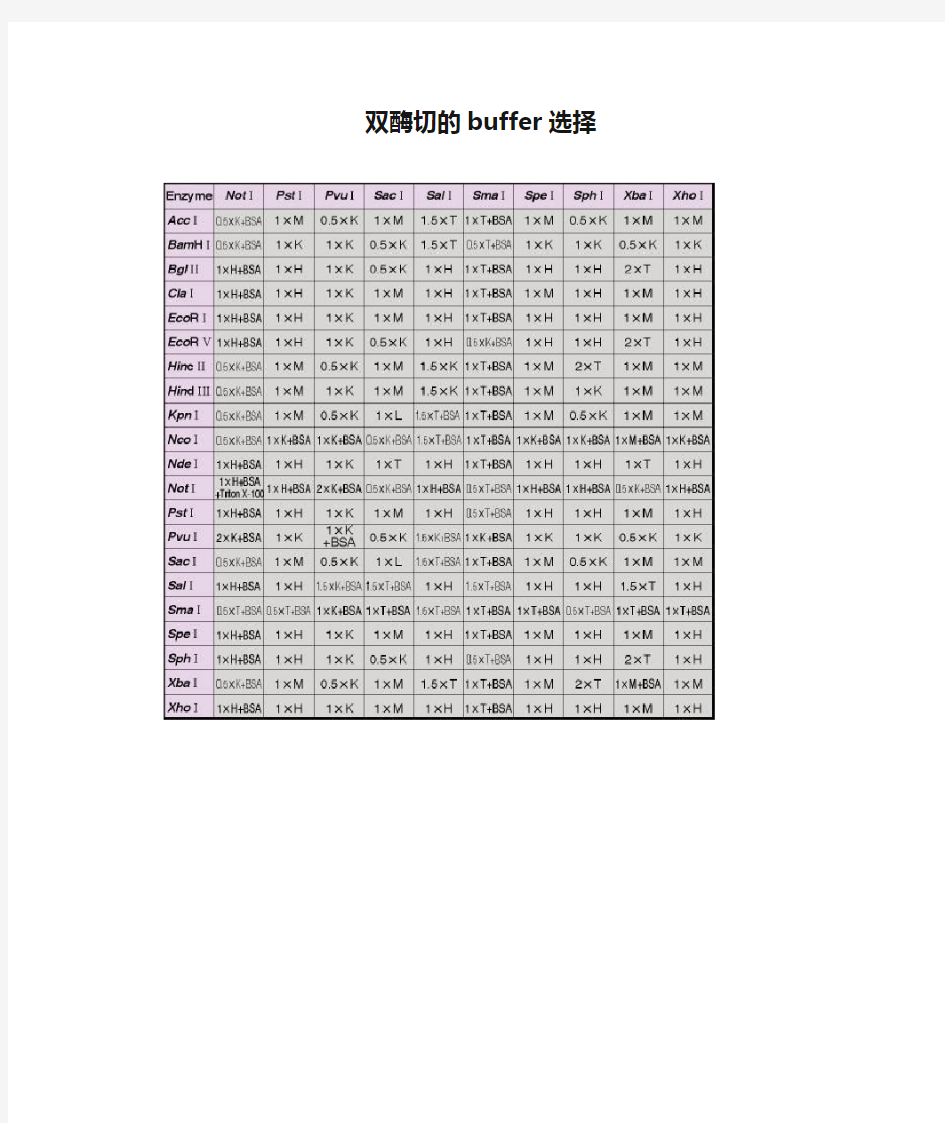
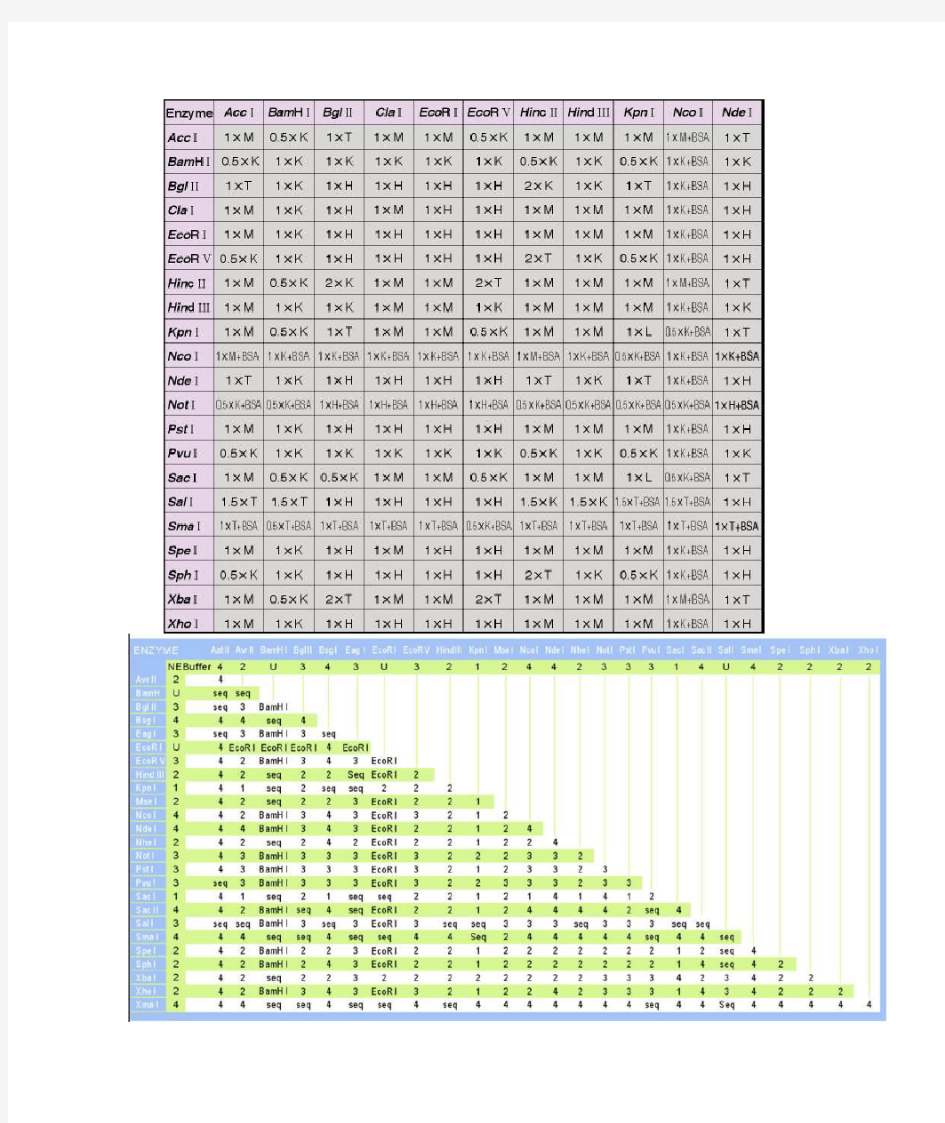
双酶切的buffer选择
注:
只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。
注意将甘油的终浓度控制在10%以下,以避免出现星号活性,可通过增加反应体系的总体积的方法实现这一要求。
某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以
"seq"标注。
双酶切概述 双酶切反应(Double Digests) 1、同步双酶切 同步双酶切是一种省时省力的常用方法。选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。NEBuffer的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。 2、分步酶切 如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。 3、使用配有特殊缓冲液的酶进行双酶切(图) 使用配有特殊缓冲液的酶进行双酶切也不复杂。在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。这保证了对缓冲液有特殊要求的酶也能良好工作。由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。 双酶切建议缓冲液 注: 只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。 注意将甘油的终浓度控制在10%以下,以避免出现星号活性,详见《星号活性》。可通过增加反应体系的总体积的方法实现这一要求。 某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以“se q”标注。 [编辑本段] 双酶切的注意事项 1、做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般连接3小时,16度。 2、对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干。 3、对照的设立:为验证双酶切是否成功,可做如下对照: 酶切反应时加各单酶分别切,两管,用同一种BUFFER,跑胶,看单切的两管是否成线性.如两管均成线性可初步判断双酶切成功.做转化时,也要进行对照。 [编辑本段] 双酶切连接反应之全攻略 1、回收PCR产物:
前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了14个质粒。现就自己的体会,谈一下质粒重组的一些个人经验。 1. 回收PCR产物: 在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶。选好酶切位点后,在各个酶的两边加上保护碱基。 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 2. 纯化问题: 纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。 3. 酶量的问题: 对1单位酶的定义如下:在50μl 反应液中,30℃温度下反应1小时,将1μg 的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml 菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。 4. 酶切、回收后的PCR产物与载体的连接: 摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PC R产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。
pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000(注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套。1pmol 1000bp DNA=0.66μg,如载体是5380b p,则0.03pmol为0.03×5.38×0.66=0.106524μg。 5. 测DNA浓度: 测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的MARKER每个条带约50ng。 6. 连接反应: TAKARA的连接酶上的说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λD NA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNa段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。 7.转化: ①全量(10 μl)加入至100μl JM109感受态细胞中,冰中放置30分钟。 ②42℃加热45秒钟后,再在冰中放置1分钟。 ③加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl 几个非常重要的问题: 1 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解。为保险起见,一般连接3小时,16度。
双酶切连接反应之全攻略 1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照: 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒 酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。 而该酶浓度约为15单位/ 微升,在除外酶降解的 因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的 DNA约 为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的 质量好,酶切完全切得动。 2、酶切、回收后的PCR产物与载体的连接 摩尔比的计算: 很多人凭经验也可以。但对于初学者从头认真计算则 非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03pmol,后者取0.3pmol。 pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为 0.03×5.38×0.66=0.106524μg。 测DNA浓度: 可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER 进行估测,如MARKER2000,5微升的 MARKER每个条带约50ng。 连接反应:TAKARA的 连接酶上的 说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段 被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连 接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。 3、转化: a、全量(10 μl)加入至100 μl JM109感受态细胞中,冰中放置30分钟。 b、42℃加热45秒钟后,再在冰中放置1分钟。 c、加入890 μl AMP阴性培养基,37℃振荡培养60分钟。 取100μl铺板。也可离心后余100μl 几个非常重要的问题 1 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般 连接3小时,16度.
【原创】双酶切连接反应之全攻略(原创) 双酶切连接反应之全攻略 前一阵子一直在做双酶切质粒重组,失败了很多次,不过很快改善了实验方法,用2周重组了 14个质粒。现就自己的体会,结合战友的宝贵经验,谈一下质粒重组的一些个人经验。 1、回收PCR产物:在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基,其原则可参照:https://www.doczj.com/doc/977918948.html,/upload/2006/08/13/31219184.pdf。 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。我用的是TAKARA的纯化柱试剂盒 酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml菌液提出的 DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。 2、酶切、回收后的PCR产物与载体的连接 摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PCR产物片段=1:10 ,一般取前者0.03pmol,后者取0.3pmol。 pmol为单位的DNA转换为为μg单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为μg,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为 0.03×5.38×0.66=0.106524μg。 测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的 MARKER每个条带约50ng。 连接反应:TAKARA的连接酶上的说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。
双酶切编辑 做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解。为保险起见,一般连接3小时,16度;对含有AMP-RESISTENCE的质粒铺板时,注意加AMP 时的温度,温度过高,会使克隆株无法筛选出来。我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干;对照的设立:为验证双酶切是否成功。 目录1简介 2连接反应 3注意事项 1简介编辑双酶切反应(Double Digests) 1、同步双酶切 同步双酶切是一种省时省力的常用方法。选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。NEBuffer 的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。 2、分步酶切 如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。 3、使用配有特殊缓冲液的酶进行双酶切(图) 使用配有特殊缓冲液的酶进行双酶切也不复杂。在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。这保证了对缓冲液有特殊要求的酶也能良好工作。由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。 双酶切建议缓冲液 注: 只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。 注意将甘油的终浓度控制在10%以下,以避免出现星号活性,详见《星号活性》。可通过增加反应体系的总体积的方法实现这一要求。 某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以“seq”标注。 2连接反应编辑1、回收PCR产物: 在进行PCR扩增时候,给引物两端设计好酶切位点,一般说来,限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,如BamHI,HindIII,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基。 双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,我一般酶切3个小时,其实1个小时已经足够。应用大体系,如100微升。 纯化问题:纯化PCR产物割胶还是柱式,我推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基
③双酶切处理目的片段和带有GFP的质粒并纯化 一、实验原理 1. 核酸限制性内切酶是在原核生物中发现的一类专一识别双链DNA中特定 碱基序列的核酸水解酶,他们的功能类似动物的免疫系统,用于抗击外来DNA的侵袭。现已发现几百种限制性内切酶,他们以内切方式水解核酸链中的磷酸二酯键,产生的DNA片段5’端为P,3’端为OH,由于限制性内切酶能识别DNA特异序列并进行切割,因而在基因重组、DNA序列分析、基因组甲基化分析、基因物理图谱绘制及分子克隆等技术上受到广泛应用。在酶切反应中,DNA的纯度、缓冲液中的离子强度、Mg2+等因素均可影响反应,一般可通过增加酶的用量,延长反应时间等措施以达到完全酶切。 2.DNA的酶切反应 II型限制性内切酶能识别双链DNA内部的特殊序列并在识别位点处将双链切断,形成粘性末端或齐平末端,通过电用酶切后的DNA混合物能够确认和分离酶切片段。 3.DNA的连接 在T4 DNA连接酶的作用下,平端或带有相同粘末端的DNA分子可以连接上。DNA连接酶的作分三步: ①T4 DNA连接酶与辅助因子ATP形成酶-AMP复合物。 ②酶-AMP复合物再结合到具有5’-磷酸基和3’-羟基切口的DNA分子上,使DNA腺苷 化 ③产生一个新的磷酸二酯键,把缺口封起来。 二、实验器材与处理方法(参照) 1、限制性内切酶酶BUFFER GFP质粒 2、T4 DNA连接酶连接酶缓冲液 3、电泳缓冲液(0.5×TBE或1×TAE)10×电泳加样缓冲液 4、溴酚蓝琼脂糖溴化乙锭(工作浓度0.5ug/ml)EcoR I 5、酚氯仿无水乙醇70%乙醇灭菌双蒸水 6、1.5ml离心管装入铝制饭盒(灭菌)、移液器吸头装入相应的吸头盒(灭 菌)电泳仪电泳槽紫外检测仪摄影设备 7、恒温水浴槽 三、实验步骤 先将步骤②扩增的目的片段进行纯化(柱层析或电泳割胶法) 1.酶切反应(按情况加大反应体系) 取GFP质粒(DNA)样品于合适的离心管中,按照表1加入试剂 ↓ 混匀;4000rpm离心30s,甩至管底 ↓ 37℃(限制性内切酶最适水温),保温1~3h酶切(酶切过夜则建议用稍大的酶切体积,以避免水分蒸发过多酶切体系各组分浓度改变过大,记得根据不同酶切位点加保护碱基)
酶切 本实验室条件下酶切连接经验之谈 1、PCR产物可以切胶回收后酶切,用Elution buffer溶解即可,不影响后续实验。 2、50ulPCR产物切胶回收后用35ul Elution buffer溶解后只需取一半体积用于后续实验即可 满足要求。 3、PCR产物可以用乙醇沉淀法获得DNA用于后续反应,但需取少于一半的量用于后续实验, 否则会不能完全切开而导致实验失败。 4、双酶切时,若2种酶不是同一厂家时,可以根据Thermo厂家该2种酶的共同buffer, 选用Tango缓冲液,一般50ul体系各加1ul酶,而快切酶只需0.5ul,切3小时即可。 5、添加酶切试剂时,应先将buffer和样品振荡均匀后再加入相应的酶,轻弹混匀即可。 6、观察酶切后的载体片段会比质粒大很多,PCR产物双酶切后的片段也比未酶切时要大, 并且酶切后产物有时会呈现稍微弥散的宽带,由此可以判断是否切开。 7、部分限制性内切酶对甲基化的DNA不能切割,如FbaI和MboI等,一般生物公司提供的内切酶说明中均有说明。 大多数酶切位点的甲基化不影响切割,而有些会影响,如XbaI, BclI等。而且甲基化只发生在特定序列,以XbaI为例,只有在位点序列旁出现GA或TC,该XbaI位才会被甲基化。 而要解除这种限制修饰作用通常有两种方法: (1)选用上述酶的同功酶,如Sau3AI,DNA识别切割位点与MboI相同;但不受甲基化影响; (2)利用甲基化酶缺失的受体细胞进行DNA的制备,如E.coli JM110和链霉菌等,前者Dam和Dcm甲基化酶已敲出,而后者细胞内本就没有甲基化酶,从这些细胞中抽提的DNA就能被上述酶切割。 8:E.coli JM110 要排除dam,dcm甲基化的影响,需要用特定的dam-,dcm-的菌株,如JM110 如果由JM110或SCS110等甲基化缺失的菌株产生的质粒,则不会被甲基化. 若酶切不成功可以考虑以下因素的影响 a)有些内切酶对PCR产物酶切效率较低 b)双酶切无共同buffer时,可以采用分步酶切 c)PCR产物直接双酶切不成功,可以选择先做TA克隆后再双酶切 d)当载体的2个酶切位点很接近,或者其中一个酶切效率很差时,可以对载体进行去磷酸 化,该酶为牛小肠碱性磷酸酶,在大多数限制酶缓冲液中均有活性 e)导致“星星活性”可能是体系中甘油浓度过高、高PH、较低的离子强度所致。 连接 1、一定要选择未经过反复冻融的连接buffer,并准确加入,确保其终浓度准确。 2、目的片段和载体比例不是影响连接成功与否的关键原因,依据经验,取相等质量的的目 的片段和载体即可,载体可以尽量少加一点。3ug 载体(5kb)相当于2pmol 线性DNA 或者4pmol双酶切产物。 3、多个片段连接时,体系中,终浓度的各个片段与载体相同即可。
双酶切连接反应的注意要点 双酶切: 1、在双酶切载体时如果2个酶切位点靠得很近,必须注意酶切顺序。因为有的限制性内切酶要求其识别序列的两端至少保留有若干个碱基才能保证酶的有效切割。有的酶要求识别序列两端有多个碱基的,则必须先切,否则就可能造成酶切失败。 2、回收PCR产物:回收的PCR产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。pmol为单位的DNA转换为为?g单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为?g,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为0.03×5.38×0.66=0.106524?g。 3、双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,一般酶切3个小时,对于PCR产物,可以过夜酶切,效果会很好。酶切体系不宜过大,会影响质粒和酶的碰撞机会,效果降低;质粒量不应该超过酶切要求的最大量,否则酶切不完全,酶的用量控制在1U酶在15-20ul体系中酶解1ugDNA。 4、两种酶切的条件不同时,分别进行两次酶切,切完一个纯化后再切:温度要求不同,先酶切低温要求的,再酶切高温要求的;若盐浓度要求不同,先酶切低盐浓度要求的,再酶切高盐浓度要求的。 5、若质粒是在TE中保存的,TE 中的EDTA可能与酶的激活因子螯合,影响酶切效果,可放大酶切体积或重新浓缩质粒。 6、限制酶的选择非常重要,尽量选择粘端酶切和那些酶切效率高的限制酶,提前看好各公司的双切酶所用公用的BUFFER,以及各酶在公用BUFFER里的效率。选好酶切位点后,在各个酶的两边加上保护碱基。 7、纯化问题:纯化PCR产物割胶还是柱式,推荐柱式,因为割胶手法不准,很容易割下大块的胶,影响纯化效率。现在的柱式纯化号称可以祛除引物,既然如此,酶切掉的几个碱基肯定也会被纯化掉了。所以,PCR产物和双酶切产物的纯化均可应用柱式纯化。 8、酶量的问题:以TAKARA的为例,其对1单位酶的定义如下:在50 μl 反应液中,30℃温度下反应1小时,将1 μg 的λDNA完全分解的酶量定义为1个活性单位(U)。而该酶浓度约为15单位/微升,在除外酶降解的因素外,该酶可分解15μg的DNA,而一般从1-4ml 菌液提出的DNA约为3μg,而PCR纯化后的产物(50体系)约为3μg,所以即便全部加进去,只要纯化的质量好,酶切完全切得动。 9、酶切、回收后的PCR产物与载体的连接:摩尔比的计算,很多人凭经验也可以。但对于初学者从头认真计算则非常有必要。回收的载体片段:回收的PCR产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。pmol为单位的DNA转换为为?g单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为?g,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol 为0.03×5.38×0.66=0.106524?g。测DNA浓度可以在专用机子上测,注意OD值,一般约1.8-2.0.另外,如果嫌麻烦,也可用MARKER进行估测,如MARKER2000,5微升的MARKER 每个条带约50ng。 10、连接反应:TAKARA的连接酶上的说明写的过夜,而其对连接酶单位的定义为:在20 μl的连接反应体系中,6 μg的λDNA-Hind III的分解物在16℃下反应30分钟时,有90%以上的DNA片段被连接所需要的酶量定义为1个活性单位(U)。而它的浓度为350 U/μl ,所以完全够用。连接酶容易失活,注意低温操作,最好在冰上。时间3个小时足已。
双酶切buffer的选择: 1、U :Supplied with its own unique reaction buffer that is different from the four standard NEBuffers. Its compatibility with the four standard NEBuffers is indicated by the chart. 2、BSA :Supplied with a separate vial of bovine serum albumin (10 mg/ml). To obtain 100% activity BSA should be added to the 1X reaction mix to a final concentration of 100 μg/ml. 3、SAM :Supplied with a separate vial of S-adenosyl-methionine (SAM). To obtain 100% activity, SAM should be added to the 1X reaction mix as specified on the product data card. 4、dd :When performing a double digest with this enzyme, this NEBuffer is recommended because it minimizes star activity. Purified by scientists at SibEnzyme and supplied with a SibEnzyme buffer (B, K, O, W or Y) which ensures 100% activity. Its compatibility with the NEBuffer System is indicated on the chart. 5、NR :This buffer is not recommended for use with with this enzyme。 6、Buffer Compositions :(1X): NEBuffer 1 (yellow): 10 mM Bis Tris Propane-HCl, 10 mM MgCl2, 1 mM DTT (pH 7.0 at 25℃). NEBuffer 2 (blue): 10 mM Tris-HCl, 10 mM MgCl2, 50 mM NaCl, 1 mM DTT (pH 7.9 at 25℃). NEBuffer 3 (red): 50 mM Tris-HCl, 10 mM MgCl2, 100 mM NaCl, 1 mM DTT (pH 7.9 at 25℃). NEBuffer 4 (green): 20 mM Tris-acetate, 10 mM magnesium acetate, 50 mM potassium acetate, 1 mM DTT (pH 7.9 at 25℃). 双酶切连接反应的经验谈: 双酶切: 1、在双酶切载体时如果2个酶切位点靠得很近,必须注意酶切顺序。因为有的限制性内切酶要求其识别序列的两端至少保留有若干个碱基才能保证酶的有效切割。有的酶要求识别序列两端有多个碱基的,则必须先切,否则就可能造成酶切失败。 2、回收PCR产物:回收的PCR产物片段=1:10,一般取前者0.03pmol,后者取0.3pmol。pmol为单位的DNA转换为为?g单位的DNA:(X pmoles×长度bp×650)/ 1,000,000 (注:长度bp×650是该双链DNA的分子量)所得数值即为?g,也可以直接用这个公式套.1pmol 1000bp DNA=0.66μg,如载体是5380bp,则0.03pmol为0.03×5.38×0.66=0.106524μg。 3、双酶切时间及其体系:需要强调的是很多人建议酶切过夜,其实完全没有必要,一般酶切3个小时,对于PCR产物,可以过夜酶切,效果会很好。酶切体系不宜过大,会影响质粒和酶的碰撞机会,效果降低;质粒量不应该超过酶切要求的最大量,否则酶切不完全,酶的用量控制在1U酶在15-20ul体系中酶解1ugDNA。
做转化的时候,进行酶连接反应时,注意保持低温状态,因为LIGASE酶很容易降解.为保险起见,一般连接3小时,16度;对含有AMP-RESISTENCE的质粒铺板时,注意加AMP时的温度,温度过高,会使克隆株无法筛选出来.我的方法是培基高温消毒后放在烤箱里,烤箱一般温度为55-60度,然后做的时候拿出来,这样好掌握温度。铺板前后注意用吹风机吹干;对照的设立:为验证双酶切是否成功。 1、同步双酶切 同步双酶切是一种省时省力的常用方法。选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。NEBuffer的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。 2、分步酶切 如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。 3、使用配有特殊缓冲液的酶进行双酶切 如图 使用配有特殊缓冲液的酶进行双酶切也不复杂。在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。这保证了对缓冲液有特殊要求的酶也能良好工作。由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。 双酶切建议缓冲液 注: 只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA 不会影响任何内切酶的活性。
双酶切系统(DDDesigner)的具体应用实例 DDDesigner group 1. 避免浪费限制性内切酶 双酶切系统能够避免限制性内切酶的浪费,因为很多限制性内切 酶在实际应用中所需的用量极少。如: 利用NEB公司的PstI-HF和EcoRV-HF两种酶消化长度为3000bp的1ug质粒DNA,当这两种酶在质粒上各只有一个切点时,完全消化质粒DNA所需要的PstI-HF仅为1.2U,需要的EcoRV-HF也仅为1.5U。 由此可见,如果按照该酶所附的使用说明用1ul酶(10或者 20IU)消化1ug质粒DNA,你将浪费了大约5-10倍的酶。这种浪费在一些特别贵的酶上面可能体现得更明显。
2.确保质粒载体的完全酶切,降低载体自连而出现的背景克隆 由于单位定义时底物选择的不同,以及酶在底物上识别位点的数 目的差别,导致在实际工作中,切割相同的质粒DNA时,不同的酶所需要的酶量(单位数)有很大的差别,有时能够相差近30倍。如: 利用NEB公司的PstI-HF和NheI-HF两种酶消化长度为3000bp的 1ug质粒DNA,当这两种酶在质粒上各只有一个切点时,完全消化质粒DNA所需要的PstI-HF仅为1.2U,需要的NheI-HF则高达32U,两种酶的用量相差近30倍。 因而,如果按照该酶所附的使用说明用1ul酶(10或者20IU)消化1ug质粒DNA,那么PstI-HF会出现浪费,但同时,NheI-HF的 用量则不能保证质粒DNA的完全酶切。此种情况下,在进行载体和外源片段的连接时,就会出现大量的载体自连,导致阳性克隆率低,或 者克隆失败。
3.确保PCR产物的完全酶切,提高克隆成功率 对同样质量(如1ug)的不同DNA样品,长度短(分子量小)的DNA样品中的分子数多,因而如果这些DNA样品分子上的酶切位点数是一样的,那么完全消化较短的DNA分子所需要的酶量将会比完全酶切较大的质粒DNA多。如: 用Takara的EcoRI和XbaI完全酶切1ug纯化的短PCR产物(300bp),且此两种酶在该分子上各有一个识别位点,那么需要大约64U的EcoRI和323U的XbaI。 因而,如果按照该酶所附的使用说明用1ul酶(10或者15IU)消化1ug DNA,那么就会导致大量的PCR产物酶切不完全,进而无法成功克隆至载体上,导致克隆失败。这个现象也提示:在进行较短的PCR产物酶切时,可以DNA的起始量,一般0.3—0.5ug就能满总克隆的需求,从而及可以节省酶的用量,也可以保证完全酶切。
Select 1st enzyme: Select 2nd Double Digests Digesting a DNA substrate with two restriction endonucleases simultaneously (double digestion)provide reaction conditions that optimize enzyme activity as well as avoid star activity associate supplied with its optimal NEBuffer to ensure 100% activity. NEBuffer compositions are listed on b Chart for Restriction Enzymes rates the percentage activity of each restriction endonuclease in t Please use NEB Double Digest Finder to perform a double digest calculation. Suggested NEBuffers for Double Digestion Enzyme AatII A vrII B amHI B glII B sgI EagI EcoRI E coRV H indIII K pnI M seI NcoI NdeI N
NEBuffer 4 4 3 3 4 3 EcoRI 3 2 1 4 3 4 AatII 4 -- 4 seq seq 4 seq seq 4 4 4 4 4 4 AvrII 4 4 -- 3 2 4 3 EcoRI 2 2 1 4 4 4 BamHI 3 seq 3 -- 3 3 3 EcoRI 3 seq seq 3 3 3 BglII 3 seq 2 3 -- 3 3 EcoRI 3 2 2 2 3 3 BsgI 4 4 4 3 3 -- seq seq 4 2 seq 4 4 4 EagI 3 seq 3 3 3 seq -- EcoRI 3 seq seq 3 3 3 EcoRI EcoRI seq EcoRI EcoRI EcoRI seq EcoRI -- EcoRI seq 1 EcoRI E coRI E coRI EcoRV 3 4 2 3 3 4 3 EcoRI -- 2 2 2 3 2 HindIII 2 4 2 seq 2 2 seq seq 2 -- 2 2 2 2 KpnI 1 4 1 seq 2 seq seq 1 2 2 -- 1 1 1 MseI 4 4 4 3 2 4 3 EcoRI 2 2 1 -- 4 4 NcoI 3 4 4 3 3 4 3 EcoRI 3 2 1 4 -- 4 NdeI 4 4 4 3 3 4 3 EcoRI 2 2 1 4 4 -- NheI 2 4 2 seq 2 4 seq 1 2 2 1 2 2 4 NotI 3 seq 3 3 3 3 3 EcoRI 3 2 2 2 3 3 PstI 3 4 3 3 3 3 3 EcoRI 3 2 1 3 3 3 PvuI 3 seq 2 3 3 3 3 EcoRI 3 2 2 3 3 3 SacI 1 4 1 seq 2 4 seq 1 2 2 1 4 1 4 SacII 4 4 4 seq 2 4 seq EcoRI 2 2 4 4 4 4 SalI 3 seq 3 3 3 3 3 EcoRI 3 seq seq 3 3 3 SmaI 4 4 4 seq seq 4 seq seq 4 4 seq 4 4 4 SpeI 4 4 4 seq 2 4 seq EcoRI 2 2 1 4 4 4 SphI 2 4 2 3 2 4 3 EcoRI 2 2 1 2 2 2 XbaI 4 4 4 3 2 4 3 seq 2 2 2 4 4 4 XhoI 4 4 4 3 3 4 3 EcoRI 3 2 1 4 4 4 XmaI 4 4 4 seq 2 4 seq seq 4 seq 4 4 4 4 Note: Enzymes in Dark Red are available in High Fidelity (HF) Format. HF Enzymes have been en 4 which may simplify your double digest. Click for more HF information. Setting up a Double Digestion ?Choose an NEBuffer that results in the most activity for both enzymes. If star activity is a using one of our High Fidelity (HF) enzymes. ?If BSA is required for either enzyme, add it to the double digest reaction (BSA does not inh enzyme). ?Set up reaction according to recommended conditions. The final concentration of glycero should be less than 5% to minimize the possibility of star activity. For example, in a 50 μl re amount of enzyme added should not exceed 5 μl. ?Incubate at recommended temperature. Overnight double digests should be avoide possibility of star activity.
Home > Tec hnic al Refer enc e > Restriction Endonucleases > NEBuffer A ctivity Chart NEBuffer Activity Chart for Restriction Enzymes For your c onvenienc e, N ew England Biolabs offers a simple 4 buffer system. A c olor-c oded 10X NEBuffer is supplied with every r e endonuclease, ensuring 100% activity. Over 170 r estriction enzymes exhibit 100% activity in NEBuffer 4, r esulting in increase d eff and ease of use especially when per for ming double digests. To help select the best c onditions for double digests, this c hart shows the optimal (supplied) NEBuffer and appr oximate activity in NEBuffer s and NEBuffer Ec oRI for eac h enzyme. If BSA is supplied with an enzyme, it is inc luded in all NEBuffer activity r eac tions table shows r ecommended reaction temperatur e, heat inactivation temper atur e, r ec ommended diluent buffer and whether the enzym qualified (i.e., cleaves substr ate in 5 m inutes under r ec ommended c onditions). N ote: The values listed in this table ar e appr oxima obtained using each enzyme's specific unit assay substr ate DNA. Please chec k out other technic al r efer enc e infor mation r elated to r estriction enzymes: Double Digestion | Heat Inactivation | Activity at 37°C | Diluent Buffer s | Time-Saver Enzymes | High Fidelity (HF) Restriction Enz Click for the Chart Legend, Ic on Description A | B | C | D | E | F | H | I | K | L | M | N | P | R | S | T | X | Z | NEBuffer 4 NEBuffer 3 NEBuffer 4 NEBuffer 3 NEBuffer 4 NEBuffer 4 NEBuffer 4 NEBuffer 4 NEBuffer 3 NEBuffer 1 NEBuffer 4 - NEBuffer 4
四、双酶切鉴定 ㈠双酶切反应(Double Digests) 1、同步双酶切 同步双酶切是一种省时省力的常用方法。选择能让两种酶同时作用的最佳缓冲液是非常重要的一步。NEB每一种酶都随酶提供相应的最佳NEBuffer,以保证100%的酶活性。NEBuffer的组成及内切酶在不同缓冲液中的活性见《内切酶在不同缓冲液里的活性表》及每支酶的说明书。能在最大程度上保证两种酶活性的缓冲液即可用于双酶切。由于内切酶在非最佳缓冲液条件下的切割速率会减缓,因此使用时可根据每种酶在非最优缓冲液中的具体活性相应调整酶量和反应时间。 2、分步酶切 如果找不到一种可以同时适合两种酶的缓冲液,就只能采用分步酶切。分步酶切应从反应要求盐浓度低的酶开始,酶切完毕后再调整盐浓度直至满足第二种酶的要求,然后加入第二种酶完成双酶切反应。 3、使用配有特殊缓冲液的酶进行双酶切(图) 使用配有特殊缓冲液的酶进行双酶切也不复杂。在大多数情况下,采用标准缓冲液的酶也能在这些特殊缓冲液中进行酶切。这保证了对缓冲液有特殊要求的酶也能良好工作。由于内切酶在非最佳缓冲液中进行酶切反应时,反应速度会减缓,因此需要增加酶量或延长反应时间。通过《内切酶在不同缓冲液里的活性表》可查看第二种酶在特殊缓冲液相应盐浓度下的作用活性。 双酶切建议缓冲液 注: 只要其中一种酶需要添加BSA,则应在双酶切反应体系中加入BSA。BSA不会影响任何内切酶的活性。 注意将甘油的终浓度控制在10%以下,以避免出现星号活性,详见《星号活性》。可通过增加反应体系的总体积的方法实现这一要求。 某些内切酶的组合不能采用同步双酶切法,只能采用分步法进行双酶切。上表中这些组合以“seq”标注。 ㈡连接反应