
四国药典有关药品微生物限度标准的比较
微生物限度规定的作用,是为药品生产提供一个标准或指导,以确保药品使用的安全。各国药典标准分为强制性的和非强制性的可达到的限度标准,这些指标正确、有效地规范了药品生产、检定和监督的程序。
药品要能反映不引起生物降解物和没有药源性污染的微生物存在是必要的,严格控制条件致病菌及致病菌。
一、CP、USP、BP、JP的微生物限度要求的特点及其发展趋势
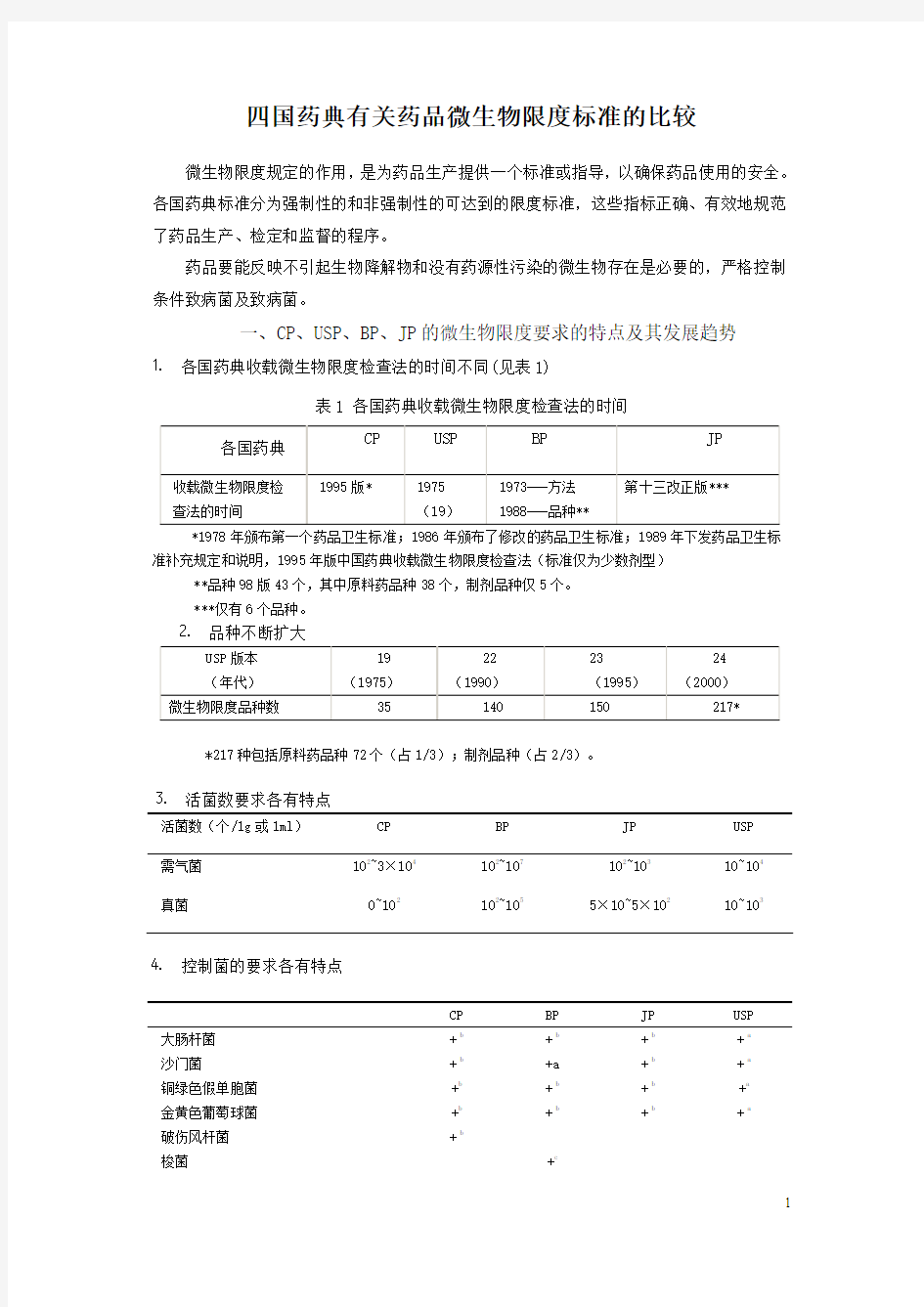
⒈各国药典收载微生物限度检查法的时间不同(见表1)
表1 各国药典收载微生物限度检查法的时间
*1978年颁布第一个药品卫生标准;1986年颁布了修改的药品卫生标准;1989年下发药品卫生标准补充规定和说明,1995年版中国药典收载微生物限度检查法(标准仅为少数剂型) **品种98版43个,其中原料药品种38个,制剂品种仅5个。
***仅有6个品种。
⒉品种不断扩大
*217种包括原料药品种72个(占1/3);制剂品种(占2/3)。
⒊活菌数要求各有特点
活菌数(个/1g或1ml)CP BP JP USP
需气菌102~3×104102~107102~10310~104
真菌0~102102~1055×10~5×10210~103
⒋控制菌的要求各有特点
CP BP JP USP
大肠杆菌+ b + b+ b+ a
沙门菌+ b+a + b+ a
铜绿色假单胞菌+b+ b+ b+a
金黄色葡萄球菌+b+ b+ b+ a
破伤风杆菌+ b
梭菌+c
肠道菌及其他某些革兰阴性杆菌+ c
支原体+
分枝杆菌+
病毒的其他因子+
活螨+ d
a 10g或10ml样品不得检出。
b 1g或1ml 样品不得检出。
c 仅为个别品种要求10~103/1g或1ml。
*d 不列在剂型项内而以说明提出,意即不作为常规检查,如有检出,以不合格处理的依据。
1000ml/L。溶解以上成份,加入1~10g 吐温-20或吐温-80。121℃高压蒸汽灭菌30min。
作用:可调节供试液pH至近中性,其中蛋白胨对菌细胞有保护作用,有利于菌数及控制菌的测定。吐温加入对含油性供试品的助溶具有作用。
** USP、BP微生物限度(污染)检查用稀释剂,除磷酸盐缓冲液、磷酸盐缓冲胨水外,尚采用以上两种培养基,直接稀释供试品并作增菌培养.。
三、稀释剂的用量
关于供试品于稀释剂的配比,国外药典普遍规定为称(量)取供试品10 g(或1 0.0 g)或10 ml(或10.0 ml),加稀释剂使成为100 ml,与我国药典的稀释剂固定加量有所不同。
四、抑菌性供试品的判定及处理原则
对供试品抑菌性的判定,应在检查前确定。
⒈国外药典一般方法是以加有供试液和不含供试液的检样中加入定量的已知菌(控制菌为10~100个、菌数测定为50~200个/ ml),经培养后检查。加入的对照菌能正常检出,加有供试液的菌落数和未加有供试液的菌落数相比较,后者不应超过前者5倍(JP13)及10倍(B P1998),否则表明供试品具有抑菌性,必须采用相应的处理方法。
方法:薄膜过滤法、稀释法和中和法。
⒉我国微生物限度检查是根据不同稀释度的菌数变化和控制菌的阳性对照试验是否正常检出来判断的。
菌数测定可根据报告规则或培养基稀释法来报告抑菌性药品的菌数,尚未提供其他方法对供试品的抑菌性成分进行处理。
控制菌的检查,需根据供试品的不同情况,适当地进行处理,以消除抑菌成分干扰。
方法:稀释法、离心沉淀法、薄膜过滤法、中和法及树脂吸附法。
五、活菌计数的方法比较
CP BP USP JP 平皿法a+ + + +d 试管法(MPN)b+ +
薄膜过滤法c+ +
a 是一种有条件的计数方法。其限制条件有三:
Ⅰ.细菌计数是以平板上生长的菌落数为基础。
Ⅱ受特定培养基和培养条件限制。
Ⅲ有繁殖能力的菌细胞才能被认定“活菌”。
b MPN适用于不完全溶解的供试品。一般采用三级3管制;此法所得的菌数与平板法所得的菌数不一定平行。
c 计数的薄膜,直径要大,最好有方格便于计数。遇到抑菌药物时,冲洗次数2~3次,冲洗量每次约50~100ml。
d 平板涂布法:将供试液涂布在琼脂平板的表面,经培养后计数表面生长的菌落数。
*有苄星青霉素、四环素等.
七、控制菌鉴定方法的比较
表1 大肠杆菌
CP BP USP 方法MI法IMViC法IMViC法
检验量(g或ml) 1 1 10
稀释剂09%氯化钠溶液
pH7.2磷酸盐缓冲液
乳糖肉汤
预增2~5小时
35~37℃
增菌胆盐乳糖增菌培养基
35~37℃18~24小时麦康凯肉汤
43~45℃18~24小时
乳糖液体培养基
30~35℃24~48小时
分离EMB、Mac.琼脂平板麦康凯琼脂平板EMB、Mac.琼脂平板
35~37℃18~24小时43~45℃18~24小时30~35℃24~48小时
生化试验培养温度37℃43.5~44.5℃37℃
生化试验判断模式+ + - - 或 - + - - + + - -或+ + - + + + - - 或 - + - -
表2 铜绿假单胞菌*
CP BP USP
检验量(g或ml) 1 1 10
稀释剂09%氯化钠溶液pH7.2
磷酸盐缓冲液pH7.0缓冲氯化钠-蛋
白胨水
大豆酪蛋白消化培养
基
增菌胆盐乳糖增菌培养基
35~37℃18~24小时大豆酪蛋白消化培养
基
35~37℃24小时
大豆酪蛋白消化培养
基
35~37℃24~48小时
分离溴化十六烷基三甲铵
琼脂平板
35~37℃18~24小时溴化十六烷基三甲铵
琼脂平板
35~37℃24小时
溴化十六烷基三甲铵
琼脂平板
35~37℃24~48小时
生化试验+ + +
*国外药典对某些原料药、口服制剂及外用制剂均作为特定菌控制。中国药典对外用制剂必检。
表3 金黄色葡萄球菌
CP BP USP(JP13) 检验量(g或ml) 1 1 10
稀释剂09%氯化钠溶液pH7.2磷
酸盐缓冲液pH7.0缓冲氯化钠-蛋白
胨
乳糖肉汤
液体大豆酪蛋白消化培
养基
增菌营养肉汤(或含亚碲酸
甲)培养基
35~37℃18~24小时大豆酪蛋白消化肉汤培
养基
35~37℃ 24~48小时
大豆酪蛋白消化液培养
基
35℃ 24~48小时
分离甘露醇氯化钠琼脂平板、
卵黄氯化钠琼脂平板
35~37℃24~48小时BP琼脂平板
35~37℃ 24~48小时
甘露醇氯化钠琼脂平板
或BP琼脂平板或VJ琼脂
平板
35~37℃24~48小时
血浆凝固酶试验+ +* +
*对过氧化氢酶阳性球菌,可用血浆凝固酶试验和脱氧核糖核酸酶试验。
表4 沙门菌
CP BP USP
检验量(g或ml) 1 10 10
稀释剂09%氯化钠溶液pH7.2磷
酸盐缓冲液
乳糖肉汤乳糖液体培养基
增菌营养肉汤培养基
35~37℃18~24小时乳糖肉汤乳糖液体培养基
35~37℃24~48小时
二次增菌TTB培养基TTB培养基SC、TTB液体培养基
35~37℃18~24小时42~~43℃18~24小时35~37℃24~48小时
分离EMB、Mac.C、DHL、SS琼
脂平板
35~37℃18~24小时DC、XLD.琼脂平板
43~45℃24~48小时
BG、XLD、BS琼脂平板
35~37℃24~48小时
生化试验+ + +
八、结果判断及复试
CP BP USP
控制菌一次检出为准一次检出为准不符合标准规定,取大于
25g的样品复试
需气菌和真菌第一次试验结果超过规定的标准,重新
抽样复试两次,三次结果平均报告
测定结果在规定的限度
标准5倍以内均为合格
同上
九、微生物限度标准的收载方式
表1 收载方式
CP BP USP
各论项下+ + + 制剂通则+ +
专论+ + +
注射剂、眼膏剂、
62 MICROBIOLOGICAL EXAMINATION OF NONSTERILE PRODUCTS: TESTS FOR SPECIFIED MICROORGANISMS 非无菌产品微生物限度检查:控制菌(USP38) 1.INTRODUCTION导言 The tests described hereafter will allow determination of the absence of, or limited occurrence of, specified microorganisms that may be detected under the conditions described. 控制菌检查法系用于在规定的试验条件下,检查供试品中是否存在特定的微生物。 The tests are designed primarily to determine whether a substance or preparation complies with an established specification for microbiological quality. When used for such purposes, follow the instructions given below, including the number of samples to be taken, and interpret the results as stated below. 当本法用于检查非无菌制剂及其原辅料是否符合相应的微生物限度标准时,应按下列规定进行检验,,包括样品的取样量,结果的判断. Alternative microbiological procedures, including automated methods, may be used, provided that their equivalence to the Pharmacopeial method has been demonstrated. 可以使用包括自动化法在内的方法,需确认与药典方法的等同性. 2.GENERAL PROCEDURES通用规程 The preparation of samples is carried out as described in Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61. 供试品制备,同USP<61> If the product to be examined has antimicrobial activity, this is insofar as possible removed or neutralized as described in Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61. 若供试品有抗菌活性,应尽可能中和或去除,中和或去除的方法同USP<61> If surface-active substances are used for sample preparation, their absence of toxicity for microorganisms and their compatibility with any inactivators used must be demonstrated as described in Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests 61. 若供试品制备过程中使用了表面活性剂,应确认其对微生物的无毒性以及与所用的中和剂/灭活剂的相容性,同USP<61> 3.GROWTH-PROMOTING AND INHIBITORY PROPERTIES OF THE MEDIA, SUITABILITY OF THE TEST AND NEGATIVE CONTROLS 培养基适用性检查,控制菌检查方法的适用性 确认,阴性对照 The ability of the test to detect microorganisms in the presence of the product to be tested must be established. Suitability must be confirmed if a change in testing performance or a change in the product that may affect the outcome of the test is introduced. 在有供试品存在的情况下,所建立的方法应能检测微生物。若检测程序或产品发生变化可能影响检测结果时,控制菌检查方法应重新进行适用性试验。 3.1.Preparation of Test Strains试验菌液的制备 Use standardized stable suspensions of test strains as stated below. Seed-lot culture maintenance techniques (seed- lot systems) are used so that the viable microorganisms used for inoculation are not more than five passages removed from the original master seed-lot.
通则1106 非无菌产品微生物限度检查:控制菌检查法控制菌检查法系用于在规定的试验条件下,检查供试品中是否存在特定的微生物。 当本法用于检查非无菌制剂及其原、辅料等是否符合相应的微生物限度标准时,应按下列规定进行检验,包括样品取样量和结果判断等。 供试品检出控制菌或其他致病菌时,按一次检出结果为准,不再复试。 供试液制备及实验环境要求同“非无菌产品微生物限度检查:微生物计数法”。 如果供试品具有抗菌活性,应尽可能去除或中和。供试品检查时, 若使用了中和剂或灭活剂,应确认有效性及对微生物无毒性。 供试液制备时如果使用了表面活性剂,应确认其对微生物无毒性以及与所使用中和剂或灭活剂的相容性。 培养基适用性检查和控制菌检查方法适用性试验 供试品控制菌检查中所使用的培养基应进行适用性检查。 供试品的控制菌检查方法应进行方法适用性试验,以确认所采用的方法适合于该产品的控制菌检查。 若检验程序或产品发生变化可能影响检验结果时,控制菌检查方法应重新进行适用性试验。 菌种及菌液制备 菌种试验用菌株的传代次数不得超过5 代(从菌种保藏中心获得的干燥菌种为第0 代),并采用适宜的菌种保藏技术进行保存,以保证试验菌株的生物学特性。 金黄色葡萄球菌(Staphylococcus aureus)〔CMCC(B)26 003〕 铜绿假单胞菌(Pseudomonas aeruginosa)〔CMCC(B)10 104〕 大肠埃希菌(Escherichia coli)〔CMCC(B)44 102〕 乙型副伤寒沙门菌(Salmonellaparatyphi B)〔CMCC(B)50 094〕 白色念珠菌(Candida albicans)〔CMCC(F)98 001〕 生孢梭菌(Clostridium sporogenes)〔CMCC(B)64 941〕 菌液制备将金黄色葡萄球菌、铜绿假单胞菌、大肠埃希菌、沙门菌分别接种于胰酪大豆胨液体培养基中或在胰酪大豆胨琼脂培养基上,30~35℃培养18~
美国微生物测试USP51 和USP61+62 介绍(2009/12/22 18:31) 美国微生物测试USP51 和USP61+62 介绍 玩具需作微生物测试的材料: 用于玩具中的化妆品、液体、糊状物、油灰(putties)、凝胶和粉末(艺术品材料,如粉笔、蜡笔、墨水等除外) 1. USP<61>微生物限量测试*1 - 测试目的: 检验材料本身受微生物污染的情况(也即微生物洁净度情况) - USP<61>包括以下六个微生物测试: (1) Total aerobic microbial Count 菌落总数(定量) (2) Mould and Yeast Count 霉菌和酵母菌数(定量) (3) Staphylococcus aureus 金黄色葡萄球菌(定性) (4) Pseudomonas aeruginosa 绿脓杆菌(定性) (5) Salmonella 沙门氏菌(定性) (6) Escherichia Coli 大肠杆菌(定性) USP61 Limit Requirement: (1)+(2) < 5000 CFU/g ( if the material was used in baby product or eye area product, the limit should be < 500 CFU/g) (3)/(4)/(5)/(6) should be “ABSENT per 10g “ 2. USP<51>防腐剂抗菌效力测试 - 测试目的 为防止的材料在保存过程中或使用过程中发生微生物*现象,须在材料中加入适量的防腐剂,而防腐剂效果如何则需通过USP《51》测试进行评价。 -USP<51>测试简述: 将标准指定编号的以下菌种接入样品,然后在第7 天,第14 天,第28 天分别检验每种菌的存活数量,存活数量越少,其防腐剂抗菌效果越好。 Staphylococcus aureus 金黄色葡萄球菌 Escherichia coli 大肠杆菌 Pseudomonas aeruginosa 绿脓杆菌 Candida albicans 白色念株菌 Aspergillus niger 黑曲霉 USP51 Limit requirement (for toy): 细菌(Staphylococcus aureus/ Escherichia coli / Pseudomonas aeruginosa) 14 天细菌减少≥2.0 LOG (99%) 28 天不再繁殖 真菌(Candida albicans /Aspergillus niger) 14 天和28 天不再繁殖 Remark: LOG REDUCTION = LOG10 (INITIAL COUNT / NO. OF MICRO-ORGANISM RECOVERED)
微生物限度检测方法验证操作规程 1 目的 确认所采用的方法适合于该产品的微生物限度检测。 2 依据 《中国药典》2015版。 3 范围 所有需进行微生物限度检测的产品。 4 责任 4.1验证小组负责检验方法验证/确认方案的起草、验证/确认方案的实施。 4.2验证委员会负责验证/确认方案的审批,验证/确认结论的审核。 5 程序 5.1 由验证小组提出验证申请,验证方案编制完成后,填写《确认和验证方案审批表》,经验证小组会签,报验证委员会审核,由生产负责人和质量负责人批准后,验证方案编制人对验证小组其余人员进行培训后,方可按验证方案试验。 5.2 试验完成后及时编制验证报告,并填写《验证报告审批表》,经验证小组会签,报验证委员会审核,由生产负责人和质量负责人批准后,验证报告结论才可实施。 6 内容 6.1 概述 通过验证以确认所采用的方法适合于该产品的需氧菌总数、霉菌和酵母菌总数的测定及控制菌的检查。根据样品特性制订检验方法和检验条件,按制定的方案进行试验,根据验证结果判断是否符合验证标准。若符合,按验证的方法和条件进行产品的微生物限度检查;若不符合,重新建立制订检验方法和检验条件,再进行验证,直至验证结果符合设立的验证标准。 6.2 需氧菌总数、霉菌和酵母菌总数计数方法的验证 6.2.1 验证用菌株
铜绿假单胞菌[CMCC(B)10 104] 金黄色葡萄球菌[CMCC(B)26 003] 枯草芽孢杆菌[CMCC(B)63 501] 黑曲霉[CMCC(F)98 003] 白色念珠菌[CMCC(F)98 001] 6.2.2 验证用菌液制备 6.2.2.1接种铜绿假单胞菌、金黄色葡萄球菌与枯草芽孢杆菌至胰酪大豆胨液体培养基中,于30~35℃培养18~24小时。取上述培养物各1ml,用0.9%无菌氯化钠溶液或pH 7.0无菌氯化钠蛋白胨缓冲液制成适宜浓度的菌悬液,备用。 6.2.2.2 接种白色念珠菌至沙氏葡萄糖液体培养基中,于20~25℃培养2~3天。取上述培养物1ml,用0.9%无菌氯化钠溶液或pH 7.0无菌氯化钠蛋白胨缓冲液制成适宜浓度的菌悬液,备用。 6.2.2.3接种黑曲霉至沙氏葡萄糖琼脂培养基上,于20~25℃培养5~7天,加入含0.05%(ml/ml)聚山梨脂80的0.9%无菌氯化钠溶液或pH 7.0无菌氯化钠蛋白胨缓冲液,将孢子洗脱。然后,用适宜方法吸出孢子悬液至无菌试管内,用含0.05%(ml/ml)聚山梨脂80的0.9%无菌氯化钠溶液或pH7.0无菌氯化钠蛋白胨缓冲液制成适宜浓度的孢子悬液,备用。 6.2.3 供试液的制备 6.2.3.1 水溶性供试试品 取供试品,用pH7.0无菌氯化钠-蛋白胨缓冲液或胰酪大豆胨液体培养基溶解或稀释制成1:10供试液。若需要,调节供试液pH值至6~8。必要时,用同一稀释液将供试液进一步10倍系列稀释。水溶性液体制剂也可用混合的供试品原液作为供试液。 6.2.3.2 水不溶性非油脂类供试品 取供试品,用pH7.0无菌氯化钠-蛋白胨缓冲液或胰酪大豆胨液体培养基溶解或稀释制成1:10供试液。分散力较差的供试品,可在稀释液中加入表面活性剂如0.1﹪的聚山梨酯80,使供试品分散均匀。若需要,调节供试液pH值至6~8。必要时,用一稀释液将
药品质量标准及答案 一、A1 1、“恒重”除另有规定外,系指供试品连续两次干燥或炽灼后的重量差异在多少以下的重量 A、0.2mg B、0.3mg C、0.4mg D、0.5mg E、0.6mg 2、我国现行的药品质量标准是 A、1995年版中国药典 B、2000年版中国药典 C、2005年版中国药典 D、2010年版中国药典 E、2015年版中国药典 3、原料药的含量测定如未规定上限时,指其上限不超过 A、99.9% B、100.0% C、100.5% D、101.0% E、102.0% 4、药物制剂的含量限度表示方法为 A、标示量 B、实际量 C、杂质量 D、实际量占标示量的百分比 E、杂质量占标示量的百分比 5、某药物注射用(标示量20ml,2.24g)用非水滴定法测定含量为每毫升实际含药物0.1100g。本品含量占标示量的百分比为 A、100.0% B、99.2% C、98.2% D、96.4% E、95.5% 6、药典中规定称取用量为“约”若干时,系指称取用量不得超过规定量的 A、±0.1% B、±1% C、±5% D、±10%
E、±20% 7、关于药品质量标准的叙述,不正确的是 A、国家对药品质量、规格及检验方法所做的技术规定 B、药品生产、供应、使用、检验和药政管理部门共同遵循的法定依据 C、体现“安全有效、技术先进、经济合埋、不断完善”的原则 D、对药品质量控制及行政管理具有重要意义 E、因生产情况不同,不必制定统一的质量标准 8、药典规定某药原料药的含量上限为102%,指的是 A、该原料药的实际含量 B、该原料药中含有干扰成分 C、用药典规定方法测定时可能达到的数值 D、方法不够准确 E、应用更准确的方法替代药典方法 9、中国药典(2015年版)中规定,称取2.00g系指 A、称取重量可为1.995-2.005g B、称取重量可为1.95-2.05g C、称取重量可为1.9995-2.0005g D、称取重量可为1.5-2.5g E、称取重量可为1-3g 10、取谷氨酸钠1.0g,加水23ml溶解后,加醋酸盐缓冲液(pH 3.5)2ml,依法检查,与标准铅溶液(10μg Pb/ml)所呈颜色相比较,不得更深。重金属限量为百万分之十,则标准铅溶液应取 A、1.0mL B、2.0mL C、3.0mL D、4.0mL E、5.0mL 11、测定结果与真实值之间的差异是 A、精密度 B、重复性 C、准确度 D、线性 E、回收率 12、回收率可用于表示 A、准确度 B、精密度 C、专属性 D、检测限 E、线性 13、在药物检测中,表示准确度的指标是
甘草四国药典比较 班级:51 学号:1045114 姓名:陈多清 一、质量标准比较 1.中国药典(CHP2010) 来源: 本品为豆科植物甘草Radix Glycyrrhiza uralensis Fisch.、胀果甘草Glycyrrhiza in flataBat.或光果甘草Glycyrrhiza glabra L.的干燥根及根茎。春、秋二季采挖,除去须根,晒干。 性状: 1)根呈圆柱形,长25~100cm,直径0.6~3.5cm。外皮松紧不一。表面红棕色或灰棕色,具显著的纵皱纹、沟纹、皮孔及稀疏的细根痕。质坚实,断面略显纤维性,黄白色,粉性,形成层环明显,射线放射状,有的有裂隙。根茎呈圆柱形,表面有芽痕,断面中部有髓。气微,味甜而特殊。 2)胀果甘草根及根茎木质粗壮,有的分枝,外皮粗糙,多灰棕色或灰褐色。质坚硬,木质纤维多,粉性小。根茎不定芽多而粗大。 3)光果甘草根及根茎质地较坚实,有的分枝,外皮不粗糙,多灰棕色,皮孔细而不明显。鉴别: 1)本品横切面:木栓层为数列棕色细胞。栓内层较窄。韧皮部射线宽广,多弯曲,常现裂隙;纤维多成束,非木化或微木化,周围薄壁细胞常含草酸钙方晶;筛管群常因压缩而变形。束内形成层明显。木质部射线宽3~5列细胞;导管较多,直径约至160μm;木纤维成束,周围薄壁细胞亦含草酸钙方晶。根中心无髓;根茎中心有髓粉末淡棕黄色。纤维成束,直径8~14μm,壁厚,微木化,周围薄壁细胞含草酸钙方晶,形成晶纤维。草酸钙方晶多见。具缘纹孔导管较大,稀有网纹导管。木栓细胞红棕色,多角形,微木化。 2)取本品粉末1g,加乙醚40ml,加热回流1小时,滤过,药渣加甲醇30ml,加热回流1小时,滤过,滤液蒸干,残渣加水40ml使溶解,用正丁醇提取3次,每次20ml,合并正丁醇液,用水洗涤3次,蒸干,残渣加甲醇5ml使溶解,作为供试品溶液。另取甘草对照药材1g,同法制成对照药材溶液。再取甘草酸铵对照品,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。照薄层色谱法试验,吸取上述三种溶液各1~2μl,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15:1:1:2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点;在与对照品色谱相应的位置上,显相同的橙黄色荧光斑点。 检查: 水分照水分测定法测定,不得过12.0% 总灰分不得过7.0% 酸不溶性灰分不得过2.0% 重金属及有害元素照铅、镉、砷、汞、铜测定法测定,铅不得过百万分之五;镉不得过千万分之三;砷不得过百万分之二;汞不得过千万分之二;铜不得过百万分之二十 有机氯农药残留量照农药残留量测定法测定,六六六(总BHC)不得过千万分之二;滴滴涕(总DDT)不得过千万分之二;五氯硝基苯(PCNB)不得过千万分之一
USP36 1117 优良微生物检测规范(中英文1/ 2) 2013-08-09 15:30:46| 分类:USP|举报|字号订阅 1117 MICROBIOLOGICAL BEST LABORATORY PRACTICES 优良微生物检测规范INTRODUCTION 介绍 Good laboratory practices in a microbiology laboratory consist of activities that depend on several principles: aseptic technique, control of media, control of test strains, operation and control of equipment, diligent recording and evaluation of data, and training of the laboratory staff. Because of the inherent risk of variability in microbiology data, reliability and reproducibility are dependent on the use of accepted methods and adherence to good laboratory practices. 优良微生物检测规范由一些活动组成,这些活动依赖于几个基本要素:无菌技术、培养基控制、检测用菌株控制、设备操作和控制、完善的记录和数据评估、化验室员工的培训。由于微生物数据具有天生的不确定性,数据的可靠性和重复性取决于是否使用被接受的方法,以及是否严格遵守化验室规范。 MEDIA PREPARATION AND QUALITY CONTROL 培养基制备和质量控制 Media Preparation 培养基制备 Culture media are the basis for most microbiological tests. Safeguarding the quality of the media is therefore critical to the success of the microbiology laboratory. Media preparation, proper storage, and quality control testing can ensure a consistent supply of high-quality media. 培养基是大多数微生物测试的基础。保证培养基的质量因而成为微生物实验室成功的关键。培养基的制备、合适的存贮和质量控制检测可以保证持续高质量培养基供应。 It is important to choose the correct media or components in making media based on the use of accepted sources or references for formulas. The manufacturer's formula and instructions for preparation routinely accompany dehydrated media and
人工牛黄甲硝唑胶囊微生物限度检查方法验证方案
1. 概述 2. 验证目的和范围 3. 组织及职责 4. 验证进度计划表 5. 验证所需要的仪器设备及相关文件的确认 6. 验证所需要的菌种、培养基、检验样品的确认 7. 验证项目和验证方法 7.1试验菌株 7.2需氧菌总数检查、霉菌和酵母菌总数检查方法验证的菌液制备 7.3需氧菌总数检查、霉菌和酵母菌总数检查方法验证--常规倾注平皿法7.4需氧菌总数检查方法验证一离心沉淀-薄膜过滤法 7.5控制菌检查方法验证一离心沉淀-薄膜过滤法 8. 偏差与漏项控制 9. 验证报告会审
1. 概述 我公司生产品种人工牛黄甲硝唑胶囊,产品微生物限度检查项目为需氧菌总数、霉菌和酵母菌 总数、以及大肠埃希菌检查和沙门菌检查。参照《中国药典》2015版四部附录1105:微生物计数 法,以及1106:控制菌检查法的规定,本公司对该产品的微生物限度检查方法予以验证。通过验证以确认所采用的微生物限度检查方法适用。 人工牛黄甲硝唑胶囊处方中含有甲硝唑、人工牛黄以及常用辅料成分,文献资料介绍甲硝唑对 细菌有抑菌特性,对霉菌和酵母菌无抑菌活性。甲硝唑在水中微溶,可以通过离心沉淀-薄膜过滤 法去除其对微生物生长的影响。本验证方案通过试验菌株的回收率测试,首先确认常规倾注平皿法是否适用于本品的微生物限度检查;如常规倾注平皿法不适用,则进一步验证可去除供试品抑菌物质的离心沉淀-薄膜过滤法是否适用于本品的微生物限度检查。 本验证方案根据样品特性制定微生物限度检查方法和检验条件,按制定的方法进行试验,根据验证结果判断是否符合验证标准。 2. 验证目的和范围 验证该产品的微生物限度检查方法的适用性,对其有效性进行评价,保证检验结果的可靠性。 本验证方案采用3批按GMF要求组织生产的人工牛黄甲硝唑胶囊,进行微生物限度检查方法的验证。 3. 组织及职责 3.1验证方案和验证报告的起草、审核、批准 验证方案由质量部QC组负责起草,由质量部审核,最终由质量负责人批准。验证方案实施完 成后,由QC组负责汇总微生物限度检查法验证的结果、撰写报告,由质量部审核,最终由质量负责人批准报告。 3.2验证方案的培训 验证方案在经质量负责人批准后,,由QC组组长对本次验证实施的相关人员组织培训工作,并将该次的培训记录归档。 3.3验证方案实施过程中的变更和偏差 验证方案实施过程中如有变更和偏差,质量负责人应当组织进行评估并采取相应的控制措施。 3.4验证工作小组成员表 4. 验证进度计划表 本次微生物限度检查方法验证的计划安排时间是2015年12月至2016年1月。 5. 验证所需要的仪器设备及相关文件的确认 5.1. 主要检验仪器设备确认表
第一章药品质量研究的内容和药典概况 一、最佳选择题 1. ICH有关药品质量的技术要求文件的标识代码是 A. E B. M C. P D. Q E. S 2. 药品标准中鉴别试验的意义在于 A.检查已知药物的纯度 B.验证已知药物与名称的一致性 C.确定已知药物的含量 D.考察已知药物的稳定性 E.确证未知药物的结构 3. 盐酸溶液(9→1000)系指 A.盐酸1.0ml加水使成1000m1的溶液 B.盐酸1.0ml加甲醇使成1000m1的溶液 C.盐酸1. 0g加水使成1000m1的溶液 D.盐酸1.0g加水1000m1制成的溶液 E.盐酸1.0ml加水1000m1制成的溶液 4. 中国药典凡例规定:称取“2.0g”,系指取重量可为 A.1.5~2.5g B.1.6~2.4g C.1.45~2.45g D.1.95~2.05g E.1.96~2.04g 5. 中国药典规定:恒重,除另有规定外,系指供试品连续两次干燥或炽灼后的重量差异在 A. 0.01lmg B. 0.03mg C. 0.lmg D. 0.3mg E. 0.5mg 6. 原料药稳定性试验的影响因素试验,疏松原料药在开口容器中摊成薄层的厚度应 A.>20cm B.≤20cm C. ≤10cm D. ≤5cm E. ≤10mm 7. 下列内容中,收载于中国药典附录的是 A.术语与符号 B.计量单位 D.准确度与精密度要求E通用检测方法 8. 下列关于欧洲药典(EP)的说法中,不正确的是 A. EP在欧盟范围内具有法律效力 B. EP不收载制剂标准 C. EP的制剂通则中各制剂项下包含:定义(Definition )、生产(Production)和检查 (Test ) D.EP制剂通则项下的规定为指导性原则 E. EP由WHO起草和出版 二、配伍选择题 [9-10] A. SFDA B. ChP C. GCP D. GLP E. GMP 下列管理规范的英文缩写是 9. 药品非临床研究质量管理规范 10.药品生产质量管理规范 [11--13] A.溶质1g(ml)在溶剂不到 ml中溶解 B.溶质1g(ml)能在溶剂1~不到10ml中溶解 C溶质1g(ml)能在溶剂10~不到30m1中溶解 D.溶质1g(ml)能在溶剂30~不到100ml中溶解 E.溶质1g(ml)能在溶剂100~不到1000m1中溶解
第十五章药品质量标准制订 [基本要求] 一、掌握药品质量标准的定义、分类与制订原则。 二、掌握药品质量标准的内容。 三、熟悉确定杂质检查项目及其限度的基本原则、选择含量测定法的基本原则。 四、熟悉溶解度测定法、熔点测定法、吸收系数的测定法。 五、了解药品稳定性试验。 [本章分配学时数] 4学时 第一节、概述 一、药品质量标准的定义:药品质量标准是国家对药品质量、规格及检验方法所作的技术规定;是药品生产、供应、使用、检验和药政管理部门共同遵循的法定依据。 二、药品质量标准的分类 1,法定的药品质量标准标准:中国药典、药品标准 2,临床研究用药品质量标准:新药研制过程中、临床试验前必须报批的药品标准。以保证临床用药安全、结论可靠。此标准仅适用于研制单位、临床试验单位和药检单位。 3,暂行或试行药品标准:1-3类新药经临床试验及国家药品监督管理局批准试生产阶段的药品标准称“试行标准”,试行期2-3年。期满后报请国家药品监督管理局,由药典委员会审批转正。
4,企业标准:由企业制定并报有关部门批准备案。也是GMP认证的必备条件。通常企业标准高于法定标准的要求。 中国药典 全称:《中华人民共和国药典》,简称中国药典,英文表示Chines Pharmacopoeia。通常写法:中国药典(××××年版)。 沿革:1949年建国以后,已出版了87版药典(1953、1963、1977、1985、1990、1995、2000、2005年版药典)。 自1963年版药典分为两部,一部和二部。 1988年正式出版了中国药典(1985年版)英文版,同年还出版了二部注释选编。 1990年版编著了《中华人民共和国药典临床用药须知》,另行出版了《药品红外光谱集》。编制出版了中国药典(1990年版)第一、第二增补本,二部注释和一部注释选编、《中药彩色图集》和《中药薄层彩色图集》。 1995年版二部药品外文名称改用英文名,取消拉丁名;中文名称只收载药品法定通用名称,不再列副名。 2000年版二部附录新增加了毛细管电泳法、热分析法和X射线粉末衍射法这三种仪器分析方法。 2005年版分三部 三、制订药品质量标准的原则 ⑴,安全性:毒副反应物质 ⑵,有效性:生物利用度、晶型等
四国药典有关药品微生物限度标准的比较 [日期:2005-2-21] 来源:作者:胡敏[1] 胡昌勤* 刘文英2 [字体:大中小] 四国药典有关药品微生物限度标准的比较 微生物限度规定的作用,是为药品生产提供一个标准或指导,以确保药品使用的安全。各国药典标准分为强制性的和非强制性的可达到的限度标准,这些指标正确、有效地规范了药品生产、检定和监督的程序。 药品要能反映不引起生物降解物和没有药源性污染的微生物存在是必要的,严格控制条件致病菌及致病菌。 一、CP、USP、BP、JP的微生物限度要求的特点及其发展趋势 ⒈各国药典收载微生物限度检查法的时间不同(见表1) 表1 各国药典收载微生物限度检查法的时间 各国药典 CP USP BP JP 收载微生物限度检查法的时间1995版* 1975 (19) 1973---方法 1988---品种** 第十三改正版*** *1978年颁布第一个药品卫生标准;1986年颁布了修改的药品卫生标准;1989年下发药品卫生标准补充规定和说明1995年版中国药典收载微生物限度检查法(标准仅为少数剂型)**品种98版43个,其中原料药品种38个,制剂品种仅5个。 ***仅有6个品种。 ⒉品种不断扩大 USP版本 (年代) 19(1975)22(1990)23(1995)24(2000)微生物限度品种数35 140 150 217* *217种包括原料药品种72个(占1/3);制剂品种(占2/3)。 ⒊活菌数要求各有特点 活菌数(个/1g或1ml)CP BP JP USP 需气菌102~3×104 102~107 102~103 10~104 真菌0~102 102~105 5×10~5×102 10~103 ⒋控制菌的要求各有特点
涂家生,中国药科大学教授国家药典委员会委员 国家药品审评中心专家 2010.5
中国药典附录《药用辅料》的解析 主要内容 前 言 2010年版中国药典收载药用辅料的过程展 望 1234
我国药用辅料现状 一、前言 药用辅料也称为赋形剂,诚如药物离不开剂型,制剂同样离不开辅料。目前,药物制剂正向以“定时、定量、定位”为目标的给药系统发展,新型给药系统的产生、发展、成熟无不与新型辅料的出现有关。 1、目前我国药用辅料的来源、生产、质量尚存在管理不理想的 状况,药用辅料的质量影响和制约了我国制剂的水平。2、另一方面,尽管辅料被认为应该生理惰性、化学惰性,但有很多药物不良事件是与药用辅料有关的,例如1930年的磺胺酏剂事件和我国广东去年的“齐二药”事件,皆因利益驱使假冒辅料(economic-motivated adulteration, EMA )二甘醇导致。3、很多辅料具有生物活性,或会改变药物的作用,例如三聚氰胺等。
据不完全统计,我国制剂使用的药用辅料大约543种,但具有药用质量标准的占少数,尤其在药典中收载较少,表1为我国药用辅料的质量标准统计情况。 表1 我国药用辅料的质量标准统计情况 标准品种数占总数的比例 药典标准(2010年版)13224.31% 部颁标准33 6.1% 地方标准31 5.7% USP和EP标准27 5.0% 国标、化工和企业标准34162.8% 总计543 可以看出,我国药用辅料质量标准符合药用标准的为少数,尤其收载于药典的仅为少数。其它标准的材料作药用辅料的现象严重。与之不同的是,美国大约有1500种辅料,大约50%已经收载于USP/NF,欧洲有药用辅料约3000种,在各种药典中收载也已经达到50%。因此,加大药用辅料收载于中国药典具有重要意义。
Chp2010、Ph.Eur.7、Jp16中药或天然药物的质量标准比较 -----以番泻叶为例阐述 班级10451 学号1045103 姓名李震
番泻叶的背景 番泻叶的功效: 功能主治:泻热行滞,通便,利水。用于热结积滞,便秘腹痛,水肿胀满。性味归经:甘、苦,寒。归大肠经。用法用量:2~6g,入煎剂宜后下,或开水泡服。注意:孕妇慎用。备注:(1)服量不宜过大,过量则有恶心、呕吐、腹痛等副作用,一般配木香、藿香等行气和中药品同用,可减少此弊。 番泻叶的药理作用: 1、泻下作用对小鼠、大鼠、家兔等多种动物及人均有显著的泻下作用,小鼠和兔于药后2-4b致泻,人口服后约6h引起泻下。本品致泻有效成分主要为番泻甙A和B,尤其是番泻甙A,番泻甙C虽致泻作用与A相近然含量很少。番泻甙A20mg/kg即可引起小鼠泻下。但倘于A中混入20%的C,则可使番泻甙A 的作用增强1.6倍。番泻甙于小肠可以有部分吸收,后经血流或胆汁进入大肠,而主要则由小肠直接进入大肠,在肠内细菌作用下经水解、还原等变化成为大黄酸蒽酮或大黄酸蒽酮-8-葡萄糖甙。由于直接注入大黄酸蒽酮的泻下作用不受影
响,且可见肠内大黄酸蒽酮的生成量显著减少,故认为大黄酸蒽酮才是番泻甙引 起泻下的真正成分。另一方面,在翻转小肠和结肠囊番泻甙可阻止葡萄糖和Na 的跨肠壁转运,表明抑制肠道对葡萄糖、钠和水的吸收,增加肠腔内容积继而刺 激肠壁反射性地使小肠和结肠蠕动增强,也可能是其致泻机制之一,且小肠也是 其泻下成分的作用部位。 2、止血作用对胃、十二指肠出血有效。用本品水浸液于胃镜下喷洒于胃出血处, 直视可见有即刻止血作用。番泻叶总甙200mg/kg腹腔注射可明显缩短小鼠出血 时间。番泻叶口服,可便血小板数及纤维蛋白原含量增加,凝血时间、凝血活酶 时间、血浆复钙时间和血块收缩时间缩短。此外,本品对盐酸和消炎痛所致大鼠 胃粘膜损伤的保护作用也有利于对胃、十二指肠出血的防治。 3、抗菌作用番泻叶浸液对多种细菌有抑制作用,如大肠杆菌、变形杆菌、痢疾 杆菌、甲型链球菌以及白色念珠和某些致病性皮肤真菌。 4、其他作用对于实验性肠梗阴大鼠,番泻甙50mg/kg腹腔注射可使降低的肠粘 膜组胺含量恢复至正常水平。此外,曾报告本品有箭毒样作用,能阻断神经-肌 肉接头冲动的传递、阻止乙酰胆碱与M受体的结合而使肌肉松弛。 5、毒性番泻叶总甙腹腔注射小鼠的LD50为1.414g/kg,折合生药为36.3g/kg。 Chp 2010Jp16Ph.eur.7药典 比较
验证文件 类别: 编号: 部门: XXXX微生 物限度检查方法适用性试验方案 XXXXX公司 2016年
XXXX微生物限度检查方法适用性试验方案审批
目录 1、适用范围 2、目的 3、概述 4、适用性所需要的仪器设备及文件 5、可接受的限度范围标准 6、测试方法 7、异常情况处理 8、测试结果 9、结论 10、再适用性周期 11、附表
1、适用范围 本适用性方案适用于XXXX微生物限度检查的方法适用性试验。 2、目的 因《中华人民共和国药典》(2015年版)颁布实施,为确保该产品的微生物限度检查方法的可靠性和可操作性,故对其检验方法进行适用性试验,以符合《中华人民共和国药典》规定。 3、概述 3.1根据《中华人民共和国药典》2015年版四部通则1105非无菌产品微生物限度检查:微生物计数法、1106非无菌产品微生物限度检查:控制菌检查法,对XXXX进行需氧菌、霉菌和酵母菌、大肠埃希菌检查方法的适用性试验,以确认供试品的抑菌活性及测定方法的可靠性。 3.2适用性试验时间及批号:年月日开始分别对、、三 批XXXX进行独立的方法适用性试验。 3.3验证小组 4、仪器设备及文件 4.1需用仪器设备
5、可接受的限度范围标准 5.1XXXX微生物限度检查质量标准 5.2需氧菌、霉菌和酵母菌计数方法采用《中华人民共和国药典》2015版四部通则1105非无菌产品微生物限度检查:微生物计数法中叙述的平皿法中的倾注法。控制菌检查方法采用1106非无菌产品微生物限度检查:控制菌检查法中大肠埃希菌检查方法 6、测试方法 6.1供试品 XXXX 批号: 6.2培养基及菌种 6.2.1采用符合中国药典规定的干燥培养基,并经过培养基适用性检查。 胰酪大豆胨琼脂培养基批号:胰酪大豆胨液体培养基批号: 沙氏葡萄糖琼脂培养基批号:沙氏葡萄糖液体培养基批号: 麦康凯琼脂培养基批号:麦康凯液体培养基批号: MUG培养基批号: 6.2.2菌种均购于:
?61? MICROBIAL LIMIT TESTS This chapter provides tests for the estimation of the number of viable aerobic microorganisms present and for freedom from designated microbial species in pharmaceutical articles of all kinds, from raw materials to the finished forms. An automated method may be substituted for the tests presented here, provided it has been properly validated as giving equivalent or better results. In preparing for and in applying the tests, observe aseptic precautions in handling the specimens. Unless otherwise directed, where the procedure specifies simply ―incubate,‖ hold the container in air that is thermostatically controlled at a temperature between 30and 35, for a period of 24 to 48 hours. The term ―growth‖ is used in a special sense herein, i.e., to designate the presence and presumed proliferation of viable microorganisms. PREPARATORY TESTING The validity of the results of the tests set forth in this chapter rests largely upon the adequacy of a demonstration that the test specimens to which they are applied do not, of themselves, inhibit the multiplication, under the test conditions, of microorganisms that may be present. Therefore, preparatory to conducting the tests on a regular basis and as circumstances require subsequently, inoculate diluted specimens of the material to be tested with separate viable cultures of Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Salmonella. This can be done by adding 1 mL of not less than 10-3 dilution of a 24-hour broth culture of the microorganism to the first dilution (in pH 7.2 Phosphate Buffer, Fluid Soybean–Casein Digest Medium, or Fluid Lactose Medium) of the test material and following the test procedure. Failure of the organism(s) to grow in the relevant medium invalidates that portion of the examination and necessitates a modification of the procedure by (1) an increase in the volume of diluent, the quantity of test material remaining the same, or by (2) the incorporation of a sufficient quantity