
制药用水制备技术
一、制药用水的种类
水是制药生产特别是注射剂制备中主要的用料和介质之一。因自然界的水中含有大量电解质、颗粒、有机物及微生物等有害物质,因此,制药生产中所用的水(简称为制药用水)必须通过复杂的生产工艺除去有害物质。制药工业中不同的生产工序对制药用水的质量要求不同,制药用水按其水质及应用范围的不同可分为饮用水、纯化水、注射用水及灭菌注射用水。
表1 制药用水的种类及应用范围
二、纯化水的制备
纯化水为饮用水经反渗透法、离子交换法、蒸馏法或其它适宜的方法制备的
制药用水,不含任何附加剂。纯化水中的电解质几乎已完全去除,通常剩余的含盐量控制在0.1 mg·L-1以下,且胶体与微生物、气体、有机物等已降至很低。根据不同的原水水质,可选择不同的纯化水的制备流程,图5-2所示的制备流程为纯化水的常见制备流程。
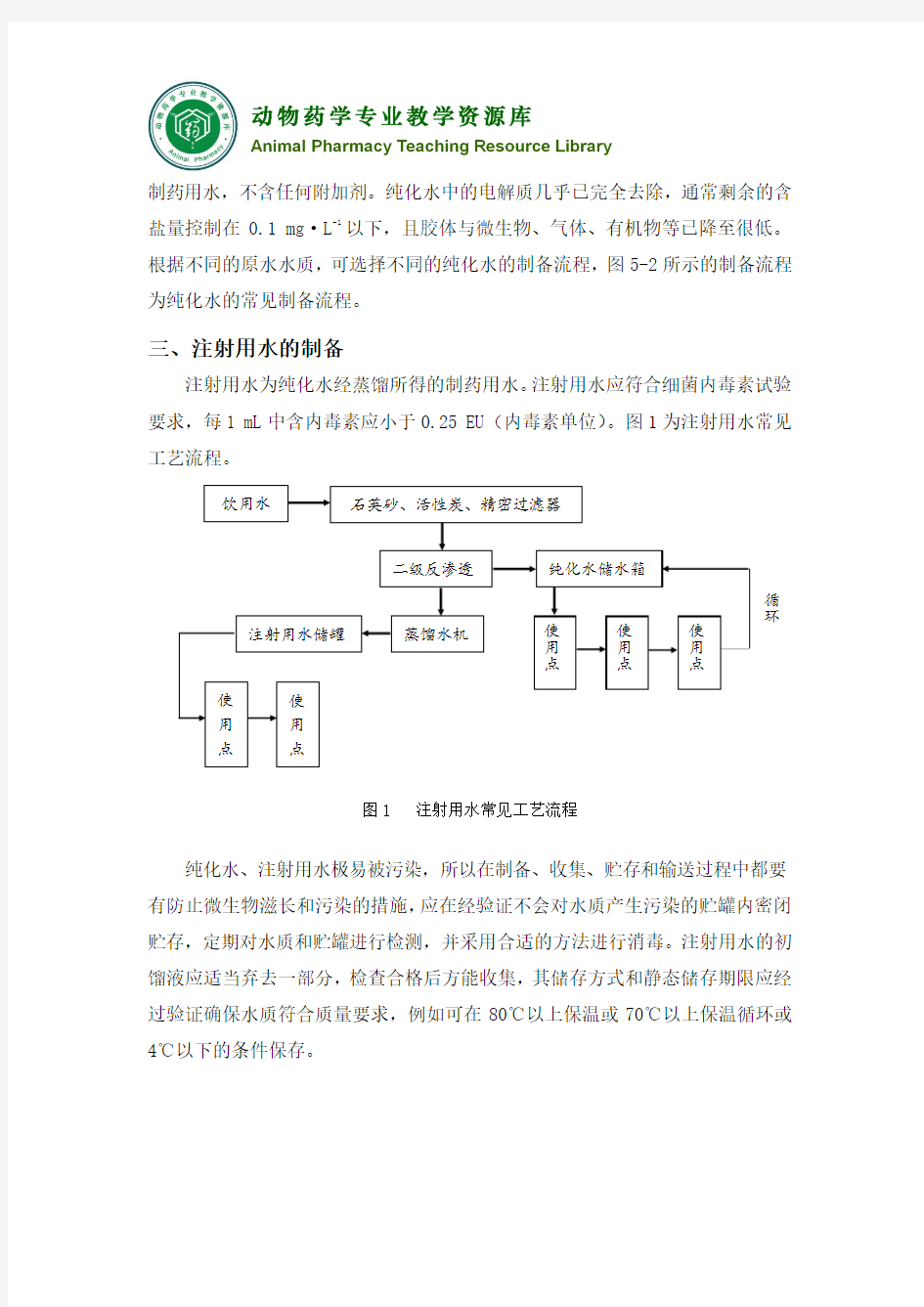
三、注射用水的制备
注射用水为纯化水经蒸馏所得的制药用水。注射用水应符合细菌内毒素试验要求,每1 mL中含内毒素应小于0.25 EU(内毒素单位)。图1为注射用水常见工艺流程。
图1 注射用水常见工艺流程
纯化水、注射用水极易被污染,所以在制备、收集、贮存和输送过程中都要有防止微生物滋长和污染的措施,应在经验证不会对水质产生污染的贮罐内密闭贮存,定期对水质和贮罐进行检测,并采用合适的方法进行消毒。注射用水的初馏液应适当弃去一部分,检查合格后方能收集,其储存方式和静态储存期限应经过验证确保水质符合质量要求,例如可在80℃以上保温或70℃以上保温循环或4℃以下的条件保存。
GMP对制药工艺用水的要求标准 一、简介 药品生产企业的工艺用水主要是指制剂生产中洗瓶、配料等工序以及原料药生产的精制、洗涤等工序所用的水。水的名称应避免和水的制造过程有关,如去离子水、除盐水、蒸馏水这样的名称,即水的制造过程与其名称脱钩,而是从化学和微生物的角度根据质量指标对水进行分类(如中国药典规定纯化水可以用三种不同方法制得,将来可能还会有更好得方法)。 二、制取方法 注射用水一般用纯化水通过蒸馏法(还有反渗透法和超滤法)制得,化学纯度高达99.999%,无热原。因纯蒸汽的制备过程与用蒸馏水制备注射用水的过程相同,可使用同一台多效蒸馏水机或单独的纯蒸汽发生器, 三、水质类别与要求 饮用水: 1、制备纯化水的水源; 2、口服剂瓶子初洗; 3、设备、容器的初洗; 4、中药材、中药饮片的清洗、浸润和提取应符合生活饮用水卫生标准(GB5749-85); 纯化水:
1、制备注射用水(纯蒸汽)的水源; 2、非无菌药品直接接触药品的设备、器具和包装材料最后一次洗涤用水; 3、注射剂、无菌药品瓶子的初洗; 4、非无菌药品的配料; 5、非无菌药品原料精制应符合中国药典标准; 注射用水: 1、无菌产品直接接触药品的包装材料最后一次精洗用水; 2、注射剂、无菌冲洗剂配料; 3、无菌原料药精制; 4、无菌原料药直接接触无菌原料的包装材料的最后洗涤用水应符合中国药典标准。 纯蒸汽: 1、无菌药品物料、容器、设备、无菌衣或其他物品需进入无菌作业区的湿热无菌处理。 2、培养基的湿热灭菌纯蒸汽冷凝水应符合中国药典注射用水标准。 四、总结 想要制取符合GMP的工艺用水那就少不了符合GMP标准的纯化水设备,也就是GMP纯化水设备,纯化水设备要GMP标准要从刚开始的设计、选材,到后来的制造、安装甚至是纯化水的运输都有着严格的要求,所以想要符合GMP要求的纯化水就要从纯化水设备抓起。 以上资料来自科瑞水处理,如需转载请标明出处!!
ISPE-CCPIE CHINA CONFERENCE 2012
制药用水系统的风险评估与质量控制
张功臣 2012-09-25
September 24-25 2012 Beijing
1
分类
? 制药用水系统是制药厂房设施的重要组成部分, 从风险评估角度,因其介质与药品直接接触,其
对药品的质量有着直接的影响,属于直接影响质 量的关键系统。
液态
制药用水系统需要调试和确认!
纯化水 高纯水
气态
纯蒸汽 无菌氮气 无菌压缩空气
注射用水
无菌氧气
无菌二氧化碳
ISPE-CCPIE CHINA CONFERENCE 2012
系统的“质量”要求
一 满足药典与法规的“质量”要 求
二 满足生产与工艺的“质量”要 求
三 满足投资与运行的“质量”要 求
ISP3E-CCPIE CHINA CONFERENCE 2012
系统的“质量”要求
一 满足药典与法规的“质量”要 求
二 满足生产与工艺的“质量”要 求
三 满足投资与运行的“质量”要 求
ISP4E-CCPIE CHINA CONFERENCE 2012
系统的“质量”要求
一 满足药典与法规的“质量”要 求
? 药典与法规 的质量要求 是什么?
ISP5E-CCPIE CHINA CONFERENCE 2012
药典对于制药用水的规定
? 制药用水的分类:
?原料水--制药生产工艺过程中使用的水。
例如:饮用水;纯化水;高纯水;注射用水; 9 工程上的制药用水特指“原料水” 。
?产品水--按制药工艺生产的包装成品水。
例如:抑菌注射用水;灭菌吸入用水;灭菌注射 用水;灭菌冲洗用水;灭菌纯化水;
ISP6E-CCPIE CHINA CONFERENCE 2012
制药用水系统验证 制药用水系统的验证,是为了证实整个工艺用水系统能够按照设计的目的进行生产和可靠操作的过程。验证工作需要从设计时期就开始,通过监按建筑、使用过程,收集和组织相关的文件资料,最终形成完善的验证文件。 通常,工艺用水系统的验证程序分为三个方面,即确认系统中采纳的所有关键的硬件和软件安装是否符合原定的要求(IQ);确认工艺用水系统中使用的设备或系统的操作是否能够满足原定的要求(OQ);确认工艺用水系统采纳的工艺是否能够按照原定的要求正常的运转(PQ)。 1 验证的预备 在针对一个指定的工艺用水系统,进行验证往常应该做好验证前的预备工作,包括下述使用文件所规定的有关内容。 使用文件是由建筑工艺用水系统的工程公司、设备制造厂、使用者共同制作的。要求这些文件必须以合适的形式组织起来,更便于同意药政治理部门(SDA、FDA等)的检查和批准。系统的使用测试和文件将满足多种资格要求。使用文件包括以下六个方面。 (1)文件清单
①系统内设备,包括设备出厂标签号、生产厂商、样品序号和设备尺寸大小; ②PC/PLC/DOS/WINDOWS输入,输出和警告; ③阀门,包括标签号、位置、类型、尺寸; ④关镀的和非关键的设施,包括标签号、位置、类型、作用/目的、范围和测定日期; ⑤管道,包括节段号、类型、尺寸和完成情况; ⑥滤膜,包括标签号、位置、品种、尺寸、制造用的材料、生产商、型号和孔径大小; ⑦工艺过程和配套公用工程,包括系统名、提供压力、温度和所需电力; ⑧采购、安装合同中所需的原材料; ⑨零部件清单; ⑩标准操作程序(适用于系统设备的操作、维护、测定,运行治理)。 (2)工厂测试程序 ①设备测试程序,测定程序和数据表; ②压力测试,PLC/PC测试; ③安全检查,制动设备的操作测试步骤。
1.适用范围 本标准适用于本公司制药生产用水(饮用水、纯化水)的管理。 2.职责 制水操作人员:严格按SOP进行操作,保证制药用水的质量。 质量部QC:负责制药用水的取样监测。 质量部QA:负责对制药用水的制备及使用进行监控。 3.内容 3.1.定义 3.1.1.饮用水 指经净化、消毒的自来水或深井水;水质必须符合GB5749-2006《生活饮用水卫生标准》。 3.1.1.纯化水 以饮用水为水源经离子交换法、反渗透法或其它适宜方法制备的制药用水,不含任何附加剂;水质必须符合《中国药典》2010年版纯化水质量标准。 3.3.制药用水的管理 3.3.1.饮用水 3.3.1.1.水源:自来水 3.3.1.2.水质维护
饮用水的管道应避免穿过垃圾堆或毒物污染区。 3.3.1.3.饮用水的使用 饮用水使用前,打开水龙头,排掉管内残留水,至清澈后使用。 3.3.1. 4.水质监测 每年至少由防疫站检定全部项目一次; 正常情况每季度由QC依据厂订饮用水质量标准,按《取样SOP》取样抽检一次。 停产7天以上,由QC在开工前,按《取样SOP》在饮用水主管网末梢及关键用水点取样,按厂订饮用水质量标准抽查一次。 发现检验结果不符合要求或异常,立即执行《检验结果超标或超常时的调查处理》、《偏差处理》。 3.3.2.纯化水 3.3.2.1.水源:饮用水。 3.3.2.2.水质维护 纯化水系统必须经过验证合格后,方可投入使用,并进行严格的变更控制。 纯化水系统正常情况每年进行一次再确认,以确认验证状态是否稳定。 纯化水系统操作人员按经确认的规程,进行制水和定期的清洗、消毒及设备维护等操作。 3.3.2.3.水质监测 每2小时由制水操作人员记录一级淡水电导率、二级淡水电导率、总送水口电导率。 每班由制水操作人员在纯化水流速、多介质过滤器出水外观、活性炭过滤器出水外观及余氯、总送水口性状、酸碱度一次。 QC每周在贮罐口、总送水口、总回水口取样,按纯化水质量标准全检一次。纯化水各用水点的抽查,可依次轮换进行,但应确保各用水点每月至少全项检测一次。 当检测指标已接近质量警戒指标时,应由QC室及时直接通知纯化水站对系统进行清洗、消毒,以确保水质符合质量要求。 检验结果不符合要求,立即执行《偏差处理SOP》,由质量部会同相关部门进行调查处理,包括已用于生产的产品、清洗的设备等方面。 3.3.2. 4.纯化水的使用 纯化水用水点应编号管理,并制作用水点编号对应图或表。 使用人员将水龙头打开至整个水龙头口四周均有出水,排水10秒钟后使用。 如使用软管或其它容器,使用前应对软管或容器用75%酒精消毒。 3.4.取样应按《取样SOP》的要求执行。 3.5.水质检验按相应检验操作规程进行检测,合格后出具检验报告。 3.6.本公司规定以质量标准中卫检指标限度的50%作为警戒限,以卫检指标限度的80%作为纠偏限。按
制药用纯化水水系统GMP验证资料 制药用水系统的验证,是为了证实整个工艺用水系统能够按照设计的目的进行生产和可靠操作的过程。验证工作需要从设计阶段就开始,通过监按建造、使用过程,收集和组织相关的文件资料,最终形成完善的验证文件。 通常,工艺用水系统的验证程序分为三个方面,即确认系统中采用的所有关键的硬件和软件安装是否符合原定的要求(IQ);确认工艺用水系统中使用的设备或系统的操作是否能够满足原定的要求(OQ);确认工艺用水系统采用的工艺是否能够按照原定的要求正常的运转(PQ)。 1 验证的准备 在针对一个指定的工艺用水系统,进行验证以前应该做好验证前的准备工作,包括下述使用文件所规定的有关内容。 使用文件是由建造工艺用水系统的工程公司、设备制造厂、使用者共同制作的。要求这些文件必须以合适的形式组织起来,更便于接受药政管理部门(SDA、FDA等)的检查和批准。系统的使用测试和文件将满足多种资格要求。使用文件包括以下六个方面。 (1)文件清单 ①系统内设备,包括设备出厂标签号、生产厂商、样品序号和设备尺寸大小; ②PC/PLC/DOS/WINDOWS输入,输出和警告; ③阀门,包括标签号、位置、类型、尺寸; ④关镀的和非关键的设施,包括标签号、位置、类型、作用/目的、范围和测定日期; ⑤管道,包括节段号、类型、尺寸和完成情况; ⑥滤膜,包括标签号、位置、品种、尺寸、制造用的材料、生产商、型号和孔径大小; ⑦工艺过程和配套公用工程,包括系统名、提供压力、温度和所需电力; ⑧采购、安装合同中所需的原材料; ⑨零部件清单; ⑩标准操作程序(适用于系统设备的操作、维护、测定,运行管理)。 (2)工厂测试程序 ①设备测试程序,测定程序和数据表; ②压力测试,PLC/PC测试; ③安全检查,制动设备的操作测试步骤。 (3)焊接文件 ①焊接管道材料的质量保证书,材料成分报告书; ②焊工证书确认,焊接质量的检查记录; ③焊接设备合格证书,焊接口抽样检查的百分比; ④焊接记录,焊接检查百分比; ⑤焊接程序,焊接检查证书和仓储。 (4)测定文件 测试仪器作为使用和验证的一部分必须进行测定校正。为了区分关键的和不关键的仪器,必须有一个仪器清单。关键测试仪器是那些为了能被药政管理部门接受,直接作用或管理水的质量和纯度的仪器。 关键仪器要在实地操作确认(OQ)前通过可迫溯的方法进行测定。非关键的测试仪器通常也要在OQ前测定。仪器的使用者决定非关键仪器维护的范围。 (5)标难操作规程(SOPs) 为组织验证文件提供一个操作的基本过程,SOPs应该尽早地起草。工艺用水系统的SOPs
一、GMP对制药用水要求 1.药品生产用水应适合其用途,应至少采用饮用水作为制药用水。各类药品生产选用的制药用水应符合《中华人民共和国药典》的相关要求。 2.饮用水应符合国家有关的质量标准,纯化水、注射用水应符合《中华人民共和国药典》的质量标准。 3.水处理设备及其输送系统的设计、安装和维护应能确保制药用水达到设定的质量标准。水处理设备的运行不得超出其设计能力。 4.纯化水、注射用水储罐和输送管道所用材料应无毒、耐腐蚀;储罐的通气口应安装不脱落纤维的疏水性除菌滤器;管道的设计和安装应避免死角、盲管。 5.应对制药用水及水源的水质进行定期监测,并有相应的记录。 6.纯化水、注射用水的制备、储存和分配应能防止微生物的滋生,如注射用水可采用70℃以上保温循环。 7.应按照操作规程定期消毒纯化水、注射用水管道、储罐以及其它必要的辅助管道(如清洁、消毒用的管道、生产用临时连接管道),并有相关记录。操作规程还应详细规定制药用水微生物污染的警戒限度、纠偏限度和应采取的措施 二、GMP对制药纯化水设备系统的要求 1.结构设计应简单、可靠、拆装简便。 2.为便于拆装、更换、清洗零件,执行机构的设计尽量采用的标准化、通用化、系统化零部件。 3.设备内外壁表面,要求光滑平整、无死角,容易清洗、灭菌。零件表面应做镀铬等表面处理,以耐腐蚀,防止生锈。设备外面避免用油漆,以防剥落。 4.制备纯化水设备应采用低碳不锈钢或其他经验证不污染水质的材料。制备纯化水的设备应定期清洗,并对清洗效果验证。 5.注射用水接触的材料必须是优质低碳不锈钢(例如316L不锈钢)或其他经验证不对水质产生污染的材料。制备注射用水的设备应定期清洗,并对清洗效果验证。 6.纯化水储存周期不宜大于24小时,其储罐宜采用不锈钢材料或经验证无毒,耐腐蚀,不渗出污染离子的其他材料制作。保护其通气口应安装不脱落纤维的疏水性除菌滤器。储罐
制药用水技术方案Purified Water and Water for Injection Treatment System 一、概述 水是药物生产中用量最大、使用最广的一种基本原料,用于生产过程及药物制剂的制备,制药用水是制药业的生命线。 随着科学技术的不断进步,有关制药用水的制备技术也发生了革命性的改变。在世界许多发达国家如美国,注射用水(Water for Injection WFI)必须由蒸馏工艺制备这一局限早已被突破,技术更先进、更节能、品质更稳定可靠的高纯水(Highly Purified Water HPW)及其制备工艺早在1975年已经得到正式确认(美国药典第19版:USP19)。现在,美国药典已经在其连续7个版本中明确确认了反渗透(RO)为基础的HPW工艺可以作为制取注射用水的法定工艺,并且,历经数十年的医药实践,HPW注射用水生产技术被证明是最先进、可靠的方法之一,以至于在美国的药物专利25条中,反渗透方法是最常用的注射用水生产工艺。由于HPW符合甚至超过WFI的各项理化参数指标,自2002年6月起正式被欧洲认可为第三水质级别。今天,以RO为基础的HPW已经为代表医药先进技术的世界主要发达国家所确认,成为医用纯化水的标准制备方法之一。 在与国际接轨过程中我国药典亦对医药用水的法定制备方法进行了重新定义。中国药典(2000年版)中所收载的制药用水,较以往有很大进步,因其使用的X围不同而分为纯化水、注射用水及灭菌注射用水,首次将过去的蒸馏水改为纯化水,并且对纯化水具体定义为“纯化水为采用蒸馏法、离子交换法、反渗透法或其它适宜的方法制得供药用的水”,实际上放弃了对生产工艺“必须为蒸馏法”的限定,为相关企业采用国际上广为流行的反渗透HPW方法制备纯化水奠定了法理基础。更为重要的是,新的国家药典将注射用水定义为“纯化水经蒸馏所得的水”,从而使RO技术进入注射用水制备过程成为可能。2000年版国家药典在制约用水技术上朝国际先进领域迈进了一大步。 与传统的蒸馏法相比较,以反渗透法为基础的联合了最新电去离子(EDI)技术的新工艺具有明显的优越性和先进性。 1.高效节能。蒸馏法系历史最为悠久的医药用水制备工艺,主要有多级蒸馏、高压分级蒸馏和离心净化蒸馏几种工艺。所有蒸馏方法均在120°C高温状态下进行,所以可以得到完全无菌的水。因此,运行当中能源的消耗相当大;同时,因为温度较高,所有设备组成部分必须耐受高温冲击,设备的造价及维护费用高昂。HPW工艺采用非常成熟的反渗透技术,结合高效臭氧消毒方法,整个系统工作于常温、低压状态,设备投资省,运行维护费用低,可靠节能:膜处理法的运行成本仅为蒸馏方法的12-15%,非常经济,极具竞争力。 2.稳定可靠。随着工业化进程的不断加快,大量而成份复杂的废物排放使世界X围的污染变得日益严重,其中水资源的污染较之以往更加严峻。易挥发有机污染物因其沸点大都低于水的汽化温度,如不加处理,蒸馏过程中极易进入产成水中,单纯蒸馏方法无法将其有效去除,必须倚重活性碳吸附等过滤办法,增加了系统和水质的不稳定性。膜法工艺采用多介质过滤器进行预处理;反渗透膜的微孔透过式工作原理保证了去除水体中所有较大的离子、分子,可以轻松去除分子直径更大的易挥发有机污染物质,从根本上保证有机物指标达到药典规定指标。 3.先进环保。膜法联合工艺替代传统纯蒸馏方法已经成为当今世界医药用水生产技术的主流。近年来代表制药用水制备工艺最高技术水平的连续电去离子技术(Continuous Electrodeionization CEDI)的出现,促使医药用水制备工艺摒弃伴生废酸、废碱污染的传统离子交换技术,令系统实现全自动计算机控
制药用水系统设备的特殊要求 制药用水系统内选用的设备,其基本特性与非制药用水系统设备的性能并无多大的区别。例如,纯化水用除盐设备的基本作用与其他行业的基本相同。制药用水系统只是在对微生物的控制上,有其特殊的要求,而系统中采用的水处理设备均围绕控制系统内微生物的要求作相应的处理。 1、对纯化水系统设备的特殊要求 对于纯化水系统来说,水处理流程中的微生物控制始终贯穿于整个处理过程。例如,系统中如果采用活性炭过滤装置或软化器,则因为活性炭的吸附作用而拦截在过滤器上流侧的有机物会不断地增多,如果没有相应的除菌措施周期性地对活性炭过滤器进行消毒处理,降低活性炭过滤器上流侧的生物负荷,则经过一段时间的使用后,尽管活性炭过滤器本身的功能(降低余氯量和去除有机物)并没有减小,但由于其上流侧的有机物的堆集,会使活性炭过滤器使用后水中微生物的指标超过处理前的进水指标。又如,纯化水的成品贮罐和配水管路要有定期进行微生物消毒的措施。因此,应该根据工艺用纯化水系统内部所采用的水处理设备的功能和特点,围绕控制和减少微生物的污染作文章。同时还应该根据所选用的消毒方法,恰当地选择设备的制造材料。例如,如果是采用热处理的方法(巴斯德消毒或蒸汽灭菌),则活性炭过滤器或软化器的制造材料应采用耐温的材料,比如不锈钢;而当采用化学消毒剂(臭氧或双氧水)时,则设备的制造材料可以不考虑耐温问题,转而考虑设备耐腐蚀的寿命问题,比如采用玻璃
钢树脂内衬PE。 纯化水设备还应具备无不流动死水段的特性,全部设备都应该具有能够将系统内部余水放空的能力,系统外部的水也不会倒流回系统而产生污染。总之,纯化水处理设备和系统管道均应有防止污染和定期消毒处理、降低生物负荷或恢复至有生物负荷水平的能力。 2、对注射用水系统设备的特殊要求 与纯化水系统的要求类似,并且更为严格。注射用水系统尤其重视微生物指标的控制。注射用水系统中的主要设备为蒸馏水机、贮罐、卫生级输送水泵、阀门和输送管道。对于这些设备或零部件,注射用水系统的特殊要求与纯化水系统相比较近乎于苛刻。主要的原则是控制蒸馏水出水的质量,蒸馏水机能够对自身进行灭菌、以防止蒸馏水机的蒸馏水出水与冷却水可能产生的交叉污染,水泵的卫生管理,系统管道对微生物的滞留和滋生情况,系统用纯蒸汽灭菌等等。 3、对纯蒸汽系统设备的特殊要求 纯蒸汽设备首先应能生产出具有注射用水同样水质指标的纯蒸汽,纯蒸汽的压力在克服管道系统阻力的基础上,能够满足灭菌设备或对系统管道进行湿热灭菌的压力与温度要求。纯蒸汽设备和管道在不使用的时候,能够与大气隔离,不会受到空气中微生物的污染。纯蒸汽设备和蒸馏水机一样,蒸馏器的换热部分应能够防止冷却水泄漏对纯蒸汽产生的污染。
制药用水技术方案 一、概述 水是药物生产中用量最大、使用最广的一种基本原料,用于生产过程及药物制剂的制备,制药用水是制药业的生命线。 随着科学技术的不断进步,有关制药用水的制备技术也发生了革命性的改变。在世界许多发达国家如美国,注射用水(Water for Injection WFI)必须由蒸馏工艺制备这一局限早已被突破,技术更先进、更节能、品质更稳定可靠的高纯水(Highly Purified Water HPW)及其制备工艺早在1975年已经得到正式确认(美国药典第19版:USP19)。现在,美国药典已经在其连续7个版本中明确确认了反渗透(RO)为基础的HPW工艺可以作为制取注射用水的法定工艺,并且,历经数十年的医药实践,HPW注射用水生产技术被证明是最先进、可靠的方法之一,以至于在美国的药物专利25条中,反渗透方法是最常用的注射用水生产工艺。由于HPW符合甚至超过WFI的各项理化参数指标,自2002年6月起正式被欧洲认可为第三水质级别。今天,以RO为基础的HPW已经为代表医药先进技术的世界主要发达国家所确认,成为医用纯化水的标准制备方法之一。 在与国际接轨过程中我国药典亦对医药用水的法定制备方法进行了重新定义。中国药典(2000年版)中所收载的制药用水,较以往有很大进步,因其使用的范围不同而分为纯化水、注射用水及灭菌注射用水,首次将过去的蒸馏水改为纯化水,并且对纯化水具体定义为“纯化水为采用蒸馏法、离子交换法、反渗透法或其它适宜的方法制得供药用的水”,实际上放弃了对生产工艺“必须为蒸馏法”的限定,为相关企业采用国际上广为流行的反渗透HPW方法制备纯化水奠定了法理基础。更为重要的是,新的国家药典将注射用水定义为“纯化水经蒸馏所得的水”,从而使RO技术进入注射用水制备过程成为可能。2000年版国家药典在制约用水技术上朝国际先进领域迈进了一大步。 与传统的蒸馏法相比较,以反渗透法为基础的联合了最新电去离子(EDI)技术的新工艺具有明显的优越性和先进性。 1.高效节能。蒸馏法系历史最为悠久的医药用水制备工艺,主要有多级蒸馏、高压分级蒸馏和离心净化蒸馏几种工艺。所有蒸馏方法均在120°C高温状态下进行,所以可以得到完全无菌的水。因此,运行当中能源的消耗相当大;同时,因为温度较高,所有设备组成部分必须耐受高温冲击,设备的造价及维护费用高昂。HPW工艺采用非常成熟的反渗透技术,结合高效臭氧消毒方法,整个系统工作于常温、低压状态,设备投资省,运行维护费用低,可靠节能:膜处理法的运行成本仅为蒸馏方法的12-15%,非常经济,极具竞争力。
制药用纯化水系统水过G M P认证资料要求 公司标准化编码 [QQX96QT-XQQB89Q8-NQQJ6Q8-MQM9N]
制药用纯化水水系统GMP验证资料 制药用水系统的验证,是为了证实整个工艺用水系统能够按照设计的目的进行生产和可靠操作的过程。验证工作需要从设计阶段就开始,通过监按建造、使用过程,收集和组织相关的文件资料,最终形成完善的验证文件。 通常,工艺用水系统的验证程序分为三个方面,即确认系统中采用的所有关键的硬件和软件安装是否符合原定的要求(IQ);确认工艺用水系统中使用的设备或系统的操作是否能够满足原定的要求(OQ);确认工艺用水系统采用的工艺是否能够按照原定的要求正常的运转(PQ)。 1 验证的准备 在针对一个指定的工艺用水系统,进行验证以前应该做好验证前的准备工作,包括下述使用文件所规定的有关内容。 使用文件是由建造工艺用水系统的工程公司、设备制造厂、使用者共同制作的。要求这些文件必须以合适的形式组织起来,更便于接受药政管理部门(SDA、FDA等)的检查和批准。系统的使用测试和文件将满足多种资格要求。使用文件包括以下六个方面。 (1)文件清单 ①系统内设备,包括设备出厂标签号、生产厂商、样品序号和设备尺寸大小; ②PC/PLC/DOS/WINDOWS输入,输出和警告; ③阀门,包括标签号、位置、类型、尺寸; ④关镀的和非关键的设施,包括标签号、位置、类型、作用/目的、范围和测定日期; ⑤管道,包括节段号、类型、尺寸和完成情况; ⑥滤膜,包括标签号、位置、品种、尺寸、制造用的材料、生产商、型号和孔径大小; ⑦工艺过程和配套公用工程,包括系统名、提供压力、温度和所需电力; ⑧采购、安装合同中所需的原材料; ⑨零部件清单; ⑩标准操作程序(适用于系统设备的操作、维护、测定,运行管理)。 (2)工厂测试程序 ①设备测试程序,测定程序和数据表; ②压力测试,PLC/PC测试; ③安全检查,制动设备的操作测试步骤。 (3)焊接文件 ①焊接管道材料的质量保证书,材料成分报告书; ②焊工证书确认,焊接质量的检查记录; ③焊接设备合格证书,焊接口抽样检查的百分比; ④焊接记录,焊接检查百分比; ⑤焊接程序,焊接检查证书和仓储。 (4)测定文件 测试仪器作为使用和验证的一部分必须进行测定校正。为了区分关键的和不关键的仪器,必须有一个仪器清单。关键测试仪器是那些为了能被药政管理部门接受,直接作用或管理水的质量和纯度的仪器。
中国药典(GMP)制药用水要求详解 制药企业的生产工艺用水,涉及到的是制剂生产过程当中容器清洗、配液及原料药精制纯化等所需要使用的水,此类用水一般分成纯化水和注射用水两大类。中国药典对此两类制药用水的制备工艺有具体的一个要求。对于注射用水,中国药典要求使用蒸馏的方法制备,通常是使用多效蒸馏器。此要求与FDA、UP和JP的要求差别较大,本文在此就不详谈制备方面的差别,下文主要谈一谈中国药典(GMP)对制药用水的各方面要求,尤其对纯化水和注射用水的TOC检测要求. 一、同制药用水的用途差别 1.1纯化水的用途: 1、制备注射用水(纯蒸汽)的水源 2、非无菌药品直接接触药品的设备、器具和包装材料最后一次洗涤用水 3、注射剂、无菌药品瓶子的初洗 4、非无菌药品的配料 5、非无菌药品原料精制 1.2注射用水的的用途 1、无菌产品直接接触药品的包装材料最后一次精洗用水 2、注射剂、无菌冲洗剂配料 3、无菌原料药精制 4、无菌原料药直接接触无菌原料的包装材料的最后洗涤用水 1.3纯蒸汽的用途 1、无菌药品物料、容器、设备、无菌衣或其他物品需进入无菌作业区的湿热无菌处理 2、培养基的湿热灭菌 二、2010年版中国药典(GMP)对注射用水中总有机碳(TOC)的新要求 2.1为什么需要检测总有机碳(TOC) 微生物超标纠正标准是指微生物污染达到某一数值,表明注射用水系统已经偏离了正常运行的条件,应采取纠偏措施,使系统回到正常的运行状态。“热原”通常是由细菌产生的,是那些能致热的微生物的代谢产物,以“细菌内霉素”指标来表示。大多数细菌和许多霉菌都能产生热,致热能力最强的是革兰阴性杆菌的产物。微生物代谢产物中的内毒素是造成热原反应的最主要因素。细菌内毒素耐热性强,其尺寸大小约在1-50μm之间,故可通过一般滤器进入滤液中,但能被活性炭、硅藻土滤器等吸附。热原本身不挥发,但能在蒸馏时被汽化的水滴带入蒸馏水中。总有机碳TOC=TC(总碳)-IC(无机碳)。 总有机碳的指标在一定意义上说明的是对水污染的监控。各种有机污染物,微生物及细菌内毒素经过催化氧化后变成二氧化碳,进而改变水的电导,电导的数据又转换成总有机碳的量。如果总有机碳控制在一个较低的水平上,意味着水中有机物、
制药用水分类及水质标准 水是药物生产中用量最大,使用最广的一种原料,用于生产过程及药物制剂的制备,而且生产过程中的用水量很大,其中工艺用水量占相当比例。水在药品生产中是保证药品质量的关键因素之一,尤其是输液生产中工艺用水显得更为重要。对于一家申报GMP认证的制药企业,其生产厂房所能达到的洁净级别及制药用水所能达到的标准,是制药企业在GMP认证中将要重点检查的两个主要项目。 一、制药用水分类及水质标准 1、制药用水分类 制药用水通常可分为:饮用水、纯化水、注射用水。按《中华人民共和国药典(2000年版)》(以下简称2000中国药典)所收载的制药用水中又另列“杀菌注射用水"一项。它们的含义是: 1.l饮用水(Potable-Water):通常为自来水公司供应的自来水,又称原水。按2000中国药典规定;饮用水不能直接用作制剂和制备或试验用水。 1.2纯化水(Purifide Water):为饮用水经蒸馏法、离子交换法、反渗透法或其他适宜的方法制得的制药用的水,不含任何附加剂。 采用离子交换法、反渗透法、超滤法等非热处理制备的纯化水,一般又称去离子水。 采用特殊设计的蒸馏器用蒸馏法制备的纯化水,一般又称蒸馏水。 纯化水可作为配制普通药物制剂用的溶剂或试验用水,不得用于注射剂的配制。 1.3注射用水(Water for Injection):是以纯化水作为原水,经特殊设计的蒸馏器蒸馏,冷凝冷却后经膜过滤制备而得的水。 目前一般的蒸馏器有多效蒸馏水机和气压式蒸馏水机等。 经蒸馏后的水需再经徽孔过滤方可作注射用水,徽孔过滤膜的孔径应为 ≤0.45μm。 注射用水可作为配制注射剂用的溶剂。 1.4 灭菌注射用水(Sterile Water for Injec-tion):为注射用水依照注射剂生产工艺制备所得的水。 灭菌注射用水用于灭菌粉末的溶剂或注射液的稀释剂。
USP33-制药用水<1231> 制药用水<1231> 引言 在药品、原料药和中间体,药典产品以及分析试剂的加工、制备和生产过程中,水被广泛用作一原料,组分和溶剂。此通用章节的信息提供了有关水的其它信息:未被包括在的水的专论中的属性,可以用以提高水质量的处理技术,以及在选择水源时应考虑最低水质量标准的描述。 此信息章节并未打算替代现有的条例或指导,现有的这些条例或指导涉及USA和国际的(ICH或WHO)的GMP问题,工程指导或其它关于水的条例(FDA、EPA或WHO)指导。其内容有有助于使用者更好地理解制药用水问题以及一些仅针对水的微生物和化学问题。此章节并不是关于制药用水的一个全面综合性的文件。它包括在水的处理、贮存和使用时需要考虑的基本信息点。保证制药用水以及生产符合相适用的政府条例、指南和各种类型的水的药典标准是使用者的职责。 这些水的化学纯度的控制是很重要的,并且是本药典中各论的主要目的。与其它药典产品不同,大批量水专论(纯化水和注射用水)也限制此产品是如何被生产的,因为认为纯化过程的本质与完善程度与(水)最终的纯度直接相关。在这些个论中所列的化学属性应被看作为一组最低的规范要求。对于某些应用来说,可能需要更加严格的规范以保证适合其特定的用途。关于这些水的适当应用的基本指导可以在正文中找到,并且在本章节中给出更进一步的解释。 对于很多水的用途来说,控制其微生物质量是很重要的。由于健康与安全的原因,所有具有药典标准的包装形式的水要求是无菌的,因为这些水的一些预期用途有此要求。USP认为大批量专论水的微生物规范是不适当的,并且未被包括在这些水的专论中。这些水可以被用于不同的用途,一些要求严格的微生物控制,而一些却未要求。对于一给定大批量水来说,所需的微生物规范取决于其用途。对于一些没有相关规范和检测的使用者来说,将不必需负担此难于控制的(水的微生物)属性的单个规范。然而,一些应用可能需要甚至更加严格的微生物控制,以避免微生物的繁殖,而微生物在水的纯化、贮存和分配过程中无处不在。当涉及“有多种用途的”或连续的水供应时,一个微生物规范将是不合适的。典型的,微生物规范用至少用48-72小时产生数据的实验方法来评估。因为制药用水是用连续的过程来生产,并且生产后立即用于产品和生产过程,在得到最后的实验结果之前,水很可能已经被使用了。如果不符合药典规范,那么要求调查对在上次取样的合格试验结果与随后取样的不合格试验
制药用水系统的验证要点 中国药典将制药用水分为饮用水(Drinking Water)、纯化水(Purified Water, PW)和注射用水(Water for Injection,WFI),本文对制药用水系统验证要点进行了总结,以提供快速了解。 在进行水系统验证前,应进行风险评估,并根据风险评估的结果,决定验证活动的深度和广度。 将影响产品质量的关键风险因素作为制药用水系统验证活动的重点,通过适当增加测试频率、延长测试周期或增加测试的挑战性等方式来证实系统的安全性、有效性、可靠性。 设计确认 在施工之前,制药用水系统的设计文件(URS、FDS、DDS等)都应逐一进行检查已确保系统能够完全满足URS及GMP中的所有要求。 设计确认应该持续整个设计阶段,从概念设计到开始采购施工,应该是一个动态的过程。 安装确认 在安装确认中,一般把制药用水的制备系统和储存分配系统分开进行。 安装确认需要的文件 (1)由质量部门批准的安装确认方案; (2)竣工文件包:工艺流程图、管道仪表图、部件清单及参数手册、电路图、材质证书、焊接资料、压力测试清洗钝化记录等; (3)关键仪表的技术参数及校准记录; (4)安装确认中用到的仪表的校准报告; (5)系统操作维护手册; (6)系统调试记录,如FAT和SAT记录。 安装确认的测试项目 (1)竣工版的工艺流程图、管道仪表图或其他图纸的确认; (2)部件的确认; (3)仪器仪表的校准; (4)部件和管路材质和表面光洁度; (5)焊接及其他管路连接方法的文件; (6)管路压力测试、清洗钝化的确认; (7)系统坡度和死角的确认;
(8)公用工程的确认; (9)自控系统的确认。 运行确认 运行确认需要的文件 (1)由质量部门批准的运行确认方案; (2)供应商提供的功能设计说明、系统操作维护手册; (3)系统操作维护标准规程; (4)系统安装确认记录及偏差报告。 运行确认的测试项目 (1)系统标准操作规程的确认; (2)检测仪器的校准; (3)储罐呼吸器确认; (4)自控系统的确认; (5)制备系统单元操作的确认; (6)制备系统的正常运行; (7)储存分配系统的确认。 性能确认 制药用水系统的性能确认一般采用3阶段法,在性能确认过程中制备和储存分配系统不能出现故障和性能偏差: 第1阶段:连续取样2~4周,按照药典检测项目进行全检。目的是证明系统能够持续产生和分配符合要求的纯化水或者注射用水,同时为系统的操作、消毒、维护SOP的更新和批准提供支持。 第2阶段:连续取样2~4周,目的是证明系统在按照相应的SOP操作后能持续生产和分配符合要求的纯化水或者注射用水。对于熟知的系统设计,可适当减少取样次数和检测项目。 第3阶段:根据已批准的SOP对纯化水或者注射用水系统进行日常监控。测试从第1阶段开始持续1年,从而证明系统长期的可靠性能,以评估季节变化对水质的影响。
、GMP对制药用水要求 1.药品生产用水应适合其用途,应至少采用饮用水作为制药用水。各类药品生产选用的制药用水应符合《中华人民共和国药典》的相关要求。 2.饮用水应符合国家有关的质量标准,纯化水、注射用水应符合《中华人民共和国药典》 的质量标准。 3.水处理设备及其输送系统的设计、安装和维护应能确保制药用水达到设定的质量标准。水处理设备的运行不得超出其设计能力。 4.纯化水、注射用水储罐和输送管道所用材料应无毒、耐腐蚀;储罐的通气口应安装不脱落纤维的疏水性除菌滤器;管道的设计和安装应避免死角、盲管。 5.应对制药用水及水源的水质进行定期监测,并有相应的记录。 6.纯化水、注射用水的制备、储存和分配应能防止微生物的滋生,如注射用水可采用 70 C以上保温循环。 7.应按照操作规程定期消毒纯化水、注射用水管道、储罐以及其它必要的辅助管道(如清洁、消毒用的管道、生产用临时连接管道),并有相关记录。操作规程还应详细规定制药用水微生物污染的警戒限度、纠偏限度和应采取的措施 二、GMP对制药纯化水设备系统的要求 1.结构设计应简单、可靠、拆装简便。 2.为便于拆装、更换、清洗零件,执行机构的设计尽量采用的标准化、通用化、系统化零部件。 3.设备内外壁表面,要求光滑平整、无死角,容易清洗、灭菌。零件表面应做镀铬等表面处理,以耐腐蚀,防止生锈。设备外面避免用油漆,以防剥落。 4.制备纯化水设备应采用低碳不锈钢或其他经验证不污染水质的材料。制备纯化水的设 备应定期清洗,并对清洗效果验证。 5.注射用水接触的材料必须是优质低碳不锈钢(例如316L不锈钢)或其他经验证不对 水质产生污染的材料。制备注射用水的设备应定期清洗,并对清洗效果验证。 6.纯化水储存周期不宜大于24小时,其储罐宜采用不锈钢材料或经验证无毒,耐腐蚀,不渗出污染离子的其他材料制作。保护其通气口应安装不脱落纤维的疏水性除菌滤器。储罐