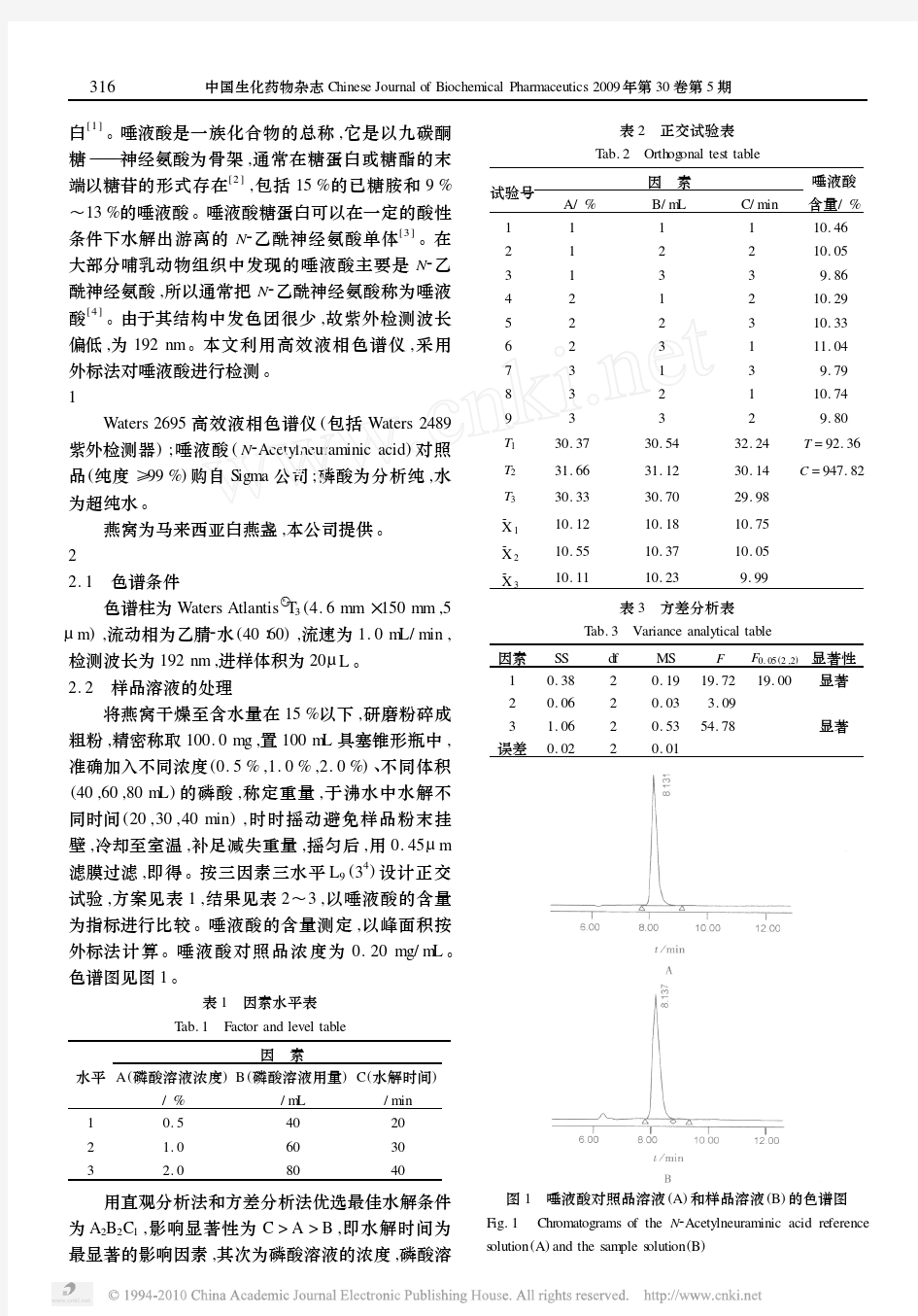
Application of Bioinformatics in Drug Molecular Design Zhao Shanrong Lin M aow ei Chen Kaixian S hanghai Institute o f M ater ia Medica,Chinese A cademy o f Sciences,Shanghai200031 Abstract With the r apid development o f bio technolo gy and Internet,bioinform atics has recently emerged.It is essential to diagnosis o f genetic diseases,pro tein structure predic-tio n,structure-based drug design,dr ug synthesis and pharmaceutical industry.The applica-tio n of bio informatics g reatly speeds up the prog ress of drug dev elo pm ent. Key Words Internet Bioinform atics Biolog ical database Drug molecular desig n (收稿:1996-12-16) 唾液酸及其衍生物的生物学研究进展 李绍顺 (第二军医大学药理教研室 上海200433) 摘 要 唾液酸广泛存在于多种生物组织中,是构成细胞表面的复合糖质的组成分。在各种生命活动调节中起着重要的作用。同时它们还是某些毒素的受体和某些病原微生物的作用部位。 近年来,以唾液酸为先导进行生物活性物质的探索成为新药研究的一个新领域。这些研究包括抗 癌转移、抗早老性痴呆、抗病毒、抗炎药物等。本文综述了该研究的一些新进展。 关键词 唾液酸 生理作用 生物学研究 药物研制 1 引言 唾液酸(Sialic acid)最早由Blix从唾液腺粘蛋白中提取分离得到,因而命名为唾液酸。而后,Klenk由神经节甙脂(Gangliosides)中分离出结晶性的多羟基氨酸,并命名为神经氨酸(Neuram inic acid)。日本的山川民夫将在红细胞膜的研究中分离得到的一种物质命名为Hemataminic acid. Kuhn等从牛的初乳中分离得到Lactaminic acid.Zilliken等从母乳中得到Gainaminic acid.山川等又从马的血清中分离得到Sero-lactaminic acid.经结构分析确认这些化合物实际与唾液酸为同一类物质。 1957年,Bliz等提出把这些氨基糖酸的基本结构(5-amino-3,5-dideox y-D-gly cero-D-g lacto-nonulo pyr anosonic acid)定名为神经氨酸,而把神经氨酸的一系列酰化物叫唾液酸。随着唾液酸衍生物的不断发现及合成研究进展,1984年小仓治夫等[1]提议将唾液酸的定义扩大,还包括神经氨酸的各种衍生物。 唾液酸广泛存在于各种生物的组织中,
炭黑含量测定方法研究 一空气环境中炭黑含量测定方法研究 1.1前言 在炭黑厂废气污染物中炭黑粉尘是主要的污染物之一,由于炭黑粉尘是疏松质轻且粒径极小的黑色粉末,在空气环境中令人讨厌,在黑炭厂周围的企业及居民反映强烈。然而在大气环境现状监测中,测定总悬浮微粒常常是不超标。即使在感觉比较严重的情况下,监测所得到的结果也只有1mg/NM3(每标准立方米)左右,浓度水平在国家大气环境质量二级标准中任何一次浓度值附近波动。这就给我们提出了以下的问题。 1.炭黑粉尘是否属于总悬浮微粒的范畴。 2.炭黑粉尘与空气中总悬浮微粒的比例关系。 对于第一个问题,水利电力部苏州热工研究所在苏州炭黑厂环境影响评价报告书中,已有结论性的意见。即炭黑中飘尘分量仅占2 0 ~ 3 0 %。炭黑粒径主要分布在总悬浮微粒区。为了准确地监测炭黑粉尘对大气环境的影响、了解炭黑粉尘与空气中总悬浮微粒的比例关系、我们以武汉炭黑厂为例、做了炭黑粉尘与空气中总悬浮微粒的比重实验与炭黑在总悬浮微粒中的含量实验。从而提出空气环境中炭黑含量的测定方法。 1.2实验部分 1.2.1实验方法选择 由于总悬浮微粒滤膜采样量少,且不容易处理。因此,考虑选用空气环境中自然降尘作为实验方法。降尘是指从空气中自然降落于地面的颗粒物,其粒径多在10um以上,而且炭黑的付聚体粒径在10-70um左右,符合总悬浮微粒的0.1-100um的粒径范畴。因此选用自然降尘作实验是可行的。 2.实验样品采集:(以武汉炭黑厂环境评价为实例) 根据常年主导风向的下风向和工厂周围的居民反映:在以炭黑厂为中心2平方公里的范围内布设采样点4个。同时在省环保所实验大楼布设实验点。按照《环境空气监测质量保证手册》中环境空气中自然沉降物的采样方法进行采集。时间周期为每月测定1次。在7,8,9,10月测定,共进行4次。 样品采集:采用集尘罐定点定时采样,每次采集前均向集尘罐内加入500ml蒸馏水,同时准确加入一定量的CuSO4溶液,防止集尘罐中微生物和藻类生长。 3.实验样品测定 (1)自然沉降量的测定:将瓷蒸发皿洗净,编号。在105℃烘箱中烘3小时,取出放在干燥器内,冷却50分钟后,在分析天平上称重,再在105℃烘50分钟,冷却5 0分钟,再称重,直至恒重(两次重量之差小于0.4 m g )。将已采集的样品,用镊子将落入缸内的异物取出,将缸内的溶液和尘粒全部转入1000ml 烧杯中,在电热板上小心蒸发,使体积浓缩至10ml左右时,转入瓷坩埚中,在电热水浴上小心蒸干后,于105℃烘箱中烘干至恒重。 计算公式:降尘量 t km2?月=W1?W a?W c S?n ×30×104 式中:W1为降尘和瓷钳锅重量(g); W a为瓷钳锅重量(g); W c为CuSO4经蒸发并烘干后的重量(g);S为集尘罐口面积(cm3); n为采样天数。 (2)自然沉降量中炭黑含量的测定:
含量测定方法学验证内容及可接受标准 1.准确度 可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。 2.线性 其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。 可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。 3.精密度 1)重复性 件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。 2)中间精密度 4.专属性 可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。 5.检测限
主峰与噪音峰信号的强度比应不得小于3。 6.定量限 主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。 7.耐用性 方法:分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、 可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。 8、系统适应性 应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。 有关物质测定方法学验证内容及可接受标准: 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至
中广核拓普(湖北)新材料有限公司李义 炭黑是一种非常优良的紫外光屏蔽剂,可以有效的抵挡紫外线对光缆护套 的照射,显著提高护套抗紫外线老化性能,是聚乙烯护套料中最常用的添 加剂。 由于炭黑在护套料中的重要作用,对于其在护套料含量要求国内外标准对 其都有明确的规定:ASTM 标准要求炭黑含量为 2.0%~3.0%,而国标 GB/T15065-2009 将炭黑含量的要求缩小为 2.35~2.85%,并规定了炭黑含量 测试的两种方法,直接燃烧法和热重分析法(TGA),但在实际操作中,这 两种方法的测试结果存在较大差异,一直饱受争议,本次我们以试验为例,对此差异大小和原因进行分析和探讨。 按 GB/T15065-2009 规定的炭黑测试条款 GB/T 2951.41-2008 标准要求,对国 标中的两种炭黑测试方法:直接燃烧法和热重分析法。选取两个不同样品 进行测试对比。 试验设备: 热重分析仪: TGA 55,美国 TA 公司;炭黑含量测试仪: HS-TH-3500 型,上海和晟仪器科技有限公司;电子分析天平:精度 0.1mg ,赛多利斯科学仪器(北京)有限公司。 测试条件: 热重分析法:称取 5~10mg 样品,氮气纯度为 99. 99 % 、流量为 50ml/min 的氮气条件下,从室温以 20℃/min 的升温速率加热至 850℃后,切换成相 同流量的氧气,继续升温至 950℃,利用设备软件计算炭黑含量。 直接燃烧法:样品重量 A(1.0±0.1)g ,氮气纯度为 99. 5 % 以上、流量为200ml/min ,设定第一个 10min 从室温加热至 350℃,第二个 10min 从 350℃
粗多糖含量测定方法学研究资料 一、仪器与试药 (1) 二、方法的研究 (2) 1.检测波长的测定 (2) 2.样品及对照制备方法 (2) 三、方法学验证 (3) 1.线性 (3) 2.精密度实验 (4) 3.稳定性实验 (4) 4.重复性试验 (5) 5.中间精密度实验 (5) 6.准确度试验 (6)
芪参颗粒粗多糖含量测定方法起草说明 标志性成分粗多糖含量测定的方法来源于《保健食品功效成分检测方法》白鸿主编(中国中医药出版社)的第二法,该方法的原理是:多糖经乙醇沉淀分离后,去除其他可溶性糖及杂质的干扰,糖与硫酸在沸水浴中加热脱水生成羟甲基呋喃甲醛(羟甲基呋喃糠醛),再与蒽酮缩合成蓝绿色化合物,其显色强度与溶液中糖的浓度成正比,在625nm波长下比色测定。 主要研究资料如下: 一、仪器与试药 1、仪器 (1) 离心机(湘南湘仪实验室仪器开发有限公司,型号TD25-WS); (2) 离心管:50ml; (3) 水浴锅(上海精宏实验设备有限公司,型号 DK-S26); (4) 旋涡混合器(DioCote,SA8); (5) SHIMADZU UV-1800 紫外可见光分光光度计; (6) JB760-68 石英比色皿(宜兴市伟鑫仪器有限公司); (7) TU-1901 双光束紫外可见光分光光度计(北京普析通用仪器有限责任公司); 2、试药 (1) 葡萄糖:广州化学试剂厂,分析纯,批号为-1; (2) 无水乙醇:西陇化学股份有限公司,分析纯,批号为160802 1; (3) 蒽酮:国药集团化学试剂有限公司,分析纯,批号为; (4) 硫酸:广州化学试剂厂,分析纯,批号为-1; (5) 葡萄糖标准液:标准称取干燥恒重的分析纯级葡萄糖,加水溶解,并定容至50ml,此溶液1ml含10mg葡萄糖,用前稀释100倍为使用液(ml)。 (6) %蒽酮硫酸溶液(W/V):准确称取蒽酮置于烧杯中,缓缓加入100ml 80%硫酸溶解,溶解后呈黄色透明溶液。现用现配。 3、试样
附录唾液酸测定法 (间苯二酚显色法) 本法系用酸水解方法将结合状态的唾液酸变成游离状态,游离状态的唾液酸与间苯二酚反应生成有色化合物,再用有机酸萃取后,测定唾液酸含量。 唾液酸对照品溶液(200μg/ml)的制备精密称取唾液酸对照品10.52mg(1μg唾液酸相当于3.24nmol),置10ml量瓶中,加水溶解并稀释至刻度,混匀,即为唾液酸贮备液(1mg/ml),按一次使用量分装,-70℃贮存,有效期1年。仅可冻融1次。4℃保存使用期为2周。精密量取唾液酸贮备液1ml,置5ml量瓶中,加水至刻度,即为每1ml含200 μg的唾液酸对照品溶液,用前配制。 测定法取供试品适量,加水稀释至蛋白质浓度约为每1ml含0.2~0.4mg,作为供试品溶液。按下表取唾液酸对照品溶液、水及供试品溶液于10ml玻璃试管中,混匀,每管再加入间苯二酚-盐酸溶液(分别量取2% 间苯二酚溶液2.5ml、0.1mol/L硫酸铜溶液62.5μl、25% 盐酸溶液20ml,加水稀释至25ml,混匀。试验前4小时内配制)1ml,加盖,沸水煮沸30分钟(水浴面高于液面约2cm),取出置冰浴中3分钟(同时振摇)后,每管加乙酸丁酯-丁醇液(取乙酸丁酯4份与丁醇1份混匀,室温下保存,12小时内使用)2ml,充分混匀, 用唾液酸对照品溶液的浓度对其相应的吸光度作直线回归(相关系数应不低于0.99),由直线回归方程求出5μg唾液酸的吸光度,再按下式计算 供试品唾液酸含量 (mol/mol 蛋白质) =A2×5×3.24×W×D A1×P×100 式中A1为5μg唾液酸的吸光度; A2为供试品的吸光度; D为供试品稀释倍数; P为供试品蛋白质含量,μg/μl; W为1nmol促红素的量,相当于 1
定量分析方法的方法学验证 定量分析方法的方法学验证 定量分析方法验证的目的是证明采用的含量测定方法适合于相应分析要求,在进行定量分析方法学研究或起草药品质量标准时,分析方法需经验证。 验证内容有:线性、范围、准确度、精密度(包括重复性和重现性)、检测限、定量限和耐用性等。 一,线性 线性是指在设计的范围内,测试结果与试样中被测物质浓度直接呈正比关系的程度。 应在规定的范围内测定线性关系。可用一贮备液经精密稀释,制备一系列供试品的方法进行测定,至少制备五份供试样品;以测得的响应信号对被测物浓度作图,观察是否呈线性,再用最小二乘法进行线性回归。必要时,响应信号可经数学转换,再进行线性回归计算。回归方程的相关系数( r ) 越接近于1 ,表明线性关系越好。 用UV 法测定时,以对照品配制一定浓度范围的对照品系列溶液,吸光度A一般在0.3 ~0.7 ,浓度点n =5 ,用浓度C 对A作线性回归,得一直线方程,方程的截距应接近于零,相关系数r 应大于0.9999 。 用HPLC 法测定时,以对照品配制一定浓度范围的对照品系列溶液,浓度点n =5 ~7 ,用浓度 C 对峰高h 或峰面积A或被测物与内标物的响应值之比进行线性回归或非线性拟合(如HPLC-ELSD ),建立方程,方程的截距应趋于零,相关系数r 应大于0.999 。 线性关系的数据包括相关系数、回归方程和线性图。 二,范围 范围系指能达到一定精密度、准确度和线性,测试方法适用的高低限浓度或量的区间。 范围应根据分析方法的具体应用和线性、准确度、精密度结果及要求确定。对于有毒的、具特殊功效或药理作用的成分,其范围应大于被限定含量的区间。 三,精确度 准确度系指用该方法测定的结果与真实值或参考值接近的程度,一般用回收率( %) 表示。准确度应在规定的范围内测试。用于定量测定的分析方法均需做准确度验证。 1. 测定方法的准确度 可用已知纯度的对照品做加样回收率测定,即于已知被测成分含量的供试品中再精密加入一定量的已知纯度的被测成分对照品,依法测定。用实测值与供试品中含有量之差,除以加入对照品量计算回收率。 在加样回率收试验中须注意对照品的加入量与供试品中被测成分含有量之和必须在标准曲线线性范围之内;加入的对照品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。 回收率% = [(C-A)/B]*100% 式中,A为供试品所含被测成分量;B 为加入对照品量;C 为实测值。 2. 数据要求 在规定范围内,取同一浓度的供试品,用 6 个测定结果进行评价;或设计 3 个不同浓度,每个浓度各分别制备 3 份供试品溶液进行测定,用9 个测定结果进行评价,一般中间浓度加入量与所取供试品含量之比控制在l ∶ 1 左右,其他两个浓度分别约为供试品含量的80% 和120% 。应报告供试品取样量、供试品中含有量、对照品加入量、测定结果和回收率( %) 计算值,以及回收率( %) 的相对标准偏差(RSD) 或可信限。 四,精密度 精密度是指在规定的测试条件下,同一个均匀供试品,经多次取样测定所得结果之间接近的程度。 1. 精密度的表示方法 气相色谱法和高效液相色谱法是对同一供试液进行至少五次以上的测定;精密度一般用相对标准偏差(relative standard deviation, RSD) 表示:RSD= 标准偏差/ 平均值′ 100 %
海藻酸钠固定酵母细胞实验资料 1实验原理 海藻酸钠是应用最广泛的水溶性海藻酸盐。海藻酸钠遇到钙离子可迅速发生离子交换,生成凝胶。利用这种性质,将海藻酸钠溶液滴入含有钙离子的水溶液中可产生海藻酸钙胶球。本实验便是将酵母细胞与海藻酸钠溶液混匀后,通过注射器或相似的滴注器将上述混合液滴入CaCl2溶液中,Ca2+从外部扩散进入海藻酸钠与细胞混合液珠内,使海藻酸钠转变为不溶于水的海藻酸钙凝胶,由此将酵母细胞包埋在其中。 2实验操作过程中的几个注意点 2.1控制好配制海藻酸钠溶液的火候、浓度,实验成败的关键步骤配置海藻酸钠溶液。加热使海藻酸钠溶化是操作中重要的一环,涉及到实验的成败。教材提示是要用小火或者间断加热,反复几次,直到完全熔化。在实验操作中,教师要提醒学生按照教材的提示进行,不然会发生焦糊现象。笔者多次实际操作发现酒精灯上隔石棉网加热,匀速搅拌基本不会发生焦糊现象,而且需要时间略长,为节省时间可选用电炉加热,加快熔化,但一定要间断加热,可以用试管夹移动烧杯。完全熔化的海藻酸钠成半透明状,最好要无颗粒无气泡,类似于平常家庭中冲调好食用的藕粉。海藻酸钠的浓度涉及到固定化细胞的质量。如果海藻酸钠浓度过高,将很难形成凝胶珠;如果浓度过低,形成的凝胶珠所包埋的酵母细胞的数目少,影响后续实验效果。教材提示是0.7 g海藻酸钠,加入10 mL蒸馏水,边加热边搅拌,直至完全熔化,用蒸馏水定容至10 mL。实际操作中,很多学生忘记定容,也有教师认为不需要定容。其实在加热搅拌的过程中,蒸馏水会蒸发,加热时间越长失去的蒸馏水越多,所以加热熔化结束后一定要定容,不然浓度肯定偏大,影响后续步骤中凝胶珠的形状。 2.2控制海藻酸钠溶液与酵母细胞混合时的温度,保证酵母细胞的活性海藻酸钠溶液与酵母细胞混合,一定要将溶化好的海藻酸钠溶液冷却至40℃以下,通常让学生自己用手感觉烧杯壁,感觉不到烫手就差不多。再加入已活化的酵母细胞,进行充分搅拌,使其混合均匀。如果不等海藻酸钠溶液冷却就混合酵母细胞,大多数酵母细胞会被烫死,这样制作的凝胶珠用来发酵,葡萄糖发酵液几乎无酒味。 2.3通过调节海藻酸钠与酵母细胞混合液浓度,从而不出现蝌蚪状凝胶珠海藻酸钠与酵母细胞混合好之后,转移至注射器中,以恒定的速度缓慢将注射器中的溶液滴加到配置好的CaCl2溶液中,形成凝胶珠。此步骤在实际操作中要做得好是有困难的,从注射器中流出的混合液不是一滴一滴的,在CaCl2溶液中就会形成蝌蚪状凝胶,甚至形成面条状。究其原因是混合液浓度过大导致,符合要求的混合液浓度不能大不能小,成流体状态,在注射器里如果要用力挤才能流出,混合液浓度肯定偏大。按照教材中的数据学生最后得到的混合液浓度都略偏大,可在混合液中滴加少量蒸馏水稀释。加多少蒸馏水才能使浓度适宜?除了观察混合液的质态,笔者觉得可以做预实验,这样指导学生的效果很好。具体是不把海藻酸钠与酵母细胞混合液全部转移到注射器中,而是用玻璃棒挑或倒少量(约0.5 mL)送到注射器的前端,滴加到CaCl2溶液中,观察形成的凝胶珠形状,如果有长尾巴或成面条状就是浓度太大,这时可在混合液中加蒸馏水搅拌均匀,然后再挑少量送到注射器的前端,滴加到CaCl2 溶液中,会发现尾巴明显变短,如仍有小尾巴或成长椭圆形,继续在混合液中加蒸馏水,直至形成的凝胶珠成圆形,再将所有混合液转移至注射器中。这一过程要注意蒸馏水千万不能加过,每次滴加量以5滴为好。另外注射器不要垂直向下,要有一定的角度,注射器距离CaCl2溶液也要保持一定的距离,30 cm左右,有利于凝胶珠成圆形。 2.4做到溶液无气泡,从而不出现漂浮在CaCl2溶液上的凝胶珠在学生制作的凝胶珠中也经常出现有漂浮在CaCl2溶液上的,原因是这些凝胶珠中含有大气泡,重量轻。这样的凝胶
唾液酸测定试剂盒(比色法) 适用范围:用于体外定量测定人血清中唾液酸的浓度。 1.1 规格 试剂盒是由试剂1和试剂2组成的液体双试剂,校准品为液体剂型,质控品为冻干粉。规格及装量见表1。 表1 规格及装 量 1.2主要组成成分
试剂1主要组分: 试剂2主要组分: 校准品主要组分: 质控品主要组分: 2.1 净含量 应不低于试剂瓶标示装量。 2.2 外观 试剂1:无色或淡黄色透明溶液;试剂2:无色或黄色透明溶液,校准品:为无色透明液体,质控品:为浅黄色至黄色冻干粉,复溶后为浅黄色至黄色液体。外包装完好、无破损,标签完好、字迹清晰。 2.3 试剂空白 2.3.1 试剂空白吸光度 在340nm处测定试剂空白吸光度,应≥0.05; 2.3.2 试剂空白吸光度变化率
试剂空白吸光度变化率△A/min≤0.8。 2.4 分析灵敏度 测试50 mg/dL的被测物时,吸光度变化率(ΔA/min)应不低于0.0005。 2.5 准确度 参照EP9-A2的方法,用比对试剂盒同时测试40例线性区间内的不同浓度的血清样本,其相关系数r≥0.975。[10,60)mg/dL区间内绝对偏差不超过±6mg/dL;[60,180]mg/dL区间内相对偏差不超过±10%。 2.6 重复性 批内变异系数(CV)应不超过10%。 2.7 线性 2.7.1在[10,180]mg/dL区间内,线性相关系数r应不低于0.990; 2.7.2[10,60)mg/dL区间内绝对偏差不超过±6mg/dL;[60,180]mg/dL区间内相对偏差不超过±10%。 2.8 批间差 对同一份样品进行重复测定,相对极差不大于10%。 2.9质控品批内瓶间差 变异系数(CV)应≤5%。 2.10溯源性 根据GB/T 21415-2008的规定,本试剂盒内校准品溯源至企业工作校准品,与已上市公司试剂盒进行比对赋值。 2.11质控品赋值有效性 质控品测值应在靶值范围内。
Q/NBHP 含海藻酸水溶性肥料 宁波海浦生物科技有限公司发布
前言 因海藻提取物叶面肥类产品现无国家标准、行业标准和地方标准,特制定本企业标准。本标准是用来代替2004年6月1日发布的原公司企业标准Q/NBHP03-2004,也为申请海藻提取物叶面肥的正式登记证而作。与原标准Q/NBHP03-2004相比,有以下几处不同:1、名称不同,由有机液肥(原标准)修改为含海藻酸水溶性肥料;2、因原料及生产方式的改变,取消了氨基酸的含量指标,海藻酸的含量由≥1.0%改为≥2.0%;微量元素由≥2.0%改为≥1.0%;PH值由4.0~8.0改为≥4.5;增加了氮磷钾的指标含量。因现无上级标准,特制定本企业标准,以作为产品大批量定型生产的依据。如发布国家标准或行业标准,本标准将自动废止或视情况修订后重新发布实施。 本标准的附录A为规范性附录。 本标准主要起草单位:宁波海浦生物科技有限公司。 本标准主要起草人:徐永安,石立峰,陈巧媛。 本标准于2004年6月1日首次发布,于2007年1月8日修订,2007年2月18日实施。
含海藻酸水溶性肥料 1范围 本标准规定了含海藻酸水溶性肥料的定义、技术要求、试验方法、检验规则、标识、包装、使用说明书及包装、运输和贮存。 本标准适用于含有海藻酸、腐植酸、大量元素、以及微量元素的水溶性肥料(以下简称水溶性肥料)。 2规范性引用文件 下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。 GB/T 601 化学试剂标准滴定溶液的制备 GB/T 602 化学试剂杂质测定用标准溶液的制备 GB/T 603 化学试剂试验方法中所用制剂及制品的制备 GB/T 1250-1989 极限数值的表示方法和判定方法 GB/T 6274 肥料和土壤调理剂术语 GB/T 6680-1986 液体化工产品采样通则 GB/T 6682 分析实验室用水规格和试验方法 GB 8172 城镇垃圾农用控制标准 GB/T 9724 化学试剂PH值测定通则 GB/T 9738 化学试剂水不溶物测定通用方法 GB/T 14540-2003 复混肥料中铜、铁、锰、锌、硼、钼含量的测定 GB/T 17767.1 有机-无机复混肥料中总氮含量的测定 GB/T 17767.2 有机-无机复混肥料中总磷含量的测定 GB/T 17767.3 有机-无机复混肥料中总钾含量的测定 国家质量监督检验检疫总局令[2005]第75号《定量包装商品计量监督管理办法》 3术语和定义 本标准采用下列定义。 3.1含海藻酸水溶性肥料 含海藻酸水溶性肥料是用海藻作主要原料加工制成的一种黑褐色无臭液体叶面肥料,主要成分是海藻中的生物活性物质,如:海藻酸等,以及腐植酸和微量元素、植物必需的矿质元素。 4要求 4.1 外观:黄褐色液体,轻微的海藻味。 4.2叶面肥的理化指标应符合表1的规定
DZ3500型碳黑含量测试仪 操作规程 1、将燃烧舟加热到灼热,然后在干燥器中冷却至少30min,称重 精确到0.0001g,将(1.0±0.1)g重的被测试样放到燃烧舟中,再一起称重,精确到0.0001g,减去燃烧舟的重量即得到试样的 重量(重量A),精确到0.0001g。 2、先用钩子勾住装有试样的燃烧舟,再把燃烧舟放到加热炉石英 管的中部(目测)。 然后将一个带温度传感器和一根进气玻璃管的橡胶塞插在石英 管的一端,使温度传感器的端头位于加热炉的中间位置。含氮 量小于0.5%的氮气按要求的流速通过石英管,并在以后的加热 过程中保持这个流速。在加热以前预先通氮气以排空石英管中 的空气为准,200ml/min的氮气流速大概需要5min将空气排净。 燃烧管的另一端插带一根排气玻璃管的橡皮塞。 3、根据待测样所适用的国家标准对参数值进行设置(详见五参数 设置),然后按“运行”键进行实验。以GB/T2951.8-1997为例,参数值的设置为: 设置---step1:max 为350℃、up为33℃/min、constant为00min (设常温为:20℃,350-20=330)---step2:max为450℃、 up 为10℃/min、constant为00min--- step3:max为500℃、 up 为5℃/min、constant为40min--- step4:max为550℃、 up
为1-15℃/min、constant为40min---返回 Step1/step2/step3均通氮气,step4通氧气。氮气是保护气,防止碳黑在裂解过程中氧化或燃烧。一般情况下氮气和氧气的 流量控制在100ml/min,如果油脂类物质多可适当调节。 GB/T13021-1991的参数设置与GB/T2951.8-1997的设置相似。 4、Step3后从排气口取出燃烧舟,在干燥器中冷却20—30min并 重新称重,测定残留物的重量(残留物重量B)精确到0.0001g。 5、再将燃烧舟放入石英管,在step4下将氧气取代氮气以适当的 流速通到石英管内,使残留碳黑燃烧完全。取出燃烧舟冷却并称重,测定残留物的重量(残留物重量C)精确到0.0001g。6、试验结果表示方法 碳黑含量=(B-C)/A×100% 7、按操作规程关闭试验仪器。 编制:审核:批准:
唾液酸测定试剂盒(NANA-醛缩酶法) 适用范围:适用于体外测定人血清中唾液酸的含量。 1.1 产品规格 1.2主要组成成分 注:校准品、质控品具有批间、赋值特异性,具体值详见靶值单。 2.1外观 2.1.1试剂盒标签标识清晰,外包装完整无破损; 2.1.2试剂1:无色或淡黄色液体,目测不得有任何沉淀及絮状悬浮物; 2.1.3试剂2:无色或淡黄色液体,目测不得有任何沉淀及絮状悬浮物;
2.1.4校准品:无色或浅黄色澄清液体,目测不得有任何沉淀及絮状悬浮物;2.1.5质控品:无色或浅黄色澄清液体,目测不得有任何沉淀及絮状悬浮物。2.2净含量 净含量不低于标示值。 2.3试剂空白 2.3.1空白吸光度 在主波长340nm、副波长405nm、37℃条件下, A≥0.5。 2.3.2空白吸光度变化率 在主波长340nm、副波长405nm、37℃条件下,△A/min≤0.01。 2.4线性范围 (2,200)mg/dL范围内,相关系数r≥0.990; (2,40]mg/dL范围内,绝对偏差不超过±4mg/dL; (40,200)mg/dL范围内,相对偏差不超过±10.0%。 2.5分析灵敏度 在产品说明书规定参数设定条件下,测定60.0mg/dl的样本, 吸光度变化率△A/min≥0.010。 2.6 精密度 2.6.1批内重复性 CV≤10.0%。 2.6.2 批间差 相对极差R≤10.0%。 2.7 准确度 与已上市产品比对:(2,200)mg/dL范围内,相关系数r≥0.990;(2,40]mg/dL范围内,绝对偏差不超过±4mg/dL;(40,200)mg/dL范围内,相对偏差不超过±10.0%。 2.8 校准品 2.8.1 均一性CV≤5.0%。 2.8.2 开瓶稳定性:开瓶后3天,相对偏差不超过±10.0%。 2.9 质控品
早在1957年,Blix就从颌下腺粘蛋白中分离出唾液酸(Sialic acid),并建立了唾液酸的命名规则[1] ,但在很长时期内人们对其生理功能了解甚少,其化学结构在1960年才被确切地测定出来。随着现代物理学、化学和生命科学技术的飞速发展,唾液酸的生理活性已逐渐被认识,并可能在不久的将来得到应用。本文就近年来唾液酸的生物活性及其应用方面的研究进展进行了较全面的综述。 1 唾液酸的来源 唾液酸(Sialic acid,SA)是九碳糖神经氨酸酰化物的总称。唾液酸在自然界分布很广,已经发现许多生物体内存在唾液酸,它通常位于细胞膜最外层的糖类部分和分泌的糖复合物(糖脂、糖蛋白和脂多糖)的关键位置,是糖复合物结构和功能多样化的重要物质基础[2,3]。随着生物进化程度的升高,唾液酸在生物体内的含量不断增加,进化程度较低的生物,如原生动物、环节动物、节肢动物等均极少有唾液酸的存在。在脊椎动物和哺乳动物体内普遍存在唾液酸,人体中脑的唾液酸含量最高,人脑灰质中的唾液酸含量是肝、肺等内脏器官的15倍[4]。唾液酸的主要食物来源是母乳,在牛奶、鸡蛋和奶酪中也存在唾液酸。至今自然界中被报道的唾液酸有30多种[5],最常见的是N-乙酰基神经氨酸(N-acetylneuraminic acid , NANA),其结构如附图所示。 2 唾液酸的测定方法 唾液酸的物理分析方法很多,有分光光度法、薄层层析法[6]、高效液相色谱法[7]、核磁共振法[8]和高效液 相色谱质谱联用技术等[9]。这些技术可用于生物样品的分离和测定,还能区分不同类型的唾液酸,为唾液酸在生物学方面的研究提供了更好的手段。2.1 分光光度法 唾液酸与一些试剂反应能产生颜色,可用于直接定性检测和定量测定。这些试剂包括硫代巴比妥酸、R-试剂和间苯二酚-过碘酸/盐酸等。游离唾液酸被过碘酸氧化生成β-甲酰丙酮酸,后者与2-硫代巴比妥酸反应生成有色物质,在549nm处有最大吸收峰[10]。由于过碘酸氧化以酮苷形式结合的唾液酸生成结合的醛,后者可与间苯二酚生成很强的颜色,其最大吸收峰在630nm。此外,唾液酸的C 8上如有三碳的侧链取代基时则过碘酸不能和该化合物发生反应[11]。但这些方法的特异性仍不太强,定量时只能测定总唾液酸含量。2.2 薄层分析法 展开剂∶正丙醇∶饱和氨水∶水=6∶1∶2.5;显色剂:间苯二酚-盐酸试剂(与唾液酸分析试剂相同)。将点好样品的层析板置于盛有展开剂的层析缸,密闭式展开,溶剂前沿距层析板上端1cm时取出,在室温下挥发干,用喷雾器喷洒显色剂,再在110℃烘箱显色,待层析板呈现蓝紫色斑点取出。 唾液酸生物活性及其应用的研究进展 魏冬旭1,2,江连洲1,王 辰1,王中江1 (1东北农业大学食品学院,哈尔滨 150030;2 黑龙江出入境检验检疫局,哈尔滨 150001) 基金项目:国家自然科学基金项目“大豆球蛋白亚基组分和空间构象与表面疏水性构效关系研究”(项目编号:C200101)。作者简介:魏冬旭(1983— ),女,河北昌黎县人,助理工程师,在读博士,研究方向为植物蛋白工程。通信作者:江连洲 摘 要:对唾液酸的来源、检测手段、生物活性及其在食品医药领域应用等方面的研究进行了综述,并对其未来的发展方向进行了展望。 关键词:唾液酸;生物活性;测定方法;应用进展附图 唾液酸(SA)的结构 OH OH OH OH OH O AcHN COOH 中国食物与营养 2011,17(7):64-68Food and Nutrition in China
聚乙烯中炭黑含量不同测试方法的探讨 摘要采用GB13021《聚乙烯管材和管体炭黑含量测定(热失重法)》和热重分析仪两种方法测定聚乙烯中炭黑含量。对两种方法的测定结果进行了比较,结果表面,两种方法均有良好的重复性和准确度,测定结果基本一致,采用不同方法得到的测定结果间可以互相参考 关键词GB13021,热重分析依法,炭黑含量 Carbon black content in polyethylene was determined by two methods of GB13021,polyethylene pipe and tube carbon black content determination (thermal gravimetric method)and thermo gravimetric https://www.doczj.com/doc/9d10876724.html,pared with the measurement results of the two methods of the surface,the two methods have good repeatability and accuracy.The measurement results are basically the same,the determination results obtained by different methods can reference each other Key words GB13021,thermal gravimetric analysis,carbon black content 近年来,聚乙烯管材已成为继PVC之后,世界消费量第二大的塑料管道品种,广泛应用于给水、农业灌溉、燃气输送、排污、油田、化工、通讯等领域。无添加剂的聚乙烯耐气候老化和日光曝晒性能很差,因而实际使用时都会添加炭黑[1]。炭黑能使材料具有足够的抗紫外老化能力,当炭黑含量为2.0%~3.0%时可确保有效地防止紫外线的影响[2]。由于炭黑含量大小对聚乙烯管材具有重要的影响,许多标准都对聚乙烯中的炭黑含量作了规定,为了研发生产和销售的目的,炭黑含量是聚乙烯管材必须进行检测的指标。目前管道用塑料中炭黑含量的测试方法主要执行GB13021–1991[3]。使用热重分析仪是现在常用的热分析手段,用来测量高聚物的成分极为方便,常用标准是ASTME1131–2008[4],热重分析仪也可以用于测定聚乙烯中的炭黑含量。目前这两种方法并存,不同实验室间经常采用不同的方法测试,存在炭黑含量分析结果无法直接比较的问题。笔者用以上两种方法测定同批聚乙烯粒料中的炭黑含量,对不同测试方法的优缺点、测量重复性以及两种方法测试结果的一致性进行了探讨,对炭黑含量测试方法的选择提供了参考。1实验部分
甲钴胺片的含量测定检测方法研究 徐丽平徐慧娟杭州康恩贝制药有限公司 摘要目的:建立甲钴胺片含量测定方法,为甲钴胺片质量标准相关检测项目提供检测方法和实验依据。方法:样品碾细,以流动相溶解,制备供试品溶液;色谱分析条件:色谱柱为Diamonsil 5u C18,250×4.6mm;流动相:乙腈-0.03mol/L 磷酸二氢钠(磷酸调pH为3.5)(19:81);流速1.0ml/min;检测波长:342nm。结果:甲钴胺线性关系良好,平均回收率100.7%。结论:本法测定结果准确,可用于甲钴胺的含量测定。 关键词:甲钴胺,高效液相色谱法 甲钴胺片活性成分甲钴胺,主要用于治疗周围神经病。由于甲钴胺对光敏感,故需包衣,包衣材料成分复杂,干扰大,现有的紫外分光光度法重现性差。本实验采用HPLC法测定甲钴胺的含量,结果准确,复现性好。 1 仪器及试药 高效液相色谱仪:安捷伦1100,VWD检测器,??工作站;电子天平:?2100;超声波清洗器。 甲钴胺对照品,浙江省药品检验所标定,批号:?,含量:?;乙腈为色谱纯,磷酸二氢钠为分析纯;甲钴胺片为本公司生产,规格0.5mg。 2 色谱条件 色谱柱:Diamonsil 5u C18(250×4.6mm);流动相:乙腈-0.03mol/L磷酸二氢钠(磷酸调pH为3.5)(19:81);流速1.0ml/min;检测波长:342nm;柱温:常温;进样量:20μl。理论塔板数按甲钴胺峰计算不低于2000,甲钴胺与相邻杂质峰的分离度应符合要求。 3溶液的制备 3.1对照品溶液 3.2供试品溶液 3.3空白溶液 4系统适用性试验 取处方量的空白辅料,照含量测定法进行测定,空白辅料不干扰测定,结果见附图2-1(图谱:c:\HPCHEM\1\DATA\JGAFFX\00009)