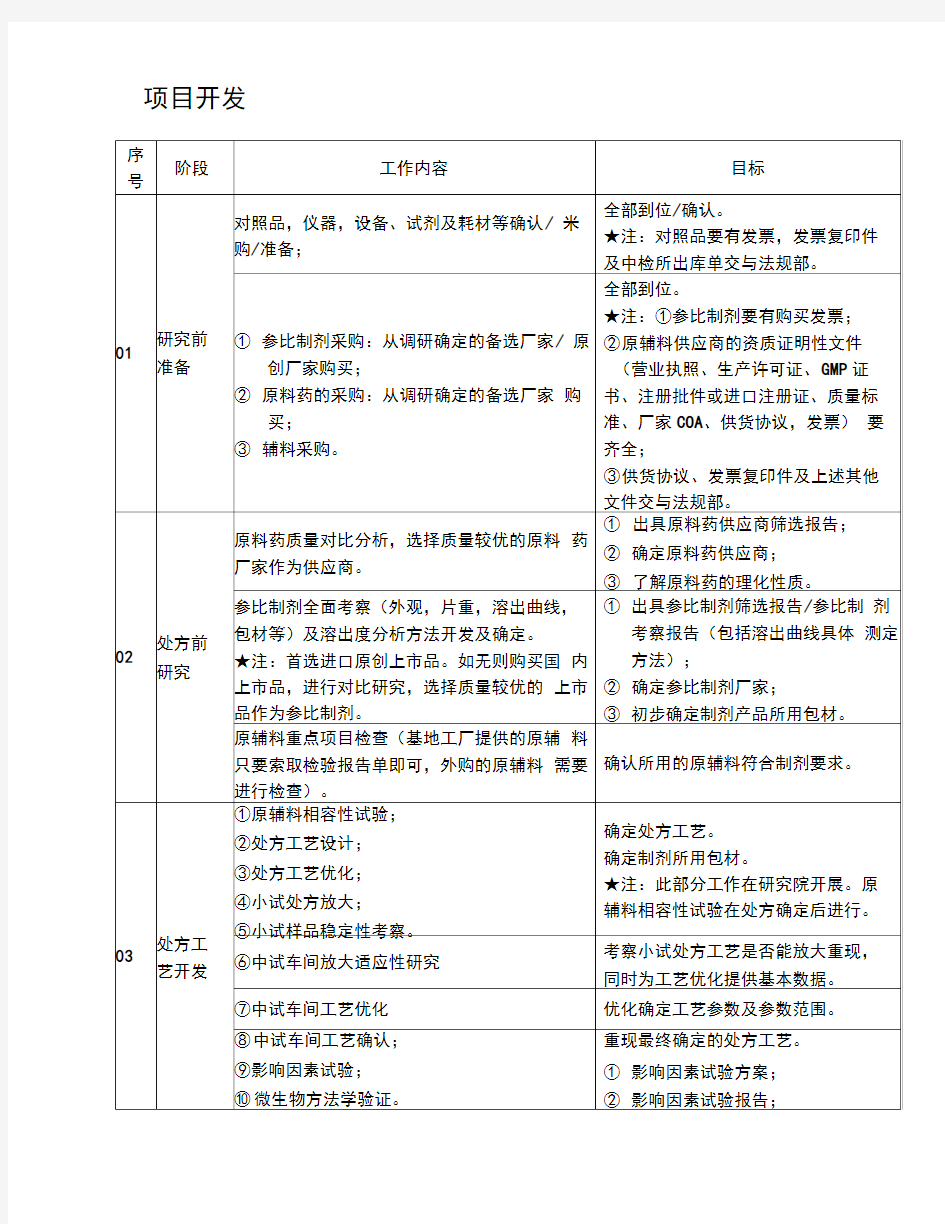

项目开发
表1项目开发计划中阶段4方法学验证”阶段工作内容
表2项目开发计划中阶段7质量研究”工作内容
*溶出度检测方法采用高效液相色谱法时需考虑进行耐用性试验,若检测方法与含量测定方法相同,则不需进行,若不同,需根据方法的差异考虑进行该项研究。
大型项目 小型项目车身结构一地板以下沿用沿用 车身结构一地板以 上,门盖 新沿用 外饰新 新 内饰新除40.1,40.5, 40.6,40.7夕卜,并可修改 动力总成修改:现有发动机/变速 箱首次应用 沿用 底盘/空调系统除沿用底盘结构20.2, 20.4, 20.5, 20.6 夕 卜, 可修改沿用 工艺变化制造一对现有主要生产 线无需调整,并有足够 调整场地 制造一对现有主要生产线 无需调整,并有足够调整 场地 设 计 / 发 布 集 成 制 造 过 程 注意:本框图仅表示必不可少交付物的关系,未表明时间尺度 提出概念 交付物责任 客户要求MD 项目设想BP DSI文件 交付物责任 SOP目标BP 目标生产场地BP 九格表BP 型谱中战略角色BP 项目赢势BP 宏观市场MD 价格范围MD 预计生产纲领MD 竞争产品MD 边际利润FN 设计要求DS 红色字体的内容不适合小型
设计主题方案CD方案分析研究 方向 设想交付物 MSS责任 MD 产品 交付物责任 产品项目规划书(PPC)BP 关键产品特性PK 质量目标确认QA 计划 交付物责任整车进度计划BP 财务评估FN 经济分析FN 项目分析BP 设计方案 交付物责任 效果图/渲染图DS 油泥模型/USB DS 竞争车对比表DS 造型主题数模DS 工程研究 交付物责任 BOM/爆炸图PK VAS结构ES 性能分析评估PK Z&L模型PK 制造策略ME 动力总成方案PT 初始装配模型VS ▼ 工程 交付物责任 BOM/爆炸图PK 油泥模型/USB DS 初始装配数模PK 项目文件 交付物责任 制造策略ME MSS MD 产品项目规划书(PPC)BP 项目章程BP 整车进度表BP 项目计划BP 市场调研报告MD 项目强制性指标BP
详解仿制药研发具体流程 目录 一、综述2 二、仿制药研发项目汇总3 三、仿制药的研发具体步骤:5 (一)产品信息调研5 (二)前期准备(约一个月完成):5 1、参比制剂的采购5 2、原料采购5 3、色谱柱及对照品采购5 4、辅料采购:6 5、包材的采购:6 (三)处方工艺研究6 1、原辅料及参比制剂的检验6 2、处方工艺摸索6 3、初步验证工艺8 4、中试生产及工艺验证8 (四)质量研究9 1、质量研究项目的选择及方法初步确定9 2、质量标准的方法学验证10 3、质量对比研究12 4、质量标准的制定14 (五)稳定性研究(中试产品)14
(六)药理毒理研究16 (七)申报资料的撰写、整理16 (八)申报临床及申报现场核查17 (九)临床研究17 (十)申报生产17 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。
详解仿制药研发具体流程目录 一、综述2 二、仿制药研发项目汇总3 三、仿制药的研发具体步骤:5 (一)产品信息调研5 (二)前期准备(约一个月完成):5 1、参比制剂的采购5 2、原料采购5 3、色谱柱及对照品采购5 4、辅料采购:6 5、包材的采购:6 (三)处方工艺研究6 1、原辅料及参比制剂的检验6 2、处方工艺摸索6
3、初步验证工艺8 4、中试生产及工艺验证8 (四)质量研究9 1、质量研究项目的选择及方法初步确定9 2、质量标准的方法学验证10 3、质量对比研究12 4、质量标准的制定14 (五)稳定性研究(中试产品)14 (六)药理毒理研究16 (七)申报资料的撰写、整理16 (八)申报临床及申报现场核查17 (九)临床研究17 (十)申报生产17 一、综述
根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”:
0概念 DSI 大型项目 车身结构—地板以 沿用 下 车身结构—地板以 新 上,门盖 外饰新 内饰新 修改:现有发动机 /变动力总成 速箱首次应用 除沿用底盘结构,,,底盘 /空调系统 外,可修改 制造一对现有主要生工艺变化产线无需调整,并有足 够调整场地 设计主题方案 方向 小型项目 沿用 沿用 新 除,,,外,并可修改 沿用 沿用 制造一对现有主要生产 线无需调整,并有足够 调整场地 1 方案分析研究 设计方案 交付物 效果图 /渲染图 油泥模型 /USB 竞争车对比表 造型主题数模 意图 设 计 / 提出概念 发 布交付物责任 客户要求MD 项目设想BP 验 / 证 学 习 红色字体的内容不适合小型 集 提成案 制 造 过 程 VPI 准备 责任 DS DS DS DS DSI 文件准备 DSI 文件 交付物责任 SOP 目标BP 目标生产场地BP 九格表BP 型谱中战略角色BP 项目赢势BP 宏观市场MD 价格范围MD 预计生产纲领MD 竞争产品MD 边际利润FN 设计要求DS 设计意图 PVC STDMD
设想 交付物 MSS 产品 交付物 产品项目规划书( PPC)关键产品特性 质量目标确认 计划 建模 交付付物物 整车SV表进面度发布计划选择颜色 /面料 财务评估 流体分析模型(CFD) 经济分析 制作 项目分析 交付物 USB —║ 外观实体模型 项目管理 项目 工程 交付物 交付物 工装设备SOR BOM DFM 计划 DTS FIVC 产品启动计划 ICD 质量着落道计划 认证 模具产能计划 交付物 虚拟 SV 评估 内外饰实体模型 责任工程 交付物责任 MD BOM/ 爆炸图开发PK PQRR 油泥模型 DS 2/USB 责任ST VPI初始装配数模PK BP AC TTA MSV ExpPT VPI DSO VCC PSVSR PK 项目文件 交付物责任 QA骡子工车程:研采究购,制造,试验制造策略ME 骡子车 /概整车集成 MSS MD 细 交付物责任化设 交付物责任 计VDS BOM/ 爆炸图PK ES产品项目规划书( PPC)BP 任 项MPL VAS 结构ES ES目管项目章程BP 责任性能分析评估PK BP DS理BP VTS/SOR EN整车进度表 DS项目Z&L Z&L 模型PK PK制造工程 FN模型责任 ME交付项物目计划责任BP DS交付物制造策略EN 初始装配数模工程 FN项目合同动力总成方案VL PT制造计划ME DFMEA EN市场调研报告MD BP3 采购计划初始装配模型PUVS产品和工装设计评估ME 100%计划T/V项100%目强制性指标BP 责任L/A/D/V 质量目标确认QA LLSVER项目质量计划QA 结CVER构车SVER LLIVER DS制造FN工程 SV 经济分析 责任成车 IV DS 集交付物VSV AC “U ”Rel 交付物集成车:采购,制造 供应商定点PU T/V责任 骡子车制造BOM EN 工程C/S/I VER 模具和样件制造 交付物责任 关键产品特性EN 产品发布 BOM EN ICD交付物EN责任责任 DTS EN认证实体模型数模DS KPC EN交付物 IDR 板金件责任 DS 责任 PQRR ME设施PQRR PQRR ME 开发ICD EN IDR 外饰 EN4DS 交付物责任交付物 骡子车试验 MVB责任 T/V STC EN EN IDR 内饰DS 生产准备虚拟Prod EN CV 评估VS MF PFMEA ME SOR EN 模具 /工装开发制造颜色 /面料ME DS EN FOVC动力总成摸底试验PS VTC ME SORP PR QA总装工艺文件ME尺寸检测系统VAL 65%PT集成车:试验“ P”Rel GD&T图EN GD&T 图ME 制造 100%PT MPCF Ca BP工艺控制文件ME总装培训计划Cal ME l责任交付物责任MVNS/MVS C/S/I VER 模具和零件制造 ME OTS 试验认证 VS 厂房设计ME 零件采购PU DS模具 /工装采购PU 产品工程 白车身制造T/V IV制造文档管理T/V 交付物责任 PMR交付物ME责任
仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料 药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安 全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产 出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要 求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关 物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要 将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制 指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用 非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如 鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不 尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳 定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月) 项目项目内容所需时间一产品信息调研质量标准、工艺处方等一周二前期准备1、参比制剂的采购:一个月
1 目的 加强设计开发的过程控制, 以保证产品设计质量。 2 适用围 规定了新产品设计、开发过程中应进行的活动容和管理程序,适用于本公司新产品的设计开 发 。 3 定义 Core Team ─核心小组,是由与设计/开发相关各部门代表组成,综合负责产品设计/开发过程中不同部门的分工与协调的组织。 PPP ─Product Program Proposal ,即产品项目建议书。 PRD ─GE Medical System Phase Review Discipline,即GE 医疗系统对产品开发指南。 SDRS ─System Design Requirement Specification,即系统设计要求。 DHF ─Design History File ,即设计开发过程文件: DHF 包括设计开发计划、PPP 、SRS 、DRS 、设计评审会议纪要,软件开发文件,分险分析,设计验证计划和报告,设计确认计划和报告,生产计划,技术支持计划,Milestone 评审文件待证明设计开发过程的文件。 首批样品─开发新品,设计更改首批及供应商变更时,供应上提供的第一批货物为首样品。设计更改首批,技术部作为协调工作进行的部门;供应商变更首批,技术部提供技术支持。 4 设计控制主要容: 4.1 设计控制流程图1 4.2设计控制(design control)的容包括 设计计划(design plan)。 详细要求见程序文件PD-E-02 设计控制方式 设计控制方式 用户需求 设计输入 设计过程 SDRS SRS DRS 等 设计输出 图形 硬件 规范 文件编制 可执行码等xx 设备 PPP PDP 等 销售的产品 SDD HDD 等
(完整)制剂仿制药研发具体流程 编辑整理: 尊敬的读者朋友们: 这里是精品文档编辑中心,本文档内容是由我和我的同事精心编辑整理后发布的,发布之前我们对文中内容进行仔细校对,但是难免会有疏漏的地方,但是任然希望((完整)制剂仿制药研发具体流程)的内容能够给您的工作和学习带来便利。同时也真诚的希望收到您的建议和反馈,这将是我们进步的源泉,前进的动力。 本文可编辑可修改,如果觉得对您有帮助请收藏以便随时查阅,最后祝您生活愉快业绩进步,以下为(完整)制剂仿制药研发具体流程的全部内容。
制剂仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查. 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一. 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同"。方法为对比研究。 1。安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。2。有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比. 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化. 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。
简述仿制药的研发流程 (1)产品信息调研:1、参比制剂的采购: 2、原料采购: 3、色谱柱及对照品采购: 4、辅料采购5、包材的采购 (2)前期准备:1、原辅料及参比制剂的检验: 2、处方工艺摸索3、初步验证工艺4、中试生产及工艺验证 (3)处方工艺研究:1、中试批量: 2、中试生产 3、工艺验证 (4)质量研究:1、质量研究项目的选择及方法初步确定2、质量标准的方法学验证3、质量对比研究(稳定性研究期间) (5)稳定性研究:1、影响因素试验 2、包材相容性试验 3、加速试验 4、长期试验 5、稳定性研究结果的评价 (6)药理毒理研究:1、药理毒理资料进行整理归纳总结 2、试验委托 (7)申报资料的撰写、整理:1、综述资料 2、药学研究资料 3、药理毒理研究资料 4、临床试验资料 (8)申报现场审核:1、将资料和电子申报表报省局,准备现场核查。 2、动态三批现场工艺核查,抽样送检省药检所复检。 (9)临床研究:1、固体口服制剂做生物等效性 2、溶液剂一般可免临床 3、局部用制剂一般需做临床试验 (10)申报生产:临床试验完成后,整理资料,申报省局。 试述申报资料撰写要求: 申报资料项目: 一、综述资料包括: 1.药品名称。 2.证明性文件。 3.立题目的与依据。 4.对主要研究结果的总结及评价。 5.药品说明书样稿、起草说明及最新参考文献。 6.包装、标签设计样稿。 二、药学研究资料 7.药学研究资料综述。 8.药材来源及鉴定依据。 9.药材生态环境、生长特征、形态描述、栽培或培植(培育) 技术、产地加工和炮制方法等。 10.药材标准草案及起草说明,并提供药品标准物质及有关资料。 11.提供植物、矿物标本,植物标本应当包括花、果实、种子等。 12.生产工艺的研究资料、工艺验证资料及文献资料,辅料来源 及质量标准。 13.化学成份研究的试验资料及文献资料。 14.质量研究工作的试验资料及文献资料。 15.药品标准草案及起草说明,并提供药品标准物质及有关资料。 16.样品检验报告书。 17.药物稳定性研究的试验资料及文献资料。 18.直接接触药品的包装材料和容器的选择依据及质量标准。 三、药理毒理研究资料 19.药理毒理研究资料综述。 20.主要药效学试验资料及文献资料。 21.一般药理研究的试验资料及文献资料。 22.急性毒性试验资料及文献资料。 23.长期毒性试验资料及文献资料。
制剂仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定
性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
仿制药研发具体流程
————————————————————————————————作者: ————————————————————————————————日期:
仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
文件类型: 程序文件 发行部门: 技术部 版本:0 页数:第 1 页 共 9 页 1 目的 加强设计开发的过程控制, 以保证产品设计质量。 2 适用范围 规定了新产品设计、开发过程中应进行的活动内容和管理程序,适用于本公司新产品的设计开 发 。 3 定义 Core Team ─核心小组,是由与设计/开发相关各部门代表组成,综合负责产品设计/开发过程中不同部门的分工与协调的组织。 PPP ─Product Program Proposal ,即产品项目建议书。 PRD ─GE Medical System Phase Review Discipline,即GE 医疗系统对产品开发指南。 SDRS ─System Design Requirement Specification,即系统设计要求。 DHF ─Design History File ,即设计开发过程文件: DHF 包括设计开发计划、PPP 、SRS 、DRS 、设计评审会议纪要,软件开发文件,分险分析,设计验证计划和报告,设计确认计划和报告,生产计划,技术支持计划,Milestone 评审文件待证明设计开发过程的文件。 首批样品─开发新品,设计更改首批及供应商变更时,供应上提供的第一批货物为首样品。设计更改首批,技术部作为协调工作进行的部门;供应商变更首批,技术部提供技术支持。 4 设计控制主要内容: 4.1 设计控制流程图1 设计控制方式 用户需求 设计输入 设计过程 SDRS SRS DRS 等 设计输出 图形 硬件 规范 文件编制 可执行码等xx 设备 PPP PDP 等 销售的产品 SDD HDD 等
详解仿制药研发具体流程
详解仿制药研发具体流程目录 一、综述2 二、仿制药研发项目汇总3 三、仿制药的研发具体步骤:5 (一)产品信息调研5 (二)前期准备(约一个月完成):5 1、参比制剂的采购5 2、原料采购5 3、色谱柱及对照品采购5 4、辅料采购:6 5、包材的采购:6 (三)处方工艺研究6 1、原辅料及参比制剂的检验6 2、处方工艺摸索6
3、初步验证工艺8 4、中试生产及工艺验证8 (四)质量研究9 1、质量研究项目的选择及方法初步确定9 2、质量标准的方法学验证10 3、质量对比研究12 4、质量标准的制定14 (五)稳定性研究(中试产品)14 (六)药理毒理研究16 (七)申报资料的撰写、整理16 (八)申报临床及申报现场核查17 (九)临床研究17 (十)申报生产17 一、综述
根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”:
制剂仿制药研发具体流 程 集团标准化工作小组 #Q8QGGQT-GX8G08Q8-GNQGJ8-MHHGN#
制剂仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。
研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
仿制药研发具体流程 凯博思2014年2月18日质量72 views暂无评论 目录 [隐藏] ?1综述 o 1.1安全性: o 1.2有效性: o 1.3晶型: ?2仿制药研发项目汇总 ?3仿制药的研发具体步骤: o 3.1产品信息调研 o 3.2前期准备 o 3.3处方工艺研究: o 3.4质量研究 o 3.5稳定性研究(中试产品) o 3.6药理毒理研究 o 3.7申报资料的撰写、整理 o 3.8申报临床及申报现场核查 o 3.9临床研究 o 3.10申报生产 综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。
如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 安全性: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 有效性: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。
制剂仿制药研发具体流程 -20170916 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型:晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药 物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明; 稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
仿制药研发具体流程模板 1
仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定, 仿制药即是已有国家药品标准的原料药或者制剂, 该类药物国内已批准生产或上市销售, 经过国内外广泛使用, 其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求, 主要是以下几点: 1、规范对被仿制药品的选择原则, 即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料, 使申报规范, 统一。 4、强调了对比研究, 是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证, 目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求, 晶型的不同, 溶解度和稳定性不同。 分析上述新要求和参考指导原则, 从而得出结论: 仿制药研发的目的是做到规模化生产, 强调本地化, 以实现”替代性”。要求是做到”同”。方法为对比研究。 1.安全性”同”: 对于安全性, 口服固体制剂控制的主要为有关物质, 而液体制剂除控制有关物质外, 还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此, 必须要将防腐剂含量测定定入质量标准。 2
研究的内容: 静态上应包括杂质谱的对比, 单个杂质的对比, 杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比, 即稳定性对比研究。 2.有效性”同”: 对于口服固体制剂, 口服混悬剂( 包括干混悬剂) , 溶出曲线是主要的控制指标[1]; 对于口服溶液剂, 防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要, 控制点为口感、渗透压、PH及有无絮凝现象; 对于局部用制剂( 如鼻喷雾剂) , 粒度分布、渗透压及黏度是主要控制指标。 研究的内容: 分别进行溶出曲线对比; 粒度分布对比; 渗透压及黏度对比。 3.晶型: 晶型的不同, 药物的溶解度及稳定性有可能不相同, 从而导致生物利用度不尽相同。而某个药物的晶型, 文献资料很少; 制剂中原料的晶型测定有一定的难度; 在做成制剂的过程中, 又不能保证晶型不产生变化。 可是, 鉴于仿制药研究的特点, 溶解度方面可经过溶出曲线对比来说明; 稳定性方面可经过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总( 从立项到申报, 时间为10—12个月) 3
通用项目管理实战操作 主讲:钟老师(曾就职于华为技术有限公司8年(高级项目经理、高级讲师)、中兴通讯有限公司1年(负责项目管理能力提升、培训授课)、(中国)2年(工程项目管理、工程外包合同谈判),具备10年以上大型通信工程项目管理经验、项目管理培训经验) 课程对象:项目经理、项目组核心成员、职能部门主管、公司管理者 授课方式:知识讲解、案例演示讲解、实战演练、小组讨论、互动交流、游戏感悟、头脑风暴、强调学员参与。 【课程价值】 1、了解理解层次模型,清晰认知项的目经理角色; 2、了解项目管理的要素,掌握项目管理常用工具和方法; 3、分享项目管理的经验; 4、分析本公司项目管理的现状,找到项目管理优化改进的方向。 【培训内容】 1、了解理解层次模型,清晰认知项的目经理角色; 2、了解项目管理的要素,掌握项目管理常用工具和方法; 3、分享项目管理的经验; 4、分析本公司项目管理的现状,找到项目管理优化改进的方向。第一天上午:培训需求汇总分析、项目经理角色认知、项目管理认知 第一天下午:项目管理要点:五大过程之启动、计划部分 第二天上午:项目管理要点:实施与监控、收尾部分 项目管理经验分享: 高效运作的典型案例分享 流程化、规范化运作的典型案例分享 精细化管理的典型案例分享 第二天下午:项目管理专题研讨: 进度管理篇、问题管理篇、项目经理管理意识与能力提升篇 头脑风暴:某公司项目管理现状分析与优化改进建议探讨
前言:培训需求汇总和分析 企业主营业务是什么? 哪些业务用项目的方式在运作? 项目运作过程中有哪些困难和问题? 项目管理培训的需求有哪些? 根据问卷调查与电话访谈的结果来定制培训的内容大家还有什么需求? 一、项目经理角色认知(这一模块可选) 前言: 作为一位管理者,你的职责是什么? 项目经理的角色(职责)应当如何界定? 1、认知工具:理解层次模型 (1)理解层次的理论模型 (2)理解层次的管理应用 管理自己 管理他人 (3)理解层次的认知应用 微软唐骏的沟通案例 2、项目经理的身份职责 (1)项目的第一责任人 (2)项目干系人的管理者 (3)项目经理是问题管理与推动解决者 (4)带领团队达成目标 案例研讨:项目经理李经理被客户投诉! 李经理是合格的项目经理吗?为什么? 李经理如何改进管理? 二、项目管理认知 前言: 项目与日常运作有哪些区别?
一、新药的研发过程 发明和研究安全有效的新药是一个长期、艰难和昂贵的进程。一种药物从最初的实验室研究到最终摆放到药柜销售平均需要花费12年的时间。进行临床前试验的5000种化合物中只有5种能进入到后续的临床试验,而仅其中的1种化合物可以得到最终的上市批准。新药研发阶段如下:(一)新药物实体的发现和确立。根据化学或生物学药物设计、天然药物、生物药物既有的经验理论、偶然的发现或现有临床的经验启发等确立研发靶标及新药物实体的来源方案。 (二)临床前试验。由制药公司进行的实验室和动物研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估。 (三)研发中新药申请(Investigational New Application, IND)。在临床前试验完成后,公司要向FDA 提请一份IND,之后才能开始进行药物的人体试验。如果30天内FDA没有发出不予批准的申明,此IND即为有效。提出的IND需包括以下内容:先期的试验结果,后续研究的方式、地点以及研究对象;化合物的化学结构;在体内的作用机制;动物研究中发现的任何毒副作用以及化合物的生产工艺。另外,IND必须得到制度审核部门(the Institutional Review Board)的审核和批准。同时,后续的临床研究需至少每年向FDA提交一份进展报告并得到准许。 (四)临床试验。
(1)Ⅰ期:此阶段大概需要1年时间,这些试验研究了药物的安全性方面,包括安全剂量范围。此阶段的研究同时确定了药物在体内的吸收、分布、代谢和排泄、以及药物的作用持续时间等项目。 (2)Ⅱ期:此阶段需要约100到300名志愿患者参与进行一些控制研究,以评价药物的疗效。这个阶段大约需要2年时间。 (3) Ⅲ期:此阶段持续约3年时间,医师通过对病患的监测以确定疗效和不良反应。 (五)新药申请(New Drug Application, NDA)。通过三个阶段的临床试验,将分析所有的试验数据。如数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律,FDA审核一份NDA的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA批准的过程都超过这个时限。 (六)批准。FDA批准一份新药申请,此种新药就可以被医师用于处方。公司必须继续向FDA提交阶段性报告,包括所有的不良反应报告和一些质量控制记录。FDA还可能对一些药物要求做进一步的研究(Ⅳ期),以评价药物的长期疗效。 二、仿制药的研发过程 仿制药是已有国家药品标准的原料药或制剂,该类药物
上海通用汽车有限公司物流解决方案 1、背景介绍 上海通用汽车有限公司(SGM)是中美两国迄今为止最大的合资企业,项目总投资15.2亿美元,被列为98年市府一号工程。中远作为中国第一大航运企业在经过了投标竞标后,承担起SGM的汽车零配件CKD 的运输任务。这是中远迄今为止最大的签约项目。1998年7月,中远集运和SGM汽车签订门到门运输协议,其中由上海中货负责SGM汽车零件CKD从上海港九区至SGM的再配送中心(RDC)的一关三检、码头提箱和内陆运输任务。 SGM拥有世界上最先进的弹性生产线,能够在一条流水线上同时生产不同型号、颜色的车辆,每小时可生产27辆汽车。在如此强大的生产力支持下,SGM在国内首创订单生产模式,紧密根据市场需求控制产量。同时,SGM的生产用料供应采用标准的JIT(JUST IN TIME)运作模式,由国际著名的RYDER物流咨询公司为其设计实行零库存管理,即所有CKD的库存存在于运输途中,不占用大型仓库,而仅在生产线旁设立RDC(再配送中心),维持288台套的最低安全库存。换言之,一旦SGM生产线满负荷运转起来,最低安全库存仅能保证11小时的零件供应,因此,系统设置至少12小时必须更新、补充零件储备。SGM每日的生产用料呈波动状态,相应的零件拉动也是呈不规则变化,以保持最低安全库存。这就要求采购、包装、海运、进口报关、检疫、陆路运输、拉动计划等一系列操作之间的衔接必须十分
密切,不能有丝毫差错。而且,生产用料供应的反应速度必须在12小时之内。 但是这个物流体系安全运作的前提是建立在市场计划(MARKETING PLAN)周期大于运输周期(TRANSPORTATION TIME)的基础上,CKD运输量才能根据实际生产需求决定。RYDER公司为SGM的整体物流设计未考虑到中国国情与北美大陆的差异,其先进的JIT运作方式反而成了不可察觉的先天隐患。事实上,SGM的市场计划周期为一周,而运输周期为四个月,这样市场计划无法指导CKD的运输安排,为了确保生产的连续性,SGM只能扩大其CKD储备量,造成大量到港的集装箱积压。 由于在计划采购等前面环节发生偏差从而产生瓶颈效应,使得海运、进口报关、内陆运输等后续工作难度大增。进口报关方面经常发生紧急报关情况;集装箱积压,RDC库存能力不够,而传统的集装箱内陆运输已无法适应SGM小批量、高频率的零件拉动。 同时,通用别克(BUICK)经历了99年销售旺势之后,2000年度市场销售大大低于预测。SGM这个汽车巨人只能压缩其产量,CKD的供应量无法紧随生产-销售的峰值变化而加以控制。所有这些情况都给SGM的物流体系造成了极大的压力: ①库存能力的压力