
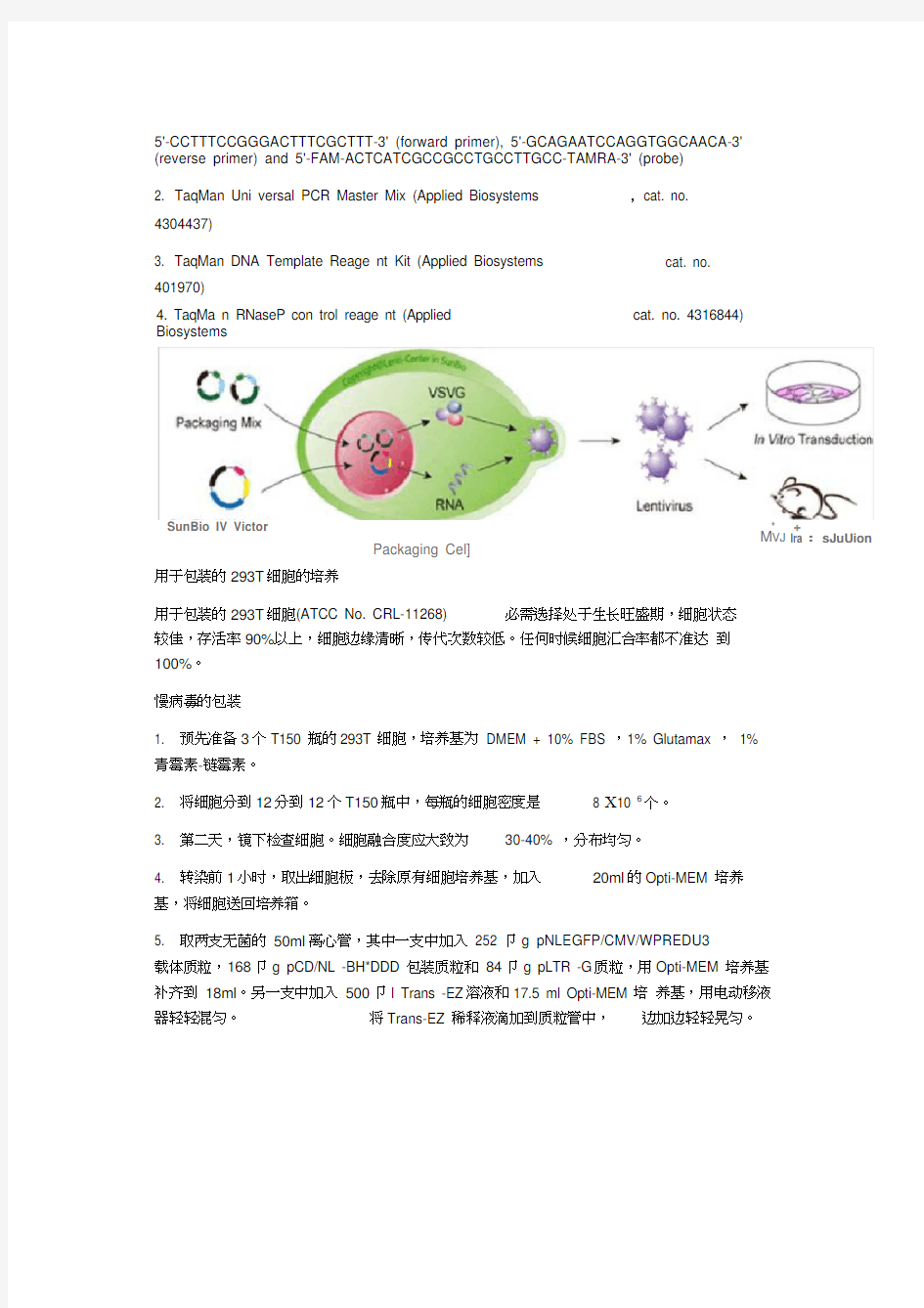
、包装细胞293T 细胞的培养
一、293T 细胞的冻存
1. 随着传代的次数增加,293T 细胞会出现生长状态下降,出现突变等。所以要在细胞购进时就进行冻存。
2. 在细胞对数生长期进行冻存,增加细胞复苏成活率。
3. 倒去细胞上清液,加入D-Hank's 液洗去残留的培养基。
4. 加入0.25% 的胰酶,消化10-20s 后倒去。
5. 镜下观察细胞变圆,细胞间间隙加大时,加入新鲜培养基吹打混匀。
6. 细胞计数。
7. 将细胞离心,1000rpm ,2min 。
8. 根据计数结果加入细胞冻存液(70% 完全培养基+20%FBS+10% DMSO )重悬细胞,密度为3X 10 6个/ml。
10. 第二天将细胞放入液氮灌,并记录。
二、293T 细胞的传代
1. 当细胞生长至汇合率达到80~90% 需要对细胞进行传代操作,以扩大细胞数量,维持细胞良好的生长状态。
2. 消化细胞,方法同上。
5
3. 细胞离心结束后,加入完全培养基重悬。密度为3 X10个/ml。
4. 分到10cm 培养皿中,10ml/ 皿。
三、293T 细胞的复苏
1. 当细胞传代次数过多(超过50 代),细胞状态变差时或细胞出现污染事故时,需要丢弃并对开始冻存的细胞进行复苏。
2. 打开水浴锅,设置温度为40 C。
3. 查看细胞库记录,根据记录从液氮灌中取出冻存的细胞(需戴上棉手套,防止被冻伤),迅速丢入水浴锅中并快速晃动,在1~2 min 内使细胞溶液完全溶解。
4. 将1ml 细胞溶液加入9 ml 完全培养基中并混匀后转入10cm 培养皿。
5. 放回37 C、3%CO 2和95%相对湿度的培养箱中培养。
6. 第二天观察细胞存活率。倒掉旧的培养基,加入10ml 新鲜培养基。
二、慢病毒的包装、浓缩和滴度测定
1. 所用病毒检测引物为WPRE 特异引物,序列如下
5'-CCTTTCCGGGACTTTCGCTTT-3' (forward primer), 5'-GCAGAATCCAGGTGGCAACA-3' (reverse primer) and 5'-FAM-ACTCATCGCCGCCTGCCTTGCC-TAMRA-3' (probe) 2. TaqMan Uni versal PCR Master Mix (Applied Biosystems , cat. no.
4304437)
3. TaqMan DNA Template Reage nt Kit (Applied Biosystems 401970)
用于包装的293T 细胞的培养
用于包装的293T 细胞(ATCC No. CRL-11268) 必需选择处于生长旺盛期,细胞状态
较佳,存活率90%以上,细胞边缘清晰,传代次数较低。任何时候细胞汇合率都不准达 到
100%。 慢病毒的包装
1. 预先准备3个T150 瓶的293T 细胞,培养基为 DMEM + 10% FBS ,1% Glutamax , 1% 青霉素-链霉素。
2. 将细胞分到12分到12个T150瓶中,每瓶的细胞密度是 8 X 10 6个。
3. 第二天,镜下检查细胞。细胞融合度应大致为
30-40% ,分布均匀。
4. 转染前1小时,取出细胞板,去除原有细胞培养基,加入 20ml 的Opti-MEM 培养
基,将细胞送回培养箱。
5. 取两支无菌的 50ml 离心管,其中一支中加入 252 卩g pNLEGFP/CMV/WPREDU3 载体质粒,168卩g pCD/NL -BH*DDD 包装质粒和 84卩g pLTR -G 质粒,用Opti-MEM 培养基补齐到 18ml 。另一支中加入 500卩l Trans -EZ 溶液和17.5 ml Opti-MEM 培 养基,用电动移液器轻轻混匀。
将Trans-EZ 稀释液滴加到质粒管中,
边加边轻轻晃匀。
cat. no. 4. TaqMa n RNaseP con trol reage nt (Applied Biosystems
cat. no. 4316844)
SunBio IV Victor
*
+
M VJ Ira :sJuUion
Packaging Cel]
关键步骤:推荐使用 Qiagen 质粒大抽试剂盒或相当的试剂盒所制备的质粒。 6. 室温孵育20分钟,使DNA 和Trans-EZ 充分结合形成转染复合体。
7. 取1支5ml 的移液管,将得到的 DNA-Trans-EZ 复合体均匀滴加入到细胞培养板 中,每板3ml 。来回晃动培养板,混匀后放回到 5%二氧化碳培养箱。每一皿细胞培养 盘不要超过6盘。 8. 6小时后,移去细胞上清,更换为 17ml 的DMEM 完全培养基。 9. 转染后一天观察细胞,所有的细胞应当都是健康的并且密度接近 60-80% 。如果所
转染质粒带有 GFP 荧光,那么这时候可以看到大于
95%的细胞都是带有荧光的。
10. 将细胞送回培养箱继续培养 2天(36-48小时)。在这个过程中,细胞会逐渐融合 形成多核体,大多数的细胞依然贴壁。
11. 收集所有的上清,分装到 50ml 离心管中。
12.4 C, 500g 离心10分钟,除去脱落的细胞和大的细胞碎片。
13.总的上清约为204 ml ,用250- ml 0.45 卩m PVD 过滤装置过滤。如果发现滤膜 被堵住,表现为过滤速度变慢,更换新的滤器。
慢病毒的浓缩与纯化 方法一超速离心沉淀法
离心管,用70%乙醇消毒后,放在超净工作台中打开紫 2. 每个 Ultra-clear SW28 离心管中加入约 32ml 的预先处理的病毒上清液。
1.取 6 个 Ultra-clear SW28 外灯继续消毒30分钟。
1供曲口的等边的转榕截停 与€11?戦体苑曾对匂餐砸
?皑箱購帚
*盯花叮刀軒魯笄再懐
3. 取一支10ml 的移液管,吸取12 ml 20% 的蔗糖溶液。将移液管一直插入到离心管的底部,缓慢将蔗糖溶液打出 4 ml 。同样地,将剩下8 ml 的蔗糖溶液分别加入到另两个离心管中。另取一支干净的移液管,对剩下 3 管进行同样处理。
4. 用PBS 调整各管的重量,使对应的离心管之间的重量相差不超过0.1g 。
5. 按次序将所有6 个离心管放入Beckman SW28 超速离心转头中。
7. 小心将管子从转头中取出。倒掉上清,将离心管倒扣在纸巾上放置10 分钟使剩余的
上清流干。吸掉剩余的液滴。在管底应当有可见的沉淀。
8. 每管中加入100ml 不含钙和镁的PBS 洗下沉淀。
9. 将SW28 超速离心管插入到50ml 锥底离心管中,盖上盖子。
10. 在4 C溶解2小时,每隔20分钟轻轻震荡。
11.4 C, 500g离心1分钟,使溶液集中于管底。
12. 用200卩I移液器轻柔吹打使沉淀重悬。避免产生泡沫。将所有管中的液体集中到一个
SW28 离心管中。
13. 集中后的病毒悬液分装成50卩I每份,保存在成品管中。用碎干冰速冻后储存在
-80 C 。
方法二PEG-8000 浓缩法
PEG (聚乙二醇)是高分子聚合物,具有高亲水性,在溶液中会吸收大量水分,减少病毒之间的距离,使病毒与病毒能够很容易的聚合在一起,病毒的相对浓度提高,达到沉淀浓缩的目的。
5X PEG8000+NaCI 配制称取NaCI 8.766 g; PEG8000 50g 溶解在200mI MiIIi-Q 纯水中;121 摄氏度30min 湿热灭绝30min ;保存在 4 C 。
1. 使用0.45 ym滤头过滤慢病毒上清液;
3. 每20?30min 混合一次,共进行3-5次;
4. 4 度放置过夜;
5. 4 度, 4000 g ,离心20min ;
6. 吸弃上清,静置管子1 ?2 分钟,吸走残余液体;
7. 加入适量的慢病毒溶解液溶解慢病毒沉淀;
8. 集中后的病毒悬液分装成50卩每份,保存在成品管中。用碎干冰速冻后储存在
-80 C
病毒滴度的测定
稀释计数法
滴度单位:TU/ml ,指每毫升中含有的具有生物活性的病毒颗粒数。”TU”
为” transducing units ”的缩写,中文为“转导单位”,表示可以感染并进入到靶细胞中的病毒基因组数。
第一天细胞准备
将生长状态良好的293T细胞消化计数后稀释至1X10 5/ml ,加入96孔板,100卩1/孔,
为每个病毒准备10个孔。放入37 C, 5%C0 2培养箱中培养。
第二天加病毒
在EP 管中做10 倍梯度稀释,连续10 个稀释度。稀释方法如下:每种病毒准备10 个
1.5ml EP 管,每管加入90卩1培养液,往第一个管中加入10卩1病毒原液,混匀后,吸
取10^1加入第二个管混匀。依此类推,做十个稀释度(10~10 -8)。
吸取96 孔板中原有的培养基,加入含稀释好的病毒液。并做好标记。第三天追加培养液
在每个孔再加入100卩1完全培养液,利于细胞的生长。
第五天观察结果并计算滴度
在荧光显微镜下观察结果,并数出最后两个有荧光的荧光细胞克隆数。假设为X 和Y, 则滴度(TU/ml )= (X+Y*10 )*1000/2/X 孔的病毒液的含量(卩)。
定量PCR 法
病毒感染 1 天前,取 6 孔板接种H0S 细胞。每孔细胞为5X10 4个。
接种细胞24 小时后, 取两个孔的细胞用血球计数板计数, 确定感染时细胞的实际数目, 记为N 。弃去其他培养板中的培养基,更换为含有5卩g/ml polybrene 的新鲜培养基。将浓缩病毒用培养基稀释200倍,也就是取1卩1病毒加入到199卩1的培养基中。在3个培养孔中分别加入0.5卩1, 5^1和50 ^1的稀释病毒。
感染开始后20小时,除去培养上清,换为500^1含DNasel (Takara Mirus Bio ,终
浓度为10 U/ml)的新鲜培养基。在37 C消化15分钟,这一步是要除去残余的质粒DNA。
然后换为2ml 正常的培养基,继续培养48 小时。
用0.5ml 0.25% 胰酶-EDTA 溶液消化细胞,在 37 C 放置1分钟。用培养基吹洗下, 离心收集细胞。按照DNeasy 试剂盒的说明抽提基因组
DNA 。每个样品管中加入 200 ^1
洗脱液洗下 DNA 。用DNA 定量试剂盒定量(Bio-Rad )。基因组 DNA 可以稳定保存 在-20 C 至少2个月。
准备PCR 所需的试剂和样品。为病毒序列检测引物配总管
I :
n = number of reactions.
例如:总反应数为 40 ,将 1ml 2 X TaqMan Universal
PCR Master Mix , 4 卩
l forward primer , 4 卩 l reverse primer , 4 卩 l probe 和 788 卩 l H 2O 混和。震荡后放在冰上。 为人基因组序列检测引物配总管
n :
n = number of reactions. 例如:总反应数为 40 ,将 1ml 2 X TaqMan Universal
PCR Master Mix , 100 卩 l 10 X RNaseP primer/probe mix 和 700 卩 l H 2O 混和。震 荡后放在
冰上。
在预冷的96孔PCR 板上完成PCR 体系建立。从总管I 中各取45卩1加入到A-D 各行 的孔中,从总管n 中各取45 ^1加入到E-G 各行的孔中。
分别取5卩1质粒标准品和待测样品基因组 DNA 加入到A-D 行中,每个样品重复 1次。 另留1个孔加入5 口1的水做为无模板对照(no-template control )。
另留1个孔加入5 口1的水做为无模板对照(no-template control )。
95 C 10分钟,然后是95 C 15秒,60 C 1分钟的40个循环。
分别取5卩1基因组标准品和待测样品基因组 DNA 加入到E-G 行中,每个样品重复1次。 所使用定量 PCR 仪为ABI PRISM 7000
定量系统。循环条件设定为:
50 C 2分钟,
数据分析:测得的DNA 样品中整合的慢病毒载体拷贝数用基因组数加以标定,得到每基因组整合的病毒拷贝数。
滴度( integration units per ml ,IU ml -1)的计算公式如下:
IU ml-1 = (C NX D X1OOO)/V
其中: C = 平均每基因组整合的病毒拷贝数
5
N = 感染时细胞的数目(约为 1 X 10 5)
D = 病毒载体的稀释倍数
V = 加入的稀释病毒的体积数
慢病毒的储存与稀释
慢病毒的储存
1. 病毒的运输采用干冰保温,收到病毒液后若几天内用于实验,可于 4 C保存(于一周
内用完)。
2. 若病毒量较大,需长期保存。则根据每次实验用量分装后放于-80 C冰箱。一般病毒可以放于-80 C约12个月以上,但若超过6个月后使用,请重新检测病毒滴度。
3. 避免反复冻融,否则会降低病毒滴度。每次冻融会降低病毒滴度10% 。
慢病毒的稀释
需要稀释病毒时,将病毒取出后置于冰上融解,用细胞培养用D-Hank's 、PBS 或培养
基稀释到所需浓度后混匀分装后4C保存,并尽快用于实验(于一周内用完)。
慢病毒在细胞水平的使用
什么是MOI ?
“ MOI为"multiplicity of infection 的缩写,中文为感染复数"或复感染指数”含义
为感染时病毒和细胞数量的比值,即平均每个细胞感染的病毒活性单位数 ( TU number /cell )。在实验中将某种细胞感染达到80% 时的MOI 定义为这种细胞的MOI 。
MOI 与整合事件以及目的基因的表达相关。一定范围内,表达水平和MOI 呈正相关。MOI 取决于多种因素,如细胞状态,目的基因的大小与性质,细胞的感染效率等。所以,实验前需查阅相关文献,确定慢病毒对目的细胞的亲嗜性、MOI 以及在体( in vivo ) 注射所需病毒量。若无文献支持,可以通过预实验得到合适的MOI 。实际上,即使有文献支持,但由于所用细胞代数、细胞状态以及目的基因的差异,实际的MOI 和文献报道也会不同,所以要安排预实验以确定所需MOI 以及病毒对细胞生长的影响。
目的细胞感染预实验及实验体系的放大