
医疗器械法规文件大全(最新权威版)
医疗器械监管法规总表
医疗器械工作程序文件目录 一、医疗器械的采购程序 二、医疗器械产品质量检查验收程序 三、医疗器械入库储存程序 四、医疗器械产品在库养护程序 五、医疗器械产品配送出库复核程序 六、医疗器械配送退回处理程序 七、不合格医疗器械的确认处理程序 八、医疗器械拆零和拼装发货程序 九、医疗器械运送程序 十、医疗器械进货退出程序 十一、证照资料的收集、审核、存档的程序 十二、质量事故上报处理程序 十三、首营品种的审批程序
一、医疗器械的采购程序 1、目的:建立一个医疗器械商品采购的标准操作程序,以保证采购行为的规范。 2、范围:适用于医药商品采购的环节与行为。 3、责任:采购员、质管员、采购资金管理员及其部门负责人对本程序负责。 4、程序: (一)、采购计划的制定程序 1、采购部门计划员根据实际库存消耗制定年度、季度或月份进货采购计划。 2、采购计划提交采购小组(采购部、营销部、质管部、财务部人员组成)讨论、修改、审定。 3、质量管理机构对计划所列商品合法性及其供货渠道的质量信誉与质量保证能力进行审核。 4、采购部门负责人审批后交各类别采购人员具体执行。 5、临时调整采购计划、审批程序同1—4条。 6、每月召开采购部门与营销部门、质管部、配送中心的联合会议,沟通商品在销售、使用、储运环节中的信息和存在的问题,以便及时调整购进计划。 (二)、合格供货单位的选择程序 1、采购部门应协助质量管理机构建立、健全和更新“合格供货方”资料档案。 2、首营企业按有关管理制度办理审批手续。 3、对拟采购的医疗器械,查看其合法的产品注册证,了解供货单位的质量保证能力和履行合同的能力。 4、对拟采购进口医疗器械,收集国外厂商在我国国家药品监督管理局已注册的证书,收集进口医疗器械注册证及进口检验报告书复印件加盖供货单位质管机构的红色印章。 5、根据购货计划表,以“合格供货方”档案中拟出需采购医疗器械的生产和供货单位。 6、相同品名、规格的产品应择厂、择优、就近进货。 (三)、采购合同的签订程序 1、各类别采购员应严格执行业务经营质量管理制度。 2、标准合同应明确签订以下质量条款:产品应符合质量标准和有关质量要求;附产品合格证;产品包装符合有关规定和货物运输要求;进口产品应提供符合规定的证书和文件。 3、与签订质量保证协议的供应商采取传真、电话等方式订货须建立非标准合同采购记录,对所订产品的质量有简明约定。
医疗器械分类目录 基础外科手术器械 医用缝合针:医用缝合针 基础外科用刀:手术刀柄皮片刀疣体剥离刀柳叶刀铲刀剃毛刀皮屑刮刀挑刀锋刀修脚刀修甲刀解剖刀手术刀片 基础外科用剪:手术剪组织剪综合组织剪拆线剪石膏剪解剖剪纱布绷带剪教育用手术剪 止血钳小血管止血钳蚊式止血钳组织钳硬质合金镶片持针钳持针钳创夹缝拆钳皮肤轧钳子弹钳纱布剥离钳海绵钳帕巾钳皮管钳基础外科用钳: 器械钳 基础外科用镊夹:小血管镊无损伤镊组织镊整形镊持针镊保健镊拔毛镊帕巾镊敷料镊解剖镊止血夹病历夹 基础外科用针、钩:动脉瘤针探针推毛针植毛针挑针直尖针静脉拉钩创口钩扁平拉钩双头拉钩皮肤拉钩解剖钩 刀片夹持器麻醉口罩麻醉开口器照明吸引器头粉刺取出器黑头粉刺压出器皮肤刮匙皮肤套刮器皮肤刮划测检器皮肤检查尺皮肤组基础外科其它器械: 织钻孔器开口器卷棉子毁形机洗瓶机 显微外科手术器械 显微外科用刀、凿:显微喉刀 显微外科用剪:显微剪显微枪形手术剪显微组织剪
显微外科用钳:显微枪形麦粒钳显微喉钳显微持针钳 显微外科用镊、夹:显微镊显微持针镊显微止血夹 显微外科用针、钩:显微耳针显微喉针显微耳钩显微喉钩 显微外科用其他器械:显微合拢器 神经外科手术器械 神经外科脑内用刀:脑神经刀可拆卸式脑膜刀脑神经刀脑膜刀 神经外科脑内用钳:肿瘤摘除钳脑组织咬除钳银夹钳U型夹钳动脉瘤夹钳 神经外科脑内用镊:脑膜镊垂体瘤镊肿瘤夹持镊 神经外科脑内用钩、刮:脑膜钩脑膜拉钩神经钩神经根拉钩交感神经钩脑刮匙脑垂体刮匙神经外科脑内用其他器械:脑活检抽吸器脑膜剥离器脑吸引器后颅凹牵开器手摇颅骨钻脑打针锤脑压板 眼科手术器械 眼科手术用剪:角膜剪眼用手术剪眼用组织剪 眼科手术用钳:晶体植入钳环状组织钳
1、目的:建立医疗器械文件管理程序,确保从合法的企业购进合法和质量可靠的医疗器械。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、适用范围:本企业质量文件的管理。 4、职责:质量管理部人员对本程序的实施负责。 5、程序: 5.1.本制度管理内容为:企业全部经营活动中的各种资料、文件、数据、票据、报表、记录等,其中包括质量监督、质量信息、意见反馈、产品质量、工作质量、规章制度、技术资料、培训纪录、人员档案等。 5.2.质管、销售二部门系企业信息中心,负担着企业所需要的各种信息反馈等情况的收集、汇总、分析、传递,等具体工作。
5.3.企业各部门要按时填报经营活动中的信息统计报表,于次月初3日内将上月资料交质管部门汇总。 5.4.各部门负责人主抓本科室的文件、资料、质量信息的收集、登记、整理、汇总工作。 5.5.全部文件、资料、纪录和信函,凭证等,一律实行借阅登记制度,规定任何人借阅资料必须签字借阅,归还核销,不得无故积压和丢失。 5.6.文件资料应与财务凭证一样注意妥善保管,一般保存10年,需销毁时,应由企业经理批准,并详细纪录文件编号、名称、发文时间等数据。 1、目的:建立医疗器械购进程序,确保从合法的企业购进合法和质量可靠的产品。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、职责:医疗器械采购人员对本程序的实施负责。
4、程序: 5.1确定供货单位合法资格和质量信誉。 5.1.1对供货单位合法资格的确定。 5.1.1.1医疗器械购进人员向供货单位销售人员索取供货单位的《器械生产(经营)许可证》和《营业执照》的复印件。 5.1.1.2医疗器械购进人员对所索取的上述“证照”复印件进行以下审核。 5.1.1.2.1“证照”复印件是否加盖了供货单位的原印章。 5.1.1.2.2“证照”是否在其注明的有效期之内。 5.1.1.2.3“证”与“照”的相关内容是否一致。 5.1.1.2.4“证照”上的注册地址是否与供货单位实际的生产或经营地址相同。 5.1.1.2.5必要时,可索取“证照”的原件进行查验。 5.1.2对供货单位质量信誉的确定。 5.1.3验明首营企业医疗器械销售人员的合法身份,并索取下列资料。 5.1.3.1加盖有企业原印章盒有企业法人代表印章或签字的企业委托授权书原件,委托书应明确授权范围和委托期限。 5.1.3.2首营企业医疗器械销售人员的身份复印件(验证原件后复印)。
二类医疗器械分类 目录大全 6801 基础外科手术器械 序 名称品名举例类别号 1医用缝合针 医用缝合针 ( 不带线 )Ⅱ(不带线) 6803 神经外科手术器械 序 名称品名举例类别号 1神经外科脑 脑神经刀、可拆卸式脑膜刀、脑神经刀、脑膜刀Ⅱ内用刀 2神经外科脑 肿瘤摘除钳、脑组织咬除钳Ⅱ内用钳 3神经外科脑 脑膜镊、垂体瘤镊、肿瘤夹持镊Ⅱ内用镊 4神经外科脑脑膜钩、脑膜拉钩、神经钩、神经根拉钩、交感神 Ⅱ内用钩、刮经钩、脑刮匙、脑垂体刮匙 神经外科脑 5内用其他器脑活检抽吸器、脑膜剥离器Ⅱ械 6804 眼科手术器械 序 名称品名举例类别号 1眼科手术用二氧化碳眼科冷冻治疗仪、白内障超声乳化系统用 Ⅱ其他器械负压导管 口腔用其它 2一次性牙科冲洗针Ⅱ器械
6807 胸腔心血管外科手术器械 序 名称 号 胸腔心血管1 外科用钳品名举例类别 心内膜心肌活组织钳、心房侧壁钳、主动脉侧壁钳、 主动脉阻断钳、主动脉止血钳、主动脉游离钳、无 损伤肺动脉止血钳、无损伤动脉止血钳、无损伤动Ⅱ 脉导管钳、动脉侧壁钳、动脉阻断钳、静脉阻断钳、 腔静脉钳、腔静脉游离钳、主肺动脉钳 2胸腔心血管大隐静脉镊、心房止血器、心耳止血器、凹凸齿止 Ⅱ外科用镊、夹血夹 胸腔心血管 3外科用其他血管打洞钳(器)、心房打洞器、二尖瓣扩张器Ⅱ器械 胸腔心血管心内吸引器(头)、左房引流管、冠状动脉吸引器、 4外科用吸引冠状动脉灌注器、大隐静脉冲洗管、静脉撑开器、Ⅱ器短柄吸引器(头) 6808 腹部外科手术器械 序 名称品名举例类别号 1腹部外科用 食道吻合器、肠道吻合夹、肠道吻合器Ⅱ其他器械 6809 泌尿肛肠外科手术器械 序 名称品名举例类别号 1泌尿肛肠科 血管阻断钳、骼血管阻断钳、骼静脉侧壁钳Ⅱ用钳 2J 型导尿管、肛肠手术用多功能小针刀、弯头负压 Ⅱ吸力式套扎枪 6810 矫形外科(骨科)手术器械 序 名称品名举例类别号 1矫形(骨科) 椎管铲刀、椎管锉刀、手锥Ⅱ外科用刀、锥 2矫形(骨科)颈椎咬骨钳、颈椎双关节咬骨钳、脊柱侧弯矫正钳、Ⅱ
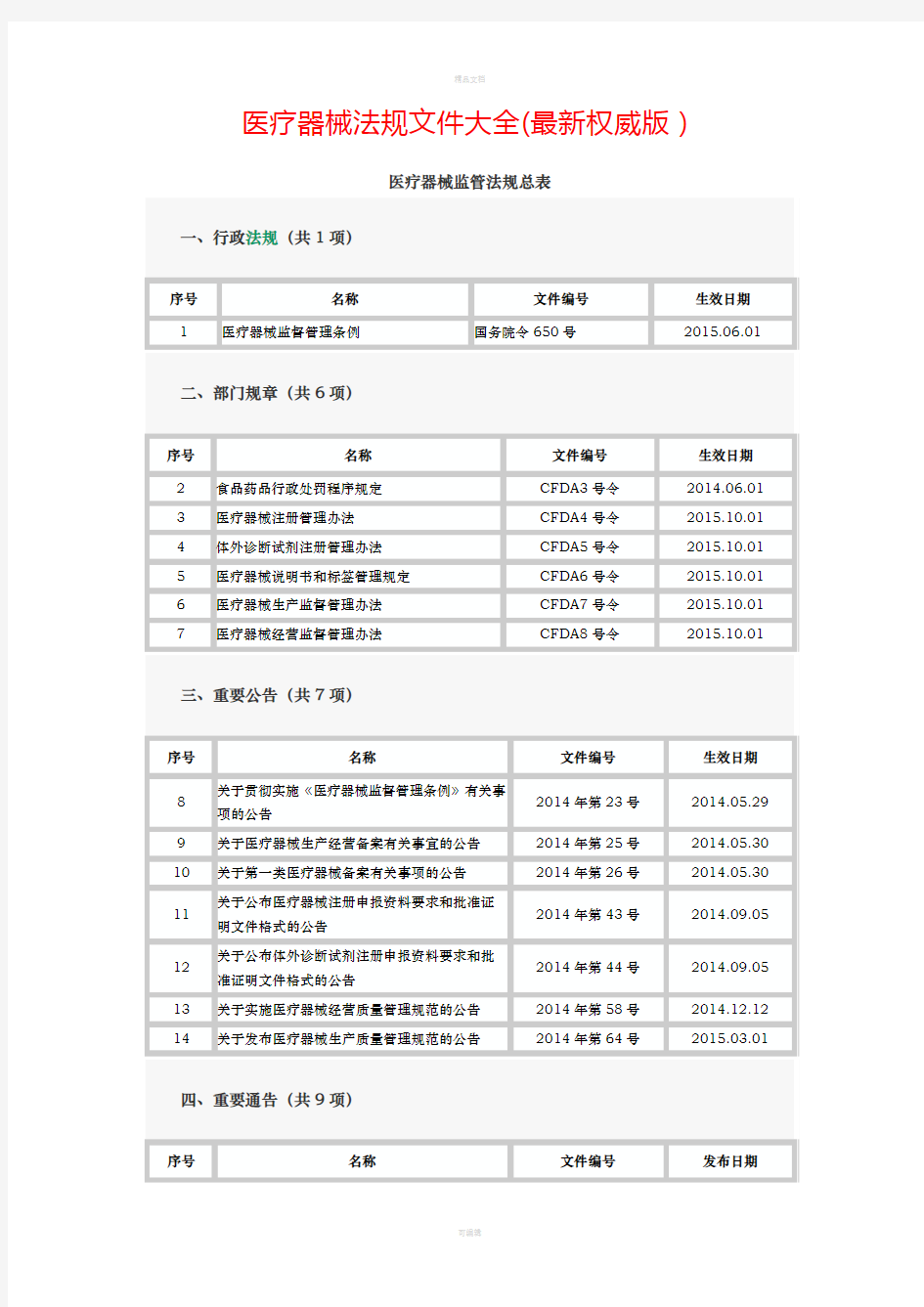
新版中国医疗器械法规清单 一、行政法规 1.《医疗器械监督管理条例》(国务院令第680号) 二、部门规章 1.医疗器械注册管理办法(CFDA局令第4号) 2.体外诊断试剂注册管理办法(CFDA局令第5号) 3.医疗器械说明书和标签管理规定(CFDA局令第6号) 4.医疗器械生产监督管理办法(CFDA局令第7号) 5.医疗器械经营监督管理办法(CFDA局令第8号) 6.药品医疗器械飞行检查办法(CFDA局令第14号) 7.医疗器械分类规则(CFDA局令第15号) 8.医疗器械使用质量监督管理办法(CFDA局令第18号) 9.医疗器械通用名称命名规则(CFDA局令第19号) 10.医疗器械临床试验质量管理规范(CFDA国家卫计委令第25号) 11.医疗器械召回管理办法(CFDA局令第29号) 12.体外诊断试剂注册管理办法修正案(CFDA局令第30号) 13.关于调整部分医疗器械行政审批事项审批程序的决定(CFDA局令第32号) 14.医疗器械标准管理办法(CFDA局令第33号) 15.医疗器械网络销售监督管理办法(CFDA局令第38号) 16.医疗器械不良事件监测和再评价管理办法(国家市场监督管理总局令第1号) 三、通告 1.关于发布第一类医疗器械产品目录的通告(CFDA通告2014年第8号) 2.关于发布医疗器械产品技术要求编写指导原则的通告(CFDA通告2014年第9号) 3.关于发布需进行临床试验审批的第三类医疗器械目录的通告(CFDA通告2014年第14 号) 4.关于发布体外诊断试剂临床试验技术指导原则的通告(CFDA通告2014年第16号) 5.关于发布体外诊断试剂说明书编写指导原则的通告(CFDA通告2014年第17号) 6.关于发布禁止委托生产医疗器械目录的通告(CFDA通告2014年第18号) 7.关于发布医疗器械生产企业供应商审核指南的通告(CFDA通告2015年第1号) 8.关于发布医疗器械临床评价技术指导原则的通告(CFDA通告2015年第14号) 9.关于发布医疗器械产品出口销售证明管理规定的通告(CFDA通告2015年第18号) 10.关于贯彻落实小微企业行政事业性收费优惠政策的通告(CFDA通告2015年第31号) 11.关于生产一次性使用无菌注、输器具产品有关事项的通告(CFDA通告2015年第71 号)
医疗器械注册检验送检须知 一、医疗器械注册检验适用范围 凡依据《医疗器械注册管理办法》及其相关文件规定,向我院提出检验申请的,属医疗器械注册检验。 二、医疗器械注册检验分类 医疗器械注册检验分类包括:国产首次注册、进口首次注册、注册变更、延续注册。 三、申请医疗器械注册检验应填写的表格 申请医疗器械注册检验应填写“检验申请表”。 如送检时同时提出医疗器械境外检验项目认可的,还应填写“医疗器械境外检测项目认可申请表”。 四、医疗器械注册检验应提交的资料 医疗器械注册检验资料由以下部分组成: (一)申报单位出具的检验申请函(必要时附注册审批有关证明文件); (二)产品技术要求(必要时附起草说明); (三)进口注册申请资料认可的,应提供自检原始记录复印件、检验报告书原文及中文译文。(四)申请人送检时同时提出医疗器械境外检验项目认可的还应提供: 1.该产品原产国政府认可的检测报告原文及中文译文; 2.批准上市的证明文件复印件; 3.相关资料(如申请认可项目的检测记录及数据分析相关资料),并加盖提供单位的公章或印签。
以上资料均须加盖申请单位公章。 提出医疗器械境外检验项目认可要符合下述两种情况: 1)安全性能指标或重要性能指标中,若某单项试验的时间超过30天的; 2)重要性能指标中,因样品数量所限,不能实施检测的。 五、申请医疗器械注册检验对样品的要求 检品数量要求: 一般情况下,检品数量应为一次检验用量的三倍。样品昂贵等特殊情况下,送检人在“申请检验登记表”中注明“不申请复验”可酌情减量,但不得少于检验及复试用量。 检品状态要求: 检品应包装完整,有完整标签,标签内容应符合国家局医疗器械标签说明书相关文件规定,无正规标签的样品,必需贴有临时标签。标签上应注明检品名称、批号或编号、型号或规格、生产日期或效期、生产单位。标签内容应与申请函(委托书)、申报资料相应内容一致。 样品效期要求: 样品剩余效期一般应满足2个检验周期,除特殊情况外(如进行稳定性考察),已过效期或效期内不能满足2个检验周期的样品不予受理。
xxx医疗器械有限公司 程序文件 文件控制程序 文件分发号:201206 文件编号:Q/SZ BR ZC(4.0)01 版号:A 拟制:年月日 审核:年月日 批准:年月日 受控状态:非受控第1页共3页
1目的 对所有与质量活动有关的文件的编制、审批、发放、使用、更改、撤换、回收、报废、保存和销毁等各项活动进行控制,确保各质量活动场所使用的文件均为有效版本,以防止使用失效或作废的文件。 2范围 适用于所有质量管理体系运行中有关质量活动的文件的控制和管理(包括适当范围内的外来文件)。 3职责 3.1办公室负责质量手册、程序文件、管理制度、考核办法及与企业经营有关的法律、法规、规章的管理控制。 3.2质管部门负责质量管理文件、验收规程的归口管理控制。 3.3业务部负责采购文件、供方及顾客的各种证照、销货记录等资料及出入库记录的归口管理控制。 4管理内容及要求 4.1文件的审定、批准和发布 4.1.1 文件:包括质量手册、程序文件、技术文件、商品标准、医疗器械经营企业许可证、医疗器械经营企业备案表、医疗器械生产企业许可证、医疗器械生产企业备案表、产品注册证、营业执照、执业许可证、管理制度、经营管理规范、验收规程、医疗器械法规规章、科技信息等。 4.1.2 《质量手册》由管代审定,总经理批准和发布。 4.1.3 《程序文件》由管代组织相关部门编写,管代审核、总经理批准和发布。 4.1.4 与商品质量有关的文件有归口管理部门负责人批准和发放。 4.2 文件的归档、发放及使用管理。 4.2.1 文件发放时填写《文件的现行修订状态的控制清单》,文件归档应填写《文件归档登记》。 4.2.2 文件的发放根据组织编制和职责范围发放;管理性文件由办公室发放,技术性文件由质管部发放。 4.2.3 因工作需要增加发放数量或扩大发放范围,填写《文件发放审批表》,公司内扩大范围或增加发放数量,由归口管理部门负责人审批;发放范围扩大至公司外的文件,必须由总经理审批。 4.2 文件编号 4.2.1 质量管理体系的编号 a)质量手册:Q—企业 SZ BR ZS—质量手册 例:本公司为Q\JN QR-ZS b) 程序文件:Q—企业 SZ BR ZC—质量程序 4.0-章节号 01-序号 例:文件控制程序SZ BR ZC(4.0)01 c) 质量记录:主要使用部门代号—记录编号 例YW—02表示业务部,编号02号 C) 各部门其他质量文件:部门代号—文件顺序 4.2.2各部门代号 总经理ZJ;管代GD;办公室BG;质管部ZG;业务部YW;储运(仓库)CY。 4.3文件编写,审核,批准,发放。 文件发布前得到评审和批准,以确保文件是充分与适宜的。 第2页共3页
医疗器械分类目录 医疗器械分类从来都不是那么简单,根据我从业的网来天下医疗器械商城网站的分析,结合行业内专业的目录参考,我总结出自己的目录模板,放在这里供行业内的朋友参考,互相学习借鉴,希望能帮助解决有关医疗器械分类目录的问题。 以下是分类图,一级类目为: 全部商品分类 配件大全 医用耗材 医疗器械 一级类目配件大全分类如下: 全部商品分类 配件大全 医用灯泡电(源)线 超声探头穿刺架 球管导联线 医用耗材 胶片灯泡口罩手套鞋罩急救包胃管 吸氧管B超打印纸 医疗器械 胎儿/母婴监护仪 麻醉监护仪心电监护仪心电图机动态血压仪常用配件 医用灯泡电(源头)线电池开关插头转换头连接器 超声配件 超声探头穿刺架球管 电子配件 稳压器涡轮机显像 自动排水系统 手术配件 双极镊双极电缆电极 止血带专业连接管 诊察配件 血氧探头心电胸吸球肢体夹 血压袖套导联线\延长线血压连接管血压充气装置体温探头 检验配件 橡胶实验用品塑料实验用品 移液器支架血压连接器 牙科配件 牙科手机配件牙科大型配件 牙科其他配件
一级类目医用耗材分类如下: 常用耗材 口罩 手套 鞋罩 医用服装 纤维毛巾PE 制品 利器盒 病历夹 硅胶垫 医用标记材料 碘伏棉棒 检验耗材 移液器吸头(盒) 胶塞 洗耳球 乳胶帽\管 乳胶球 其他 一次性管包/护理 胃管 护理管 吸氧管 导尿管 换药包 产包 手术包 急救包 导尿包 橡胶布 护理袋 造口袋 肛门袋 引流袋 肛门圈 备皮刀 大便球妇科冲洗器 胶片/试剂(纸) 医用胶片 血糖试纸 尿液试纸 生化试剂 B 超打印纸 采血/消毒 微量血气采血器 采血管 手消毒液 吸氧/雾化 吸氧面罩 一次性吸氧管 一次性雾化器 冰敷/降温 医用冰毯 医用冰帽 医用冰带 医用冰枕 退热凝胶 一次性冰袋 全部商品分类 配件大全 医用灯泡 电(源)线 超声探头 穿刺架 球管 导联线 医用耗材 胶片 口罩 手套 鞋罩 急救包 胃管 吸氧管 B 超打印纸 医疗器械 胎儿/母婴监护仪 心电监护仪 麻醉监护仪 心电图机 动态血压仪
医疗器械相关法律法规知识培训 医疗器械监督管理条例 第一章总则 第一条为了加强对医疗器械的监督管理,保证医疗器械的安全、有效、保障人体健康的生命安全,制定本条例。 第二条在中华人民共和国境内从事医疗器械的研制、生产、经营、使用、监督管理的单位或者个人,应当遵守本条例。 第三条本条例所称医疗器械,是指单独或者组合使用于人体的仪器、设备、器具、材料或者其他物品,包括所需要的的软件;其用于人体体表及体内 的作用不是用药理学、免疫学或者代谢的手段获得,但是可能有这些手 段参与并一定的辅助作用;其作用旨在达到目的下更预期目的;(一)对疾病的预防、诊断、治疗、监护、缓解; (二)对操作或者残疾的诊断、治疗、监护、缓解、补偿; (三)对解剖或者生理过程的研究、替代、调节; (四)妊娠控制。 第四条国务院药品监督管理部门负责全国的医疗器械监督管理工作。 第五条国家对医疗器械实行分类管理。 第一类是指,通过常规管理足以保证其安全性、有效性的医疗器械。 第二类是指,对其安全性、有效性应当加以控制的医疗器械。 第三类是指,植入人体;用于支持、维持生命;对人体具有潜在危险,对其安全性、有效性必须严格控制的医疗器械。 第六条生产和使用以提供具体量值为目的的医疗器械,应当符合计量法的规
定。具体产品目录由国务院药品监督管理部门会同国务院计量行政管理 部门制定并公布。 第二章医疗器械的管理 第七条国家鼓励研制医疗器械新产品,医疗器械新产品,是指国内市场尚未出现过的或者安全性、有效性及产品机理未得到国内认可的全新的品种。第八条国家对医疗器械实行产品生产注册制度。 生产第一类医疗器械,由设区的市级人民政府药品监督管理部门审查批准,并发给产品生产注册证书。 生产第二类医疗器械,由省、自治区、直辖市人民政府药品监督管理部门审查批准,并发给产品生产注册证书。 生产第三类医疗器械,由国务院药品监督管理部门审查批准,并发给产品生产注册证书。 生产第二类、第三类医疗器械,应当通过临床验证。 第九条省、自治区、直辖市人民政府药品监督管理部门负责审批本行政区域内的第二类医疗器械的临床试用或者临床验证。国务院药品监督管理部门负责审批第三类医疗器械的临床试用或者临床验证。 临床试用或者临床验证应当在省级以上人民政府药品监督管理部门指定的医疗机构进行。医疗机构进地临床试用或者临床验证,应当符合国务院药品监督管理部门的规定。 进行临床试用或者临床验证的医疗机构的资格,由国务院药品监督管理部门会同国务院卫生行政部门认定。 第十条医疗机构根据本单位的临床需要,可以研制医疗器械,在执业医师
ZD-QP-4.2.3-01 文件控制程序 1 目的 对与质量管理体系有关的文件和资料进行控制,确保公司各相关部门使用的文件为有效版本。 2 范围 适用于本公司质量管理体系文件的编制、评审、批准、发放、使用、更改、标识、作废等全过程的控制,包括外来文件。 3 职责 3.1总经理办公室负责组织各类文件的发放、修订及归档管理,组织编制质量手册及各类文件,并负责组织对现有体系文件的定期评审。 3.2 各部门负责本部门相关文件的编制、归档等并正确使用文件。 4 程序要求 4.1 文件控制范围 4.1.1公司质量管理体系控制的文件范围包括: a)公司的质量方针和质量目标; b)质量手册; c)标准所要求的程序文件; d)为确保过程有效策划、运行和控制所要求的文件,即技术文件、管理文件和外来文件; e)质量管理体系所要求的记录; f)国家或地区颁发的有关医疗器械的法律法规; 4.1.2根据文件的内在联系可将它们分为三个层次的文件:质量手册(含质量方针、目标)为第一层次文件;程序文件为第二层次的文件;操作规程、作业指导书、检验规范、记录、外来文件等为第三层次文件。 4.1.3本公司的文件可采用任何的媒体和类型的形式,如纸张、光盘、磁盘、照片样件、磁带等,多媒体文件的控制按照《多媒体和电子文件管理规定》执行。
4.1.4 记录是一种特殊类型的文件,本公司的记录应按QP4.2.4《记录控制程序》中的相关规定进行控制。 4.2文件的编写、审核、批准 各类文件应按规定编制、审核或进行评审,发布前必须经授权人批准。 4.2.1质量手册由总经理办公室负责组织编写,管理者代表审核,总经理批准后发布。质量手册中的程序文件、管理规定由相关部门负责人审核,管理者代表批准。 4.2.2 公司各类管理文件(操作规程、作业指导书、检验规范等)由相关部门负责人审核,管理者代表批准后实施。 4.3 文件的发放、修订和管理 4.3.1 文件的发放 a)公司质量管理体系文件为受控文件,受控清单由总经理办公室提出,文件发放前应由管理者代表批准,以确保文件是充分与适宜的; b)必须保证各部门都能得到相应有效的文件,发放的文件应填写《文件收发登记表》,并在该文件封面加盖“受控”印章和注明分发号。 4.3.2 文件的修订 a)当文件需要修订时,由提出部门说明原因,原编制部门根据评审结论对文件及时更改确认,更改后的文件需经管理者代表再次批准后,由总经理办公室下达《文件更改申请(通知)单》,通知文件发放部门,若指定其它部门审批时,应获得审批所需依据的有关背景资料; b)所有被修订的原文件必须由总经理办公室统一收回,以确保有效文件的唯一性,对需要销毁的作废文件,由总经理办公室填写《文件销毁申请》,经批准后在适当的时候可统一销毁; c)质量手册每经修改换页后,修改状态都应随之改变,如修改状态1 等,经多次修改后或文件需要大幅度更改时,应进行文件换版,版序以英文字母表示,如版号A 、版号B 等,公司的受控文件清单应随文件的修订而及时修订,便于文件使用者随时识别文件的现行修改状态。 4.3.3 文件的管理 a)各类文件应制定特有的名称及文件编号,质量管理体系文件经审核批准
国家食品药品监督管理局令 第15号 《医疗器械经营企业许可证管理办法》于2004年6月25日经国家食品药品监督管理局局务会审议通过,现予公布,自公布之日起施行。 医疗器械经营企业许可证管理办法 第一章总则 第一条为加强对医疗器械经营许可的监督管理,根据《医疗器械监督管理条例》,制定本办法。 第二条《医疗器械经营企业许可证》发证、换证、变更及监督管理适用本办法。 第三条经营第二类、第三类医疗器械应当持有《医疗器械经营企业许可证》,但是在流通过程中通过常规管理能够保证其安全性、有效性的少数第二类医疗器械可以不申请《医疗器械经营企业许可证》。不需申请《医疗器械经营企业许可证》的第二类医疗器械产品名录由国家食品药品监督管理局制定。 第四条国家食品药品监督管理局主管全国《医疗器械经营企业许可证》的监督管理工作。 省、自治区、直辖市(食品)药品监督管理部门负责本辖区内《医疗器械经营企业许可证》的发证、换证、变更和监督管理工作。 设区的市级(食品)药品监督管理机构或者省、自治区、直辖市(食品)药品监督管理部门直接设置的县级(食品)药品监督管理机构负责本辖区内《医疗器械经营企业许可证》的日常监督管理工作。 第五条国家食品药品监督管理局逐步推行医疗器械经营质量规范管理制度。医疗器械经营质量管理规范由国家食品药品监督管理局组织制定。 第二章申请《医疗器械经营企业许可证》的条件 第六条申请《医疗器械经营企业许可证》应当同时具备下列条件: (一)具有与经营规模和经营范围相适应的质量管理机构或者专职质量管理人员。质量管理人员应当具有国家认可的相关专业学历或者职称; (二)具有与经营规模和经营范围相适应的相对独立的经营场所; (三)具有与经营规模和经营范围相适应的储存条件,包括具有符合医疗器械产品特性要求的储存设施、设备; (四)应当建立健全产品质量管理制度,包括采购、进货验收、仓储保管、出库复核、质量跟踪制度和不良事件的报告制度等; (五)应当具备与其经营的医疗器械产品相适应的技术培训和售后服务的能力,或者约定由第三方提供技术支持。 第七条申请《医疗器械经营企业许可证》的,必须通过(食品)药品监督管理部门的检查验收。 省、自治区、直辖市(食品)药品监督管理部门应当依据本办法,结合本辖区实际,制定医疗器械经营企业检查验收标准,报国家食品药品监督管理局备案。
精品文档 YZB 标准名称 (企业单位名称)发布
前言 XXX产品是我公司开发的新产品。目前该产品尚无国家标准、行业标准,为规范产品的技术特性,确保产品的安全有效,特制订本注册产品标准,做为生产质量控制的依据。 XXX产品注册产品标准引用了GB191《包装储运图示标志》,GBT14710《医用电气设备环境要求及试验方法》,GB9706.1《医用电气设备第一部分:安全通用要求》等标准中的内容。本标准的安全要求全面贯彻了GB9706.1—1995。并将其列入本标准附录A(规范性附录)。 本标准的附录A是规范性附录 本注册标准由天津市XXXXXXX公司提出并负责起草。 本注册标准主要起草人:XXX。 本注册标准首次发布于200X年XX月。 本注册标准由天津市XXXX(企业名称)法定代表人批准,并对所规定的内容负责。
标准名称 1 范围 本标准规定了XXX产品的结构和基本参数、要求、试验方法、检验规则、标志、包装、运输、贮存。 本标准适用于XXX产品(以下简称XX)。该产品供医疗单位_____( 用途)___________________。 2 规范性引用文件 下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。 GB/T 191—2000 包装储运图示标本 GB 9706.1—1995 医用电器设备第一部分:安全通用要求 GB/T 14710—1993 医用电器设备环境要求及试验方法 3 结构和基本参数 3.1 结构:XXX产品由主机、控制部分、输出部分组成。 3.2 XXX产品的型号为:XX-XX-XX XX-XX-XX (对符号的解释)设计序号(产品规格)(罗马字母) (对符号的解释)产品分类(英文字母) (对符号的解释)企业名称简称(以汉字拼音字头) 3.3 XXX产品的基本参数 3.3.1 3.3.2 4 要求 4.1 XXX产品的工作条件 a)温度:5℃—40℃ b)相对湿度:不大于80% c)电源电压:AC220V±22V d)频率:50Hz±1Hz 4.2 性能指标: 4.2.1 XXX产品工作时噪声不大于60dB(A) 4.2.2 牵引力设置范围: 4.2.3 牵引治疗时间: 4.2.4 预置指示力示值与实际作用力误差: 4.2.5 预置指示时间与实际有效时间误差不大于±5% 4.3 安全
程序文件 采购控制程序 文件编号 版本/修订号: 编制人/日期: 编制人/日期: 编制人/日期: 年月日发布实施发放部门:
1.目的:对供方和采购过程进行有效的控制,确保所采购的产品符合规定的要求。 2.范围:适用于对生产所需的原辅料、包材采购、外协加工及供方提供服务的控制;对供方进行评价、选择和控制。 3.职责: 3.1.采购部组织对供方进行评价,选择合格供方,编制《合格供方名录》,并对供方业绩定期进行评价,建立供方档案;编制采购计划,执行采购作业,负责原辅包材采购并就采购原辅包材的质量和数量与供方沟通。 3.2技术部组织编制并确定采购材料分类明细及原辅料的标准,对外购外协件实施进货验证。 3.3质控部负责采购材料的进货检验,协助采购部对供方进行资质审查、评价和监控; 3.4.管理者代表批准对供方的评价记录、《外购外协件分类清单及技术要求》、《合格供方名录》。 4.内容: 4.1采购物资分类 4.1.1依据采购物资对随后的产品实现或最终产品的影响,将采购原材料分为关键物资、主要物资和一般原材料三类,即A、B、C三类,其中每一类物资中包含外购件和外协件。 关键物资(A类):对产品质量、性能起关键重要作用的物料; 重要物资(B类):对产品的质量、性能起主要作用的物料; 一般物资(C类):对产品起辅助作用的物料。 4.2供方审核 供方的审核由采购部先进行文件审核,通过后再由采购部根据采购物资分类和重要程度,组织技术部、质量部和生产部进行采购物资的样件审核和供方现场审核。关键物资和重要物资(A和B类)必须进行样件审核;其中关键物资和重要物资(A和B类)中外协件的供方必须进行现场审核。 4.2.1供方文件审核 采购部负责对供方进行初步调查,向供方收集相应资料,进行供方文件审核,文件审核通过的供方建立《供应商档案》,同时在《供应商档案》中保持供方文件
法律法规试题 一、单选题(15题,每题1分) 1、省、自治区、直辖市(食品)药品监督管理部门应当在受理之日起( )个工作日内作出是否核发《医疗器械经营企业许可证》的决定。 A、10 B、15 C、20 D、30 答案: D 2、从事医疗器械生产活动,非必须具备的条件是:( ) A、有与生产的医疗器械相适应的生产场地、环境条件、生产设备以及专业技术人员; B、有对生产的医疗器械进行质量检验的机构或者专职检验人员以及检验设备; C、产品标准 D、有保证医疗器械质量的管理制度; 答案:C 3、第一类医疗器械产品备案和申请第二类、第三类医疗器械产品注册,不用提交的资料( ) A、仓库地址的地理位置图、平面图、房屋产权证明(或租赁协议)复印件产品、技术要求; B、产品检验报告; C、注册产品标准 答案:A 4、医用中心制氧系统按医疗器械的结构特征、使用状态分类,为( ) A、有源医疗器械、接触或进入人体器械 B、有源医疗器械、非接触人体器械 C、无源医疗器械、接触或进入人体器械 D、无源医疗器械、非接触人体器械 答案:B 5、涂改、倒卖、出租、出借医疗器械注册证书,或者以期货形式非法转让医疗器械注册证书的,由县组以上(食品)药品监督管理部门责令改正,可以并处( )罚款。 A、5000元以上10000万以下 B、5000元以上20000万以下 C、3万元以下 答案: C 6、医疗器械注册产品标准的法律责任主体是( ) A、医疗器械制造商 B、技术监督管理部门 C、医疗器械注册产品标准复核备案部门 答案: A 7、医疗器械使用状态可分为接触或进入人体器械和非接触人体器械,属于非接触人体器械的内容是:( ) A、对医疗效果的影响,其程序分为:基本不影响;有间接影响;有重要影响 B、使用时限分为:暂时使用;短期使用;长期使用
医疗器械工作程序文件 Hessen was revised in January 2021
1、目的:建立医疗器械文件管理程序,确保从合法的企业购进合法和质量可靠的医疗器械。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、适用范围:本企业质量文件的管理。 4、职责:质量管理部人员对本程序的实施负责。 5、程序: .本制度管理内容为:企业全部经营活动中的各种资料、文件、数据、票据、报表、记录等,其中包括质量监督、质量信息、意见反馈、产品质量、工作质量、规章制度、技术资料、培训纪录、人员档案等。 .质管、销售二部门系企业信息中心,负担着企业所需要的各种信息反馈等情况的收集、汇总、分析、传递,等具体工作。 .企业各部门要按时填报经营活动中的信息统计报表,于次月初3日内将上月资料交质管部门汇总。 .各部门负责人主抓本科室的文件、资料、质量信息的收集、登记、整理、汇总工作。 .全部文件、资料、纪录和信函,凭证等,一律实行借阅登记制度,规定任何人借阅资料必须签字借阅,归还核销,不得无故积压和丢失。
.文件资料应与财务凭证一样注意妥善保管,一般保存10年,需销毁时,应由企业经理批准,并详细纪录文件编号、名称、发文时间等数据。 1、目的:建立医疗器械购进程序,确保从合法的企业购进合法和质量可靠的产品。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、职责:医疗器械采购人员对本程序的实施负责。 4、程序: 确定供货单位合法资格和质量信誉。 对供货单位合法资格的确定。 医疗器械购进人员向供货单位销售人员索取供货单位的《器械生产(经营)许可证》和《营业执照》的复印件。 医疗器械购进人员对所索取的上述“证照”复印件进行以下审核。 .1“证照”复印件是否加盖了供货单位的原印章。 .2“证照”是否在其注明的有效期之内。 .3“证”与“照”的相关内容是否一致。 .4“证照”上的注册地址是否与供货单位实际的生产或经营地址相同。 .5必要时,可索取“证照”的原件进行查验。
公司工作程序目录
文件 1、目的:建立医疗器械文件管理程序,确保从合法的企业购进合法和质量可靠的医疗器械。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、适用范围:本企业质量文件的管理。 4、职责:质量管理部人员对本程序的实施负责。 5、程序: 5-1、本制度管理内容为:企业全部经营活动中的各种资料、文件、数据、报表、纪录等,其中包括质量监督、质量信息、意见反馈、产品质量、工作质量、规章制度、技术资料、培训纪录、人员档案等。 5-2、质管、销售二部门系企业信息中心,负担着企业所需要的各种信息反馈等情况的收集、汇总、分析、传递,等具体工作。 5-3、企业各部门要按时填报经营活动中的信息统计报表,于次月初3日内将上月资料交质管部门汇总。 5-4、各部门负责人主抓本科室的文件、资料、质量信息的收集、登记、整理、汇总工作。 5-5、全部文件、资料、纪录和信函,凭证等,一律实行借阅登记制度,规定任何人借阅资料必须签字借阅,归还核销,不得无故积压和丢失。
5—6、文件资料应与财务凭证一样注意妥善保管,一般保存10年,需销毁时,应由企业经理批准,并详细纪录文件编号、名称、发文时间等数据。
文件 1、目的:建立医疗器械购进程序,确保从合法的企业购进合法和质量可靠的产品。 2、依据:《医疗器械监督管理条例》《医疗器械经营企业检查验收标准通知》。 3、职责:医疗器械购进人员对本程序的实施负责。 4、程序: 5.1确定供货单位合法资格和质量信誉。 5.1.1对供货单位合法资格的确定。 5.1.1.1医疗器械购进人员向供货单位销售人员索取供货单位的《器械生产(经营)许可证》和《营业执照》的复印件。 5.1.1.2医疗器械购进人员对所索取的上述“证照”复印件进行以下审核。 5.1.1.2.1“证照”复印件是否加盖了供货单位的原印章。 5.1.1.2.2“证照”是否在其注明的有效期之内。 5.1.1.2.3“证”与“照”的相关内容是否一致。 5.1.1.2.4“证照”上的注册地址是否与供货单位实际的生产或经营地址相同。 5.1.1.2.5必要时,可索取“证照”的原件进行查验。
1、格式要求:(1)申请材料的同一项目的填写应一致;(2)申请材料应使用A4规格纸张打印;(3)申请材料应清晰、整洁,每份申请材料均应装订并加盖企业公章,并按照申请材料目录的顺序装订成册;(4)在每项文件的第一页作一标签,或用带标签的隔页纸分隔,并标明项目编号;(5)用档案袋将报送的材料装好,档案袋需使用封面(格式见“档案袋封面格式.doc”),在袋面标明生产企业名称、地址、产品名称、联系人及电话,并加注申请材料审核的医疗器械注册申请事务人员姓名(需亲笔签名),联系方式,如是医疗器械注册专员请提供姓名(需亲笔签名)、联系方式及备案凭证号。 2、医疗器械注册申请表、产品标准一式两份,其他资料各一份。(附件1~附件5另附,无需与整套申请材料一起装订) 3、各项(上市批件、标准、检测报告、说明书)申报资料中的产品名称应与申请表中填写的产品名称实质性内容相对应。若有商品名,应标注商品名。申报资料应当使用中文,根据外文资料翻译的申报资料,应当同时提供原文。 4、申报资料受理后,企业不得自行补充申请,但属于《医疗器械注册管理办法》第三十八条规定情形的,可以补充申请。 5、生产企业在提交注册申报资料时,应同时提交医疗器械注册申请表、产品注册标准及备案说明书、标签和包装标识的电子文本(Word格式,具体要求见《关于调整医疗器械说明书备案内容的通知》(食药监办[2008]125号),其内容须与纸质文件的内容相一致。电子文本可通过移动存储设备(U盘或光盘)形式提交。 6、办理医疗器械注册申请事务的人员应当受生产企业委托,应提交生产企业负责人身份证明原件与复印件(生产企业负责人办理时);或者生产企业出具的本企业注册申请事务办理人员的授权书及该办 理人身份证明原件与复印件(非生产企业负责人办理时),身份证明原件经核对后退回。 7、如某项申请材料符合国家食品药品监督管理局《医疗器械注册管理办法》中相关豁免条款的规定,或符合国家局其他相关文件规定的,应提交相应的说明文件。 8、本指南已明确要求提交原件的,不得提交复印件。凡申请材料需提交复印件的,申请人(单位)须在复印件上注明“此复印件与原件相符”字样或者文字说明,注明日期,加盖单位公章。注1:企业应自行保存1份注册申报资料复印件以便注册核查用。(若注册申请未获批准,整套注册申报资料不予退还,申请人可凭复印件申请换回原件。) (二)申报资料的具体要求: 1、医疗器械注册申请表(1)应有法定代表人签字并加盖公章,所填写项目应齐全、准确; (2)“生产企业名称”、“注册地址”与《工商营业执照》相同; (3)“产品名称”、“规格型号”与所提交的产品标准、检测报告等申请材料中所用名称、规格型号一致。
与售后服务相关的法规要求 一、中华人民共和国国务院令(第680号)《医疗器械监督管理条例》 第三十八条发现使用的医疗器械存在安全隐患的,医疗器械使用单位应当立即停止使用,并通知生产企业或者其他负责产品质量的机构进行检修;经检修仍不能达到使用安全标准的医疗器械,不得继续使用。 二、《医疗器械使用质量监督管理办法》(国家食品药品监督管理总局令第18号) 第十七条医疗器械使用单位可以按照合同的约定要求医疗器械生产经营企业提供医疗器械维护维修服务,也可以委托有条件和能力的维修服务机构进行医疗器械维护维修,或者自行对在用医疗器械进行维护维修。 医疗器械使用单位委托维修服务机构或者自行对在用医疗器械进行维护维修的,医疗器械生产经营企业应当按照合同的约定提供维护手册、维修手册、软件备份、故障代码表、备件清单、零部件、维修密码等维护维修必需的材料和信息。 第十八条由医疗器械生产经营企业或者维修服务机构对医疗器械进行维护维修的,应当在合同中约定明确的质量要求、维修要求等相关事项,医疗器械使用单位应当在每次维护维修后索取并保存相关记录;医疗器械使用单位自行对医疗器械进行维护维修的,应当加强对从事医疗器械维护维修的技术人员的培训考核,并建立培训档案。 第十九条医疗器械使用单位发现使用的医疗器械存在安全隐患的,应当立即停止使用,通知检修;经检修仍不能达到使用安全标准的,不得继续使用,并按照有关规定处置。
第二十三条食品药品监督管理部门对医疗器械使用单位建立、执行医疗器械使用质量管理制度的情况进行监督检查,应当记录监督检查结果,并纳入监督管理档案。 食品药品监督管理部门对医疗器械使用单位进行监督检查时,可以对相关的医疗器械生产经营企业、维修服务机构等进行延伸检查。 医疗器械使用单位、生产经营企业和维修服务机构等应当配合食品药品监督管理部门的监督检查,如实提供有关情况和资料,不得拒绝和隐瞒。 三、《医疗器械经营监督管理办法》(国家食品药品监督管理总局局令第8号) 第三十三条医疗器械经营企业应当从具有资质的生产企业或者经营企业购进医疗器械。 医疗器械经营企业应当与供货者约定质量责任和售后服务责任,保证医疗器械售后的安全使用。 与供货者或者相应机构约定由其负责产品安装、维修、技术培训服务的医疗器械经营企业,可以不设从事技术培训和售后服务的部门,但应当有相应的管理人员。 第三十八条医疗器械经营企业应当配备专职或者兼职人员负责售后管理,对客户投诉的质量问题应当查明原因,采取有效措施及时处理和反馈,并做好记录,必要时应当通知供货者及医疗器械生产企业。 四、国家食品药品监督管理总局关于发布医疗器械生产质量管理规范的公告(2014年第64号)
二类医疗器械注册所需资料及详细步骤 所需资料: 1、医疗器械注册申请表 2、医疗器械生产企业资格证明 3、产品技术报告 4、安全风险分析报告 5、适用的产品标准及说明 6、产品性能自测报告 7、有承检资质的医疗器械检测机构出具的产品注册检测 报告 8、医疗器械临床试验资料 9、医疗器械说明书 10、产品生产质量体系考核(认证)的有效证明文件 11、所提交材料真实性的自我保证声明 注册步骤: a)产品技术报告(产品名称、产品型号/规格及其划分说明; 性能指标:能客观判定的成品的功能性、安全性指标及质量控制相关指标,符合国家标准/行业标准,但不包括产品设计开发的评价性内容;检验方法:具有可重现性和可操作性。) b)注册检测(要求检测公司具有相应的检测资质,我方提供
技术相关资料、注册检验的样品及产品技术;检测公测出检测报告和预评价报告) c)临床试验(临床试验;临床试验合同或协议、临床试验方 案、临床试验报告) d)产品生产质量体系考核(认证)的有效证明文件 e)安全风险分析报告、产品性能自测报告…….. 备注: 医疗器械注册申请表(包括电子版和纸质版,已有表格)、医疗器械生产企业资格证明(生产许可证和营业执照)、 安全风险分析报告(包括能量危害、生物学危害、环境危害、有关使用的危害和由功能失效、维护不周及老化引起的危害等方面的风险分析、风险控制与防范措施等,有相应的表格)、 适用的产品标准及说明(产品的标准可为国家标准、行业标准或注册产品标准文本;采用国家和行业标准的,要提交所采纳的国家标准或行业标准的有效文本及采标说明,采用注册产品标准作为产品标准的,提交注册产品标准正式文本及其编制说明) 产品性能自测报告(产品标准中要求的出厂检测,包括产品名称、规格型号、产品编号或批号、生产日期、样品数量、抽样基数;检测依据、检测项目、标准要求、检测结果、结