guide-annex_3_en
- 格式:pdf
- 大小:325.10 KB
- 文档页数:86

ANNEX 1附录ⅠMANUFACTURE OF STERILE MEDICINAL PRODUCTS无菌药品的生产Principle原则The manufacture of sterile products is subject to special requirements in order to minimize risks of microbiological contamination, and of particulate and pyrogen contamination. Much depends on the skill, training and attitudes of the personnel involved. Quality Assurance is particularly important, and this type of manufacture must strictly follow carefully established and validated methods of preparation and procedure. Sole reliance for sterility or other quality aspects must not be placed on any terminal process or finished product test.为将微生物、微粒和热原污染的风险降低至最小限度,无菌药品的生产应有各种特殊要求。
这在很大程度上取决于生产人员的技能、所接受的培训及其工作态度。
质量保证极为重要,无菌药品的生产必须严格按照精心制订并经验证的方法及规程进行。
产品的无菌或其它质量特性绝不能仅依赖于任何形式的最终操作或成品检验。
Note:注:This guidance does not lay down detailed methods for determining the microbiological and particulate cleanliness of air, surfaces etc. Reference should be made to other documents such as the EN/ISO Standards.本指南没有制定详细的空气和物体表面等的微生物和微粒的测定方法,相关事项可以参照其他文件,诸如欧洲/国际标准。

CAPE PARROTIDENTIFICATION GUIDECompiled on behalf of the Scientific Authority of SouthAfricaMay 2015(English only / Únicamente en inglés / Seulement en anglais)AC28 Doc. 21.1Annex 5A guide to distinguish the Cape Parrot (Poicephalusrobustus) and the Grey-headed Parrot (Poicephalusfuscicollis suahelicus)BackgroundUntil recently the Cape parrot (Poicephalus robustus robustus) was not recognised as a separate species from the grey-headed parrot (Poicephalus robustus fuscicollis) and both these taxa were therefore included together in Appendix II to CITES and traded under the name Poicephalus robustus. The Cape parrot, a South African endemic, has a restricted and fragmented distribution with an estimated global population size of 1000 – 1500 individuals representing considerably less than 500 breeding pairs. The species is listed as Endangered in the South African Red Data Book for Birds, as critically endangered on the current Threatened or Protected species list (section 56 of the National Environmental Management: Biodiversity Act No. 10 (NEMBA) of 2004), as specially protected in the province of KwaZulu-Natal and is a protected species in the Eastern Cape province of South Africa. However, due to the grey-headed parrot and Cape parrot not being recognised as two separate species, both taxa are together considered as Least Concern at a global level.However, genetic work recently conducted by the University of KwaZulu-Natal (paper submitted and accepted by PLOSONE – revision in progress) indicates clear genetic differentiation between P.r. robustus and the P.r. suahelicus - P.r. fuscicollis cluster, supporting previous recommendations that the Cape parrot should be viewed as a separate species, namely P. robustus, and that the grey-headed parrot and brown-necked parrot should be grouped under the P. fuscicollis species complex as P.f. suahelicus and P.f. fuscicollis respectively.The Cape parrot (Poicephalus robustus) has been the focus of much debate over the past few years, from its taxonomic position, to habitat use and population declines. Various factors have contributed to population declines in the species such as the illegal harvest of individuals from the wild for the pet trade, habitat destruction and diseases, specifically psittacine beak and feather virus (Wirminghaus et al. 1999).In preparation for the Cape parrot being treated as a separate species under CITES, the South African National Biodiversity Institute developed an identification guide to assist law enforcers with identifying the Cape parrot and distinguishing it from the grey-headed parrot. The guide highlights the main morphological and colour differences between the two species. It consists of1. A brief description of the two species,2. Tables listing the main ecological, morphological and biological differencesbetween the two species (tables 1 & 2 and fig. 1),3. Photographs of both sexes annotated to show the colour differences betweenthe two species (fig. 2, 3, & 4), and4. A colour palette (table 3) to assist in recognising the various coloursdescribed.Brief description of the Cape parrotThe Cape parrot has small body dimensions, a narrow bill; the head colour is olive, yellow to golden.The head, throat and neck are olive yellow to golden brown. The body and the wings are dark green. Thighs and outer edges of the wings orange-red and the tail and flight feathers are bottle-green to black. Females typically have an orange-red forehead. Males have a dark-earth fore crown and no colouration on the forehead. Juveniles resemble the female but without red on the tibia and wing edges.Brief description of the grey-headed parrotThe grey-headed parrot has large body dimensions; the bill is heavier basally with the apex tapered and longer. Head colour is silver grey.The grey-headed parrot is generally larger than the Cape parrot and has a short tail. The body is green. Adults normally have orange-red patches on shoulders and thighs. Just like the Cape parrot, females have a well-defined orange-red forehead. Similar to the Cape parrot, juveniles resemble females with the orange-red colour on the forehead.Table 1: Ecological, morphological and biological differences between the Cape and the grey-headed parrot.5Table 2: Morphometric comparison of the Cape and grey-headed parrot.678Figure 1:Different body parts of a bird (bird anatomy).9Figure 2: Male Cape parrot (P. robustus) (left) and male grey-headed parrot (P. fuscicollis suahelicus ) (right)Fore crown colour is rusty grey-brownFore crown is dark earth-brownNape and Collar colours are warm-rusty brown Nape and Collar yellow green Upper chest is yellowgreenUpper chest colour isrusty grey brown10 Figure 3:Male grey-headed parrot.11Figure 4:Female grey-headed parrot (left) and female Cape parrot (right).Table 3: Colour palette showing the different colours occurring on the head and body of the Cape and the grey-headed parrotOlive green Olive green Rusty grey-brown Silvery grey-brown12Orange brown Orange brown Warm grey-brown Yellow green Yellow green Rusty grey-brown Grey-green13Dark oily-green Dark green Olive-greenBlue-green Blue-green Leaf-green Blue-green1415ReferencesDOWNS, C. T. 2005. Abundance of the endangered Cape parrot, Poicephalus robustus, in South Africa: implications for its survival. African Zoology 4(1): 15-24.PERRIN, M. 2012. Parrots of Africa, Madagascar and the Mascarene Islands. Wits University Press. 1 Jan Smuts Avenue. Johannesburg.WIRMINGHAUS, J.O., DOWNS, C.T., SYMES, C.T. & PERRIN, M.R. 1999. Conservation of the Cape parrot in southern Africa. South African Journal of Wildlife Research 29: 118–129.WIRMINGHAUS. J. O., DOWNS. C. T., PERRIN. M. R. and SYMES. C. T. 2002. Taxonomic relationships of the subspecies of the Cape Parrot Poicephalus robustus (Gmelin).Journal of Natural History. 36: 361-378.16。

NuMicro®FamilyArm® ARM926EJ-S BasedNuMaker-HMI-N9H30User ManualEvaluation Board for NuMicro® N9H30 SeriesNUMAKER-HMI-N9H30 USER MANUALThe information described in this document is the exclusive intellectual property ofNuvoton Technology Corporation and shall not be reproduced without permission from Nuvoton.Nuvoton is providing this document only for reference purposes of NuMicro microcontroller andmicroprocessor based system design. Nuvoton assumes no responsibility for errors or omissions.All data and specifications are subject to change without notice.For additional information or questions, please contact: Nuvoton Technology Corporation.Table of Contents1OVERVIEW (5)1.1Features (7)1.1.1NuMaker-N9H30 Main Board Features (7)1.1.2NuDesign-TFT-LCD7 Extension Board Features (7)1.2Supporting Resources (8)2NUMAKER-HMI-N9H30 HARDWARE CONFIGURATION (9)2.1NuMaker-N9H30 Board - Front View (9)2.2NuMaker-N9H30 Board - Rear View (14)2.3NuDesign-TFT-LCD7 - Front View (20)2.4NuDesign-TFT-LCD7 - Rear View (21)2.5NuMaker-N9H30 and NuDesign-TFT-LCD7 PCB Placement (22)3NUMAKER-N9H30 AND NUDESIGN-TFT-LCD7 SCHEMATICS (24)3.1NuMaker-N9H30 - GPIO List Circuit (24)3.2NuMaker-N9H30 - System Block Circuit (25)3.3NuMaker-N9H30 - Power Circuit (26)3.4NuMaker-N9H30 - N9H30F61IEC Circuit (27)3.5NuMaker-N9H30 - Setting, ICE, RS-232_0, Key Circuit (28)NUMAKER-HMI-N9H30 USER MANUAL3.6NuMaker-N9H30 - Memory Circuit (29)3.7NuMaker-N9H30 - I2S, I2C_0, RS-485_6 Circuit (30)3.8NuMaker-N9H30 - RS-232_2 Circuit (31)3.9NuMaker-N9H30 - LCD Circuit (32)3.10NuMaker-N9H30 - CMOS Sensor, I2C_1, CAN_0 Circuit (33)3.11NuMaker-N9H30 - RMII_0_PF Circuit (34)3.12NuMaker-N9H30 - RMII_1_PE Circuit (35)3.13NuMaker-N9H30 - USB Circuit (36)3.14NuDesign-TFT-LCD7 - TFT-LCD7 Circuit (37)4REVISION HISTORY (38)List of FiguresFigure 1-1 Front View of NuMaker-HMI-N9H30 Evaluation Board (5)Figure 1-2 Rear View of NuMaker-HMI-N9H30 Evaluation Board (6)Figure 2-1 Front View of NuMaker-N9H30 Board (9)Figure 2-2 Rear View of NuMaker-N9H30 Board (14)Figure 2-3 Front View of NuDesign-TFT-LCD7 Board (20)Figure 2-4 Rear View of NuDesign-TFT-LCD7 Board (21)Figure 2-5 Front View of NuMaker-N9H30 PCB Placement (22)Figure 2-6 Rear View of NuMaker-N9H30 PCB Placement (22)Figure 2-7 Front View of NuDesign-TFT-LCD7 PCB Placement (23)Figure 2-8 Rear View of NuDesign-TFT-LCD7 PCB Placement (23)Figure 3-1 GPIO List Circuit (24)Figure 3-2 System Block Circuit (25)Figure 3-3 Power Circuit (26)Figure 3-4 N9H30F61IEC Circuit (27)Figure 3-5 Setting, ICE, RS-232_0, Key Circuit (28)Figure 3-6 Memory Circuit (29)Figure 3-7 I2S, I2C_0, RS-486_6 Circuit (30)Figure 3-8 RS-232_2 Circuit (31)Figure 3-9 LCD Circuit (32)NUMAKER-HMI-N9H30 USER MANUAL Figure 3-10 CMOS Sensor, I2C_1, CAN_0 Circuit (33)Figure 3-11 RMII_0_PF Circuit (34)Figure 3-12 RMII_1_PE Circuit (35)Figure 3-13 USB Circuit (36)Figure 3-14 TFT-LCD7 Circuit (37)List of TablesTable 2-1 LCD Panel Combination Connector (CON8) Pin Function (11)Table 2-2 Three Sets of Indication LED Functions (12)Table 2-3 Six Sets of User SW, Key Matrix Functions (12)Table 2-4 CMOS Sensor Connector (CON10) Function (13)Table 2-5 JTAG ICE Interface (J2) Function (14)Table 2-6 Expand Port (CON7) Function (16)Table 2-7 UART0 (J3) Function (16)Table 2-8 UART2 (J6) Function (16)Table 2-9 RS-485_6 (SW6~8) Function (17)Table 2-10 Power on Setting (SW4) Function (17)Table 2-11 Power on Setting (S2) Function (17)Table 2-12 Power on Setting (S3) Function (17)Table 2-13 Power on Setting (S4) Function (17)Table 2-14 Power on Setting (S5) Function (17)Table 2-15 Power on Setting (S7/S6) Function (18)Table 2-16 Power on Setting (S9/S8) Function (18)Table 2-17 CMOS Sensor Connector (CON9) Function (19)Table 2-18 CAN_0 (SW9~10) Function (19)NUMAKER-HMI-N9H30 USER MANUAL1 OVERVIEWThe NuMaker-HMI-N9H30 is an evaluation board for GUI application development. The NuMaker-HMI-N9H30 consists of two parts: a NuMaker-N9H30 main board and a NuDesign-TFT-LCD7 extensionboard. The NuMaker-HMI-N9H30 is designed for project evaluation, prototype development andvalidation with HMI (Human Machine Interface) function.The NuMaker-HMI-N9H30 integrates touchscreen display, voice input/output, rich serial port serviceand I/O interface, providing multiple external storage methods.The NuDesign-TFT-LCD7 can be plugged into the main board via the DIN_32x2 extension connector.The NuDesign-TFT-LCD7 includes one 7” LCD which the resolution is 800x480 with RGB-24bits andembedded the 4-wires resistive type touch panel.Figure 1-1 Front View of NuMaker-HMI-N9H30 Evaluation BoardNUMAKER-HMI-N9H30 USER MANUAL Figure 1-2 Rear View of NuMaker-HMI-N9H30 Evaluation Board1.1 Features1.1.1 NuMaker-N9H30 Main Board Features●N9H30F61IEC chip: LQFP216 pin MCP package with DDR (64 MB)●SPI Flash using W25Q256JVEQ (32 MB) booting with quad mode or storage memory●NAND Flash using W29N01HVSINA (128 MB) booting or storage memory●One Micro-SD/TF card slot served either as a SD memory card for data storage or SDIO(Wi-Fi) device●Two sets of COM ports:–One DB9 RS-232 port with UART_0 used 75C3232E transceiver chip can be servedfor function debug and system development.–One DB9 RS-232 port with UART_2 used 75C3232E transceiver chip for userapplication●22 GPIO expansion ports, including seven sets of UART functions●JTAG interface provided for software development●Microphone input and Earphone/Speaker output with 24-bit stereo audio codec(NAU88C22) for I2S interfaces●Six sets of user-configurable push button keys●Three sets of LEDs for status indication●Provides SN65HVD230 transceiver chip for CAN bus communication●Provides MAX3485 transceiver chip for RS-485 device connection●One buzzer device for program applicationNUMAKER-HMI-N9H30 USER MANUAL●Two sets of RJ45 ports with Ethernet 10/100 Mbps MAC used IP101GR PHY chip●USB_0 that can be used as Device/HOST and USB_1 that can be used as HOSTsupports pen drives, keyboards, mouse and printers●Provides over-voltage and over current protection used APL3211A chip●Retain RTC battery socket for CR2032 type and ADC0 detect battery voltage●System power could be supplied by DC-5V adaptor or USB VBUS1.1.2 NuDesign-TFT-LCD7 Extension Board Features●7” resolution 800x480 4-wire resistive touch panel for 24-bits RGB888 interface●DIN_32x2 extension connector1.2 Supporting ResourcesFor sample codes and introduction about NuMaker-N9H30, please refer to N9H30 BSP:https:///products/gui-solution/gui-platform/numaker-hmi-n9h30/?group=Software&tab=2Visit NuForum for further discussion about the NuMaker-HMI-N9H30:/viewforum.php?f=31 NUMAKER-HMI-N9H30 USER MANUALNUMAKER-HMI-N9H30 USER MANUAL2 NUMAKER-HMI-N9H30 HARDWARE CONFIGURATION2.1 NuMaker-N9H30 Board - Front View Combination Connector (CON8)6 set User SWs (K1~6)3set Indication LEDs (LED1~3)Power Supply Switch (SW_POWER1)Audio Codec(U10)Microphone(M1)NAND Flash(U9)RS-232 Transceiver(U6, U12)RS-485 Transceiver(U11)CAN Transceiver (U13)Figure 2-1 Front View of NuMaker-N9H30 BoardFigure 2-1 shows the main components and connectors from the front side of NuMaker-N9H30 board. The following lists components and connectors from the front view:NuMaker-N9H30 board and NuDesign-TFT-LCD7 board combination connector (CON8). This panel connector supports 4-/5-wire resistive touch or capacitance touch panel for 24-bits RGB888 interface.Connector GPIO pin of N9H30 FunctionCON8.1 - Power 3.3VCON8.2 - Power 3.3VCON8.3 GPD7 LCD_CSCON8.4 GPH3 LCD_BLENCON8.5 GPG9 LCD_DENCON8.7 GPG7 LCD_HSYNCCON8.8 GPG6 LCD_CLKCON8.9 GPD15 LCD_D23(R7)CON8.10 GPD14 LCD_D22(R6)CON8.11 GPD13 LCD_D21(R5)CON8.12 GPD12 LCD_D20(R4)CON8.13 GPD11 LCD_D19(R3)CON8.14 GPD10 LCD_D18(R2)CON8.15 GPD9 LCD_D17(R1)CON8.16 GPD8 LCD_D16(R0)CON8.17 GPA15 LCD_D15(G7)CON8.18 GPA14 LCD_D14(G6)CON8.19 GPA13 LCD_D13(G5)CON8.20 GPA12 LCD_D12(G4)CON8.21 GPA11 LCD_D11(G3)CON8.22 GPA10 LCD_D10(G2)CON8.23 GPA9 LCD_D9(G1) NUMAKER-HMI-N9H30 USER MANUALCON8.24 GPA8 LCD_D8(G0)CON8.25 GPA7 LCD_D7(B7)CON8.26 GPA6 LCD_D6(B6)CON8.27 GPA5 LCD_D5(B5)CON8.28 GPA4 LCD_D4(B4)CON8.29 GPA3 LCD_D3(B3)CON8.30 GPA2 LCD_D2(B2)CON8.31 GPA1 LCD_D1(B1)CON8.32 GPA0 LCD_D0(B0)CON8.33 - -CON8.34 - -CON8.35 - -CON8.36 - -CON8.37 GPB2 LCD_PWMCON8.39 - VSSCON8.40 - VSSCON8.41 ADC7 XPCON8.42 ADC3 VsenCON8.43 ADC6 XMCON8.44 ADC4 YMCON8.45 - -CON8.46 ADC5 YPCON8.47 - VSSCON8.48 - VSSCON8.49 GPG0 I2C0_CCON8.50 GPG1 I2C0_DCON8.51 GPG5 TOUCH_INTCON8.52 - -CON8.53 - -CON8.54 - -CON8.55 - -NUMAKER-HMI-N9H30 USER MANUAL CON8.56 - -CON8.57 - -CON8.58 - -CON8.59 - VSSCON8.60 - VSSCON8.61 - -CON8.62 - -CON8.63 - Power 5VCON8.64 - Power 5VTable 2-1 LCD Panel Combination Connector (CON8) Pin Function●Power supply switch (SW_POWER1): System will be powered on if the SW_POWER1button is pressed●Three sets of indication LEDs:LED Color DescriptionsLED1 Red The system power will beterminated and LED1 lightingwhen the input voltage exceeds5.7V or the current exceeds 2A.LED2 Green Power normal state.LED3 Green Controlled by GPH2 pin Table 2-2 Three Sets of Indication LED Functions●Six sets of user SW, Key Matrix for user definitionKey GPIO pin of N9H30 FunctionK1 GPF10 Row0 GPB4 Col0K2 GPF10 Row0 GPB5 Col1K3 GPE15 Row1 GPB4 Col0K4 GPE15 Row1 GPB5 Col1K5 GPE14 Row2 GPB4 Col0K6GPE14 Row2GPB5 Col1 Table 2-3 Six Sets of User SW, Key Matrix Functions●NAND Flash (128 MB) with Winbond W29N01HVS1NA (U9)●Microphone (M1): Through Nuvoton NAU88C22 chip sound input●Audio CODEC chip (U10): Nuvoton NAU88C22 chip connected to N9H30 using I2Sinterface–SW6/SW7/SW8: 1-2 short for RS-485_6 function and connected to 2P terminal (CON5and J5)–SW6/SW7/SW8: 2-3 short for I2S function and connected to NAU88C22 (U10).●CMOS Sensor connector (CON10, SW9~10)–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11)–SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensorconnector (CON10)Connector GPIO pin of N9H30 FunctionCON10.1 - VSSCON10.2 - VSSNUMAKER-HMI-N9H30 USER MANUALCON10.3 - Power 3.3VCON10.4 - Power 3.3VCON10.5 - -CON10.6 - -CON10.7 GPI4 S_PCLKCON10.8 GPI3 S_CLKCON10.9 GPI8 S_D0CON10.10 GPI9 S_D1CON10.11 GPI10 S_D2CON10.12 GPI11 S_D3CON10.13 GPI12 S_D4CON10.14 GPI13 S_D5CON10.15 GPI14 S_D6CON10.16 GPI15 S_D7CON10.17 GPI6 S_VSYNCCON10.18 GPI5 S_HSYNCCON10.19 GPI0 S_PWDNNUMAKER-HMI-N9H30 USER MANUAL CON10.20 GPI7 S_nRSTCON10.21 GPG2 I2C1_CCON10.22 GPG3 I2C1_DCON10.23 - VSSCON10.24 - VSSTable 2-4 CMOS Sensor Connector (CON10) FunctionNUMAKER-HMI-N9H30 USER MANUAL2.2NuMaker-N9H30 Board - Rear View5V In (CON1)RS-232 DB9 (CON2,CON6)Expand Port (CON7)Speaker Output (J4)Earphone Output (CON4)Buzzer (BZ1)System ResetSW (SW5)SPI Flash (U7,U8)JTAG ICE (J2)Power ProtectionIC (U1)N9H30F61IEC (U5)Micro SD Slot (CON3)RJ45 (CON12, CON13)USB1 HOST (CON15)USB0 Device/Host (CON14)CAN_0 Terminal (CON11)CMOS Sensor Connector (CON9)Power On Setting(SW4, S2~S9)RS-485_6 Terminal (CON5)RTC Battery(BT1)RMII PHY (U14,U16)Figure 2-2 Rear View of NuMaker-N9H30 BoardFigure 2-2 shows the main components and connectors from the rear side of NuMaker-N9H30 board. The following lists components and connectors from the rear view:● +5V In (CON1): Power adaptor 5V input ●JTAG ICE interface (J2) ConnectorGPIO pin of N9H30Function J2.1 - Power 3.3V J2.2 GPJ4 nTRST J2.3 GPJ2 TDI J2.4 GPJ1 TMS J2.5 GPJ0 TCK J2.6 - VSS J2.7 GPJ3 TD0 J2.8-RESETTable 2-5 JTAG ICE Interface (J2) Function●SPI Flash (32 MB) with Winbond W25Q256JVEQ (U7); only one (U7 or U8) SPI Flashcan be used●System Reset (SW5): System will be reset if the SW5 button is pressed●Buzzer (BZ1): Control by GPB3 pin of N9H30●Speaker output (J4): Through the NAU88C22 chip sound output●Earphone output (CON4): Through the NAU88C22 chip sound output●Expand port for user use (CON7):Connector GPIO pin of N9H30 FunctionCON7.1 - Power 3.3VCON7.2 - Power 3.3VCON7.3 GPE12 UART3_TXDCON7.4 GPH4 UART1_TXDCON7.5 GPE13 UART3_RXDCON7.6 GPH5 UART1_RXDCON7.7 GPB0 UART5_TXDCON7.8 GPH6 UART1_RTSCON7.9 GPB1 UART5_RXDCON7.10 GPH7 UART1_CTSCON7.11 GPI1 UART7_TXDNUMAKER-HMI-N9H30 USER MANUAL CON7.12 GPH8 UART4_TXDCON7.13 GPI2 UART7_RXDCON7.14 GPH9 UART4_RXDCON7.15 - -CON7.16 GPH10 UART4_RTSCON7.17 - -CON7.18 GPH11 UART4_CTSCON7.19 - VSSCON7.20 - VSSCON7.21 GPB12 UART10_TXDCON7.22 GPH12 UART8_TXDCON7.23 GPB13 UART10_RXDCON7.24 GPH13 UART8_RXDCON7.25 GPB14 UART10_RTSCON7.26 GPH14 UART8_RTSCON7.27 GPB15 UART10_CTSCON7.28 GPH15 UART8_CTSCON7.29 - Power 5VCON7.30 - Power 5VTable 2-6 Expand Port (CON7) Function●UART0 selection (CON2, J3):–RS-232_0 function and connected to DB9 female (CON2) for debug message output.–GPE0/GPE1 connected to 2P terminal (J3).Connector GPIO pin of N9H30 Function J3.1 GPE1 UART0_RXDJ3.2 GPE0 UART0_TXDTable 2-7 UART0 (J3) Function●UART2 selection (CON6, J6):–RS-232_2 function and connected to DB9 female (CON6) for debug message output –GPF11~14 connected to 4P terminal (J6)Connector GPIO pin of N9H30 Function J6.1 GPF11 UART2_TXDJ6.2 GPF12 UART2_RXDJ6.3 GPF13 UART2_RTSJ6.4 GPF14 UART2_CTSTable 2-8 UART2 (J6) Function●RS-485_6 selection (CON5, J5, SW6~8):–SW6~8: 1-2 short for RS-485_6 function and connected to 2P terminal (CON5 and J5) –SW6~8: 2-3 short for I2S function and connected to NAU88C22 (U10)Connector GPIO pin of N9H30 FunctionSW6:1-2 shortGPG11 RS-485_6_DISW6:2-3 short I2S_DOSW7:1-2 shortGPG12 RS-485_6_ROSW7:2-3 short I2S_DISW8:1-2 shortGPG13 RS-485_6_ENBSW8:2-3 short I2S_BCLKNUMAKER-HMI-N9H30 USER MANUALTable 2-9 RS-485_6 (SW6~8) FunctionPower on setting (SW4, S2~9).SW State FunctionSW4.2/SW4.1 ON/ON Boot from USB SW4.2/SW4.1 ON/OFF Boot from eMMC SW4.2/SW4.1 OFF/ON Boot from NAND Flash SW4.2/SW4.1 OFF/OFF Boot from SPI Flash Table 2-10 Power on Setting (SW4) FunctionSW State FunctionS2 Short System clock from 12MHzcrystalS2 Open System clock from UPLL output Table 2-11 Power on Setting (S2) FunctionSW State FunctionS3 Short Watchdog Timer OFFS3 Open Watchdog Timer ON Table 2-12 Power on Setting (S3) FunctionSW State FunctionS4 Short GPJ[4:0] used as GPIO pinS4Open GPJ[4:0] used as JTAG ICEinterfaceTable 2-13 Power on Setting (S4) FunctionSW State FunctionS5 Short UART0 debug message ONS5 Open UART0 debug message OFFTable 2-14 Power on Setting (S5) FunctionSW State FunctionS7/S6 Short/Short NAND Flash page size 2KBS7/S6 Short/Open NAND Flash page size 4KBS7/S6 Open/Short NAND Flash page size 8KBNUMAKER-HMI-N9H30 USER MANUALS7/S6 Open/Open IgnoreTable 2-15 Power on Setting (S7/S6) FunctionSW State FunctionS9/S8 Short/Short NAND Flash ECC type BCH T12S9/S8 Short/Open NAND Flash ECC type BCH T15S9/S8 Open/Short NAND Flash ECC type BCH T24S9/S8 Open/Open IgnoreTable 2-16 Power on Setting (S9/S8) FunctionCMOS Sensor connector (CON9, SW9~10)–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11).–SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensorconnector (CON9).Connector GPIO pin of N9H30 FunctionCON9.1 - VSSCON9.2 - VSSCON9.3 - Power 3.3VCON9.4 - Power 3.3V NUMAKER-HMI-N9H30 USER MANUALCON9.5 - -CON9.6 - -CON9.7 GPI4 S_PCLKCON9.8 GPI3 S_CLKCON9.9 GPI8 S_D0CON9.10 GPI9 S_D1CON9.11 GPI10 S_D2CON9.12 GPI11 S_D3CON9.13 GPI12 S_D4CON9.14 GPI13 S_D5CON9.15 GPI14 S_D6CON9.16 GPI15 S_D7CON9.17 GPI6 S_VSYNCCON9.18 GPI5 S_HSYNCCON9.19 GPI0 S_PWDNCON9.20 GPI7 S_nRSTCON9.21 GPG2 I2C1_CCON9.22 GPG3 I2C1_DCON9.23 - VSSCON9.24 - VSSTable 2-17 CMOS Sensor Connector (CON9) Function●CAN_0 Selection (CON11, SW9~10):–SW9~10: 1-2 short for CAN_0 function and connected to 2P terminal (CON11) –SW9~10: 2-3 short for CMOS sensor function and connected to CMOS sensor connector (CON9, CON10)SW GPIO pin of N9H30 FunctionSW9:1-2 shortGPI3 CAN_0_RXDSW9:2-3 short S_CLKSW10:1-2 shortGPI4 CAN_0_TXDSW10:2-3 short S_PCLKTable 2-18 CAN_0 (SW9~10) Function●USB0 Device/HOST Micro-AB connector (CON14), where CON14 pin4 ID=1 is Device,ID=0 is HOST●USB1 for USB HOST with Type-A connector (CON15)●RJ45_0 connector with LED indicator (CON12), RMII PHY with IP101GR (U14)●RJ45_1 connector with LED indicator (CON13), RMII PHY with IP101GR (U16)●Micro-SD/TF card slot (CON3)●SOC CPU: Nuvoton N9H30F61IEC (U5)●Battery power for RTC 3.3V powered (BT1, J1), can detect voltage by ADC0●RTC power has 3 sources:–Share with 3.3V I/O power–Battery socket for CR2032 (BT1)–External connector (J1)●Board version 2.1NUMAKER-HMI-N9H30 USER MANUAL2.3 NuDesign-TFT-LCD7 -Front ViewFigure 2-3 Front View of NuDesign-TFT-LCD7 BoardFigure 2-3 shows the main components and connectors from the Front side of NuDesign-TFT-LCD7board.7” resolution 800x480 4-W resistive touch panel for 24-bits RGB888 interface2.4 NuDesign-TFT-LCD7 -Rear ViewFigure 2-4 Rear View of NuDesign-TFT-LCD7 BoardFigure 2-4 shows the main components and connectors from the rear side of NuDesign-TFT-LCD7board.NuMaker-N9H30 and NuDesign-TFT-LCD7 combination connector (CON1).NUMAKER-HMI-N9H30 USER MANUAL 2.5 NuMaker-N9H30 and NuDesign-TFT-LCD7 PCB PlacementFigure 2-5 Front View of NuMaker-N9H30 PCB PlacementFigure 2-6 Rear View of NuMaker-N9H30 PCB PlacementNUMAKER-HMI-N9H30 USER MANUALFigure 2-7 Front View of NuDesign-TFT-LCD7 PCB PlacementFigure 2-8 Rear View of NuDesign-TFT-LCD7 PCB Placement3 NUMAKER-N9H30 AND NUDESIGN-TFT-LCD7 SCHEMATICS3.1 NuMaker-N9H30 - GPIO List CircuitFigure 3-1 shows the N9H30F61IEC GPIO list circuit.Figure 3-1 GPIO List Circuit NUMAKER-HMI-N9H30 USER MANUAL3.2 NuMaker-N9H30 - System Block CircuitFigure 3-2 shows the System Block Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-2 System Block Circuit3.3 NuMaker-N9H30 - Power CircuitFigure 3-3 shows the Power Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-3 Power Circuit3.4 NuMaker-N9H30 - N9H30F61IEC CircuitFigure 3-4 shows the N9H30F61IEC Circuit.Figure 3-4 N9H30F61IEC CircuitNUMAKER-HMI-N9H30 USER MANUAL3.5 NuMaker-N9H30 - Setting, ICE, RS-232_0, Key CircuitFigure 3-5 shows the Setting, ICE, RS-232_0, Key Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-5 Setting, ICE, RS-232_0, Key Circuit3.6 NuMaker-N9H30 - Memory CircuitFigure 3-6 shows the Memory Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-6 Memory Circuit3.7 NuMaker-N9H30 - I2S, I2C_0, RS-485_6 CircuitFigure 3-7 shows the I2S, I2C_0, RS-486_6 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-7 I2S, I2C_0, RS-486_6 Circuit3.8 NuMaker-N9H30 - RS-232_2 CircuitFigure 3-8 shows the RS-232_2 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-8 RS-232_2 Circuit3.9 NuMaker-N9H30 - LCD CircuitFigure 3-9 shows the LCD Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-9 LCD Circuit3.10 NuMaker-N9H30 - CMOS Sensor, I2C_1, CAN_0 CircuitFigure 3-10 shows the CMOS Sensor,I2C_1, CAN_0 Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-10 CMOS Sensor, I2C_1, CAN_0 Circuit3.11 NuMaker-N9H30 - RMII_0_PF CircuitFigure 3-11 shows the RMII_0_RF Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-11 RMII_0_PF Circuit3.12 NuMaker-N9H30 - RMII_1_PE CircuitFigure 3-12 shows the RMII_1_PE Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-12 RMII_1_PE Circuit3.13 NuMaker-N9H30 - USB CircuitFigure 3-13 shows the USB Circuit.NUMAKER-HMI-N9H30 USER MANUALFigure 3-13 USB Circuit3.14 NuDesign-TFT-LCD7 - TFT-LCD7 CircuitFigure 3-14 shows the TFT-LCD7 Circuit.Figure 3-14 TFT-LCD7 CircuitNUMAKER-HMI-N9H30 USER MANUAL4 REVISION HISTORYDate Revision Description2022.03.24 1.00 Initial version NUMAKER-HMI-N9H30 USER MANUALNUMAKER-HMI-N9H30 USER MANUALImportant NoticeNuvoton Products are neither intended nor warranted for usage in systems or equipment, anymalfunction or failure of which may cause loss of human life, bodily injury or severe propertydamage. Such applications are deemed, “Insecure Usage”.Insecure usage includes, but is not limited to: equipment for surgical implementation, atomicenergy control instruments, airplane or spaceship instruments, the control or operation ofdynamic, brake or safety systems designed for vehicular use, traffic signal instruments, all typesof safety devices, and other applications intended to support or sustain life.All Insecure Usage shall be made at customer’s risk, and in the event that third parties lay claimsto Nuvoton as a result of customer’s Insecure Usage, custome r shall indemnify the damagesand liabilities thus incurred by Nuvoton.。

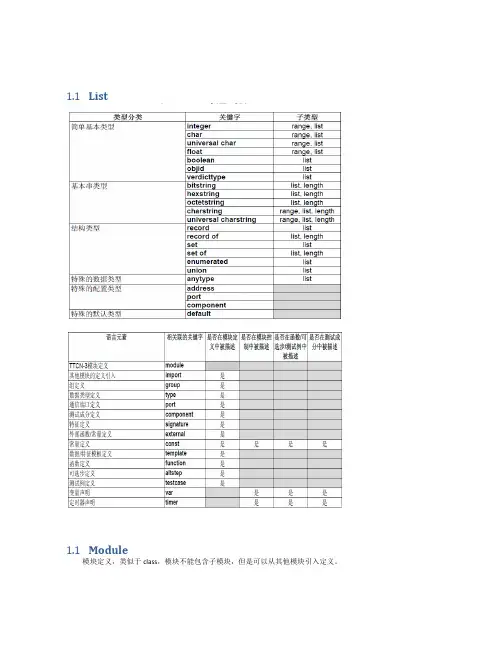
1.1List1.1Module模块定义,类似于class,模块不能包含子模块,但是可以从其他模块引入定义。
定义部分:定义测试成分、通信端口、数据类型、常数、测试数据模板、函数、测试端口上调用的过程特征(signatures)、测试例等等。
控制部分:分调用测试例并控制它们的执行。
控制部分也可以声明(局部)变量等,程序语句(如ifelse和do-while)可以用于各个测试例的选择和执行顺序。
TTCN-3不支持全局变量的概念。
1.2Type用户自定义数据类型type float pi (3.1415926);type set MySetType{integer field1,charstring field2}1.3Template模板(Templates)用于传送一个特定值的集合或是测试接收的值的集合是否与模板说明匹配。
a) 模板提供了一种组织和重复使用测试数据的方法b) 模板能够被参数化;c) 模板允许匹配机制;d) 模板既能够用于基于消息的通信,也能用于基于过程的通信1.4ComponentThe component type defines which ports are associated with a component.•It is also possible to declare constants, variables and timers local to a particular component type.•These declarations are visible to all testcases, functions and altsteps that run on an instance of the given component type. This shall be explicitly stated using the runs on keyword (see clause 16) in the testcase, function or altstep header.1.5Port && messagePort:端口可以基于消息(message标识),和基于过程(procedure)的,甚至是二者混合(mixed)的。

COMMISSION IMPLEMENTING DECISIONof 25 November 2013on Guidelines on Annex I to Regulation (EC) No 1223/2009 of the European Parliament and of the Council on cosmetic products(Text with EEA relevance)(2013/674/EU)THE EUROPEAN COMMISSION,Having regard to the Treaty on the Functioning of the European Union,Having regard to Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products ( 1 ), and in particular the third subparagraph of Article 10(1) thereof,Whereas:(1) It is essential that cosmetic products made available on the Union market be safe for human health when used under normal and reasonably foreseeable conditions of use. To that end, Regulation (EC) No 1223/2009 requires that, in order to establish that a cosmetic product is safe under those conditions, cosmetic products undergo a safety assessment. (2) The operator designated as the responsible person in accordance with Regulation (EC) No 1223/2009 is to ensure that, for each cosmetic product which is to be placed on the Union market, a cosmetic product safety report is drawn up on the basis of the relevant information and in accordance with the requirements laid down in Annex I to Regulation (EC) No 1223/2009. (3) In order to facilitate the understanding of the requirements of Annex I to Regulation (EC) No 1223/2009 by all undertakings, and especially small and medium-size enterprises, the Regulation requires that the Commission adopts appropriate guidelines. (4) This Decision sets out appropriate guidelines on Annex I to Regulation (EC) No 1223/2009. They were developedwith the contribution of the relevant stakeholders, including representatives of small and medium-sized enterprises. (5) The guidelines should assist responsible persons in complying with their regulatory obligations. However, they are not meant to replace the knowledge and expertise of the qualified safety assessor, as required by Article 10(2) of Regulation (EC) No 1223/2009, who should remain the only professional allowed to carry out the cosmetic product safety assessment as described in Part B of Annex I. (6) The measures provided for in this Decision are in accordance with the opinion of the Standing Committee on Cosmetic Products,HAS ADOPTED THIS DECISION: Article 1 The guidelines to enable undertakings to comply with the requirements laid down in Annex I to Regulation (EC) No 1223/2009 on cosmetic products are set out in the Annex to this Decision. Article 2 This Decision shall enter into force on the twentieth day following that of its publication in the Official Journal of the European Union .Done at Brussels, 25 November 2013. For the CommissionThe President José Manuel BARROSO ( 1 ) OJ L 342, 22.12.2009, p. 59.ANNEXGUIDELINES ON ANNEX I TO REGULATION (EC) No 1223/2009 ON THE COSMETIC PRODUCT SAFETYREPORT1. INTRODUCTIONArticle 11 of Regulation (EC) No 1223/2009 requires that a product information file is drawn up for each product before it is placed on the market. The product information file should be updated when necessary and kept readily accessible, in electronic or other format, at the address of the responsible person given on the label, to the competent authorities for market surveillance purposes for a period of 10 years following the placing on the market of the last batch of the product.The most important element of the product information file, from a safety point of view, is the cosmetic product safety report referred to in Article 10(1). The other elements are a clear description of the cosmetic product, a description of the method of manufacturing and a statement on compliance with good manufacturing practice, the proof of the effects claimed, and data on animal testing (1).Where the responsible person drawing up the cosmetic product safety report is not the manufacturer of the product, they should ensure they have access to all the technical and scientific skills necessary to obtain reliable cosmetic product safety information and an appropriate safety assessment to demonstrate that the product they are responsible for is safe, in accordance with Article 3 of Regulation (EC) No 1223/2009. They may therefore need to involve not only the safety assessor, but also the manufacturer, the suppliers of raw materials, and other technical experts.In any case, the responsible person is to ensure that the intended use of the cosmetic product and the anticipated systemic exposure to individual ingredients in a final formulation are taken into account in the safety assessment;an appropriate weight-of-evidence approach is used in the safety assessment for reviewing data from all existing sources; the cosmetic product safety report is kept up to date in view of additional relevant information generated subsequent to placing the product on the market (2).The cosmetic product safety assessment, as set out in Part B of Annex I to Regulation (EC) No 1223/2009, is to be carried out by a qualified safety assessor. The responsible person and the safety assessor should work closely together to ensure that the safety of the product is properly assessed and documented and that the assessment is kept up to date. The responsible person and the safety assessor should gather all the necessary information as required by Part A of Annex I to Regulation (EC) No 1223/2009.The cosmetic product safety report should be drawn up in a transparent way and should be well-argued and easily understood.The Cosmetic Product Safety Report is an expert piece of work made up of different modules and where the information required under Part A may be stored in different databases. The report, which should contain, as a minimum, all the information indicated in Annex I to Regulation (EC) No 1223/2009, should appear under the same or similar headings for ease of reference of the competent authorities. However, it may be sufficient to provide under each heading a clear reference to a document containing the information and readily available in electronic or printed format.1223/2009—COSMETICPRODUCTREPORTSAFETY(EC)2. ANNEXITOREGULATIONNoIn accordance with Annex I to Regulation (EC) No 1223/2009, the Cosmetic Product Safety Report is to contain, ‘as a minimum’, the information required under each of the headings of Part A and Part B.Part A aims to gather all the data necessary for the safety assessment of the product, while Part B sets out the reasoning, starting from the data, for drawing conclusions as to the safety of the product.(1) Article 11(2) of Regulation (EC) No 1223/2009.(2) Article 10(1) of Regulation (EC) No 1223/2009.The structure and content of the safety report should reflect the requirements of Annex I to Regulation (EC) No 1223/2009. However, if the report does not directly contain the required information, it should provide a reference to another readily available source.The responsible person is to ensure that the Cosmetic Product Safety Report is kept up to date in the light of additional relevant information emerging after the product has been placed on the market (1).PRODUCTCOSMETICSAFETYINFORMATION—3. PARTAPart A of the cosmetic product safety report is intended to gather the data necessary to prove that the cosmetic product is safe. The information should enable the safety assessor to clearly identify and quantify, based on the identified hazards, the risks a cosmetic product may present to human health.A hazard may arise, for example, from the raw materials, the manufacturing process, the packaging, theconditions of use of the product, the microbiological specifications, the quantities used, the toxicological profile of the substances, etc.As Part A of Annex I to Regulation (EC) No 1223/2009 requires that the data listed under its headings are provided as a minimum, any discrepancy with regards to the requirements of Part A should be justified.Part A of Annex I to Regulation (EC) No 1223/2009 lists the data that is to be available, ‘as a minimum’, for the safety assessor to be able to carry out the safety assessment.In addition to the minimum data listed in Part A of Annex I to Regulation (EC) No 1223/2009, the safety assessor can use any additional data, where relevant. On the other hand, they, or the responsible person, may consider that, depending on the type of product, some of the required data are not relevant or necessary to assess the safety of the product (e.g. preservation challenge test). In this case, the absence of specific data is to be clearly justified in Part A and the justification is to be repeated and validated by the safety assessor in their reasoning in Part B. The responsible person should check the presence of the required data or the justification for their absence.The data required by Part A can be drawn from any reliable source. Examples include: data from suppliers, scientific literature, experience gained with similar or other product categories, results of studies on the product itself or on the substances it contains, available data on similar formulations, or computer models. The safety report should highlight the relevance of the data in relation to the product.The guidance published by the EU scientific committees concerned with risk assessment (2), as well as the recommendations of national competent authorities or professional organisations, may provide further helpful support.3.1. Quantitative and qualitative composition of the cosmetic productThe aim of that section of the cosmetic product safety report is to provide the exact quantitative and qualitative composition of the finished product, starting from the raw materials. Raw materials are substances or mixtures used in the manufacturing of the cosmetic product. The intended function of each substance is to be indicated.The complete product composition is to be specified, stating the name and identity (qualitative) of each raw material (including chemical name, INCI, CAS, Einecs/ELINCS, where possible), and the amount of each raw material, stating the weight percentage (quantitative). Ranges should not be used, unless this can be justified(e.g. viscosity or pH adjusters). If concentration ranges are unavoidable, toxicological considerations and calculations should be based on the highest concentration figure. It may also be useful to indicate the supplier(s) of the raw materials.(1) Article 10(1)(c) of Regulation (EC) No 1223/2009.(2) The SCCS’s Notes of Guidance for the Testing of Cosmetic Ingredients and their Safety Evaluation, 8th Revision, SCCS/1501/12, and itssubsequent updates.All substances entering into the composition of commercial mixtures supplied as raw materials (including directly added preservatives, antioxidants, chelators, buffering agents, solvents, other additives, etc.) are to be identified and quantified in the formula of the finished product. This also applies to all substances indirectly added to the product, such as preservatives used for preserving raw materials. The intended function of each substance is to be indicated.When chemically well-defined substances are present, their quantity and molecular formula should be given together with their analytical specifications (degree of purity, identification of major impurities, criteria and test methods used).When complex ingredients are present, their nature and quantity together with a clear definition of the mixture and the material(s) used should be given in order to identify the substances with regard to their composition and effects (manufacturing and purification processes, including physical, chemical, enzymatic, biotechnological and microbiological steps). The purity criteria and test methods used should be provided. Examples of complex ingredients include those of mineral, botanical, animal or biotechnological origin. The scope of the information needed on complex ingredients, depending on their nature and origin, is explicitly listed in the Scientific Committee for Consumer Safety (SCCS) Note of Guidance (1).When a mixture of both chemically well-defined substances and complex ingredients is present, the above guidance also applies.Where any fragrance (or flavour) compound comprising a mixture of fragrance (or flavour) ingredients and functional components with olfactory, odour-enhancing, odour-protecting or blending properties is formulated and intentionally added to a cosmetic product to impart a scent (or flavour) or to cover a malodour, its identification is to include the name and code number as well as the identity of the supplier. Qualitative and quantitative information about regulated substances in the fragrance (or flavour) compound and information relevant for a safety assessment should be disclosed to the responsible person and the safety assessor, and should be included in the safety report.3.2. Physical/chemical characteristics and stability of the cosmetic productThe aim of that section of the cosmetic product safety report is to describe the relevant physical and chemical specifications of the substances or mixtures used and the cosmetic product itself. These specifications are crucial for an appropriate safety assessment, as they may influence the safety of a cosmetic product. For example, physico-chemical properties, in combination with other information, can help the safety assessor determine the need to investigate relevant toxicological parameters.In addition, the physico-chemical characteristics of the substances or mixtures and finished products set the benchmark against which the products and the raw materials can be considered acceptable from a quality point of view (2).That section of the cosmetic product safety report also requires an assessment of the stability of the cosmetic product, under reasonably foreseeable storage conditions. The aim is to evaluate if the stability of the cosmetic product affects the safety and quality of the product, and to use the information to determine its minimum durability and period-after-opening (PAO).3.2.1. Physical/chemical characteristics of substances or mixturesThis description is to include the most relevant physico-chemical properties of each substance and mixture contained in the product, for example: chemical identification, physical form, molecular weight, solubility, partition coefficient, substance purity, other parameters relevant for the characterisation of specific substances and mixtures, and, for polymers, the average molecular weight and range.(1) SCCS Notes of Guidance, para. 3-6.2, pp. 35-36.(2) This point is relevant in the context of Good Manufacturing Practices, and is explicitly addressed by the relevant standard EN ISO22716:2007. More specifically, it matches the requirements for the release of raw materials and the finished product.Where relevant, the particle-size distribution curve of substances should be included in the physico-chemical characteristics, especially for nanomaterials.Cosmetics manufacturers should ensure that the specifications of raw materials are properly documented by their suppliers. Specifications should be available for each raw material actually used in the product. Based on function, additional specifications may be needed. For UV absorbers, for instance, the absorption spectra should be provided.For each description of physico-chemical properties and specifications (for each substance and mixture contained in the product), the reference methods should be stated in the safety report.3.2.2. Physical/chemical characteristics of the finished cosmetic productThis description is to contain the specifications of the finished product. Each specification should be given with relevant limits, e.g. pH between 5.5 and 6.5.For each description of physico-chemical properties and specifications of the finished product, the reference methods should be stated in the cosmetic product safety report.3.2.3. Stability of the cosmetic productAs the requirement is to assess the stability of the cosmetic product under reasonably foreseeable storage conditions, if stability is dependent on storage conditions, information about these conditions should be passed on throughout the supply chain, and, if relevant for the end user, it should be indicated on the labelling of the product.The methodology used to determine the product’s minimum durability should be described. Any specific preservation precautions should be mentioned.All available data used to justify the indicated minimum durability should be listed in the safety report. In order to determine the coherence of the stability study conducted, and to check the relevance of the date of minimum durability chosen for the product, the description of the tests specific to the stability study and the results of those tests should be included in the cosmetic product safety report. In addition, the following should also be provided:(1) evidence that the composition of the product used for stability testing corresponds to the product actuallyplaced on the market;(2) the results of the preservative efficacy study, e.g. challenge test, if applicable (1);(3) when applicable, the period-after-opening (PAO) (2) and its justification.The SCCS has recommended that ‘relevant stability tests, adapted to the type of cosmetic product and its intended use, should be carried out. To make sure that no stability problems are induced by the type of container and packaging used, physical stability tests are currently carried out with inert containers and those intended to be used on the market.’ (3)(1) See section 3.3 on Microbiological quality.(2) See ‘Practical implementation of Article 6(1)(c) of the Cosmetics Directive (76/768/EEC): Labelling of product durability: “period of timeafter opening” ’ (Council Directive 76/768/EEC, OJ L 262, 27.9.1976, p. 169) http://ec.europa.eu/consumers/sectors/cosmetics/files/doc/ wd-04-entr-cos_28_rev_version_adoptee20040419_en.pdf(3) SCCS Notes of Guidance, para. 4-3.3, p. 74.3.3. Microbiological qualityThe aim of that section of the cosmetic product safety report is to determine the acceptable microbiological specifications of the raw materials (substances or mixtures) and finished product from a microbiological point of view. In accordance with Annex I to Regulation (EC) No 1223/2009, particular attention is to be paid to the microbiological specifications of cosmetic products intended to be used on sensitive body parts and on specific populations. In addition, information regarding microbiological quality is essential in order to justify the effectiveness of the preservation system and justify the indicated minimum durability of the cosmetic product stored under appropriate conditions and period-after- opening (PAO) (1) of the finished product in terms of safety.The microbiological specifications of the raw materials (substances or mixtures) and cosmetic product are to form part of the safety assessment. Particular attention is to be paid to the microbiological specifications of cosmetic products intended to be used around the eyes, on mucous membranes in general, on damaged skin (e.g. skin care products suitable for atopic or irritated skin), on children under three years of age, on elderly people or on persons with compromised immune responses.3.3.1. Microbiological quality of substances and mixturesThe main parameters for microbiological quality are the original level of contamination and the possibility of microbial growth. Particular attention should be paid to the raw materials (substances and mixtures) most susceptible to microbial growth (e.g. water-based mixtures, protein-rich materials, plant or animal raw materials).On the other hand, there are raw materials which do not support microbial growth, e.g. organic solvents.3.3.2. Microbiological quality of the finished cosmetic productConcerning microbiological susceptibility, there is a difference between three product categories:(1) low microbiological risk products (e.g. products with an alcohol content > 20 %, products based on organicsolvents, high/low-pH products), for which neither a preservation challenge test nor microbiological quality tests on the finished product are necessary. A scientific justification is to be provided, however;(2) single-use products, and products which cannot be opened (e.g. for which the packaging allows dosing theproduct without it coming in contact with the air), for which only microbiological quality tests on the finished product are necessary. A scientific justification is to be provided, however;(3) all other products, for which both a preservation challenge test and microbiological quality tests on thefinished product are necessary.Specific ‘Guidelines on Microbiological Quality of the Finished Product’ are provided in the SCCS Notes of Guidance (2).3.4. Impurities, traces, information about the packaging materialThe aim of that section of the cosmetic product safety report is to assess whether the cosmetic product contains substances that have not been intentionally added to the formulation, and which may have an impact on its safety.Impurities are unintended substances in raw materials.(1) The ‘date of minimum durability’ is the date until which the cosmetic product, stored under appropriate conditions, will continue tofulfil its initial function and, in particular, will remain safe; the PAO is the period of time after opening for which the product can be used without any harm to the consumer. See ‘Practical implementation of Article 6(1)(c) of the Cosmetics Directive (76/768/EEC): Labelling of product durability: “period of time after opening” ’.(2) SCCS Notes of Guidance, para 4-4, pp. 75–76.A trace is a small quantity of an unintended substance in the finished product. Traces are to be evaluatedwith regard to safety of the finished product. When traces of prohibited substances are present, evidence of their technical unavoidability are also to be provided.Traces can originate from the following sources: impurities in the raw materials/substances; the manufacturing process; potential chemical evolution/interaction and/or migration of substances in the product that could occur under normal storage conditions and/or through contact with the packaging material.Because substances may migrate from the packaging to the formulation, the relevant characteristics of the packaging material are to be considered.In accordance with point 4 of Annex I to Regulation (EC) No 1223/2009, the section on ‘Impurities, traces, information about the packaging material’ is to address three specific issues:(a) the purity of substances and mixtures;(b) in case of traces of prohibited substances, evidence of their technical unavoidability;(c) the relevant characteristics of the packaging material, in particular purity and stability.In practical terms, those elements may be interpreted as follows:(a) precise definition of impurities and traces (see 3.4.1);(b) evidence of technical unavoidability of prohibited substances (see 3.4.2);(c) potential release of substances from the packaging or possible deterioration of the product in contact with thepackaging (see 3.4.3).For the analysis of impurities and packaging material, data from suppliers are of crucial importance and should be preferred.3.4.1. Purity of substances and mixturesThe presence of unintended substances, such as impurities and traces, can have an impact on the safety of the finished product. The cosmetic product safety report is to include data on the purity of raw materials (substances and mixtures) and the identification of the toxicologically relevant unintended substances. These substances should be taken into account in the safety assessment of the product.Impurities are unintended substances in raw materials.A trace is a small quantity of an unintended substance in the finished product.The presence of traces in the finished product can be evaluated in two ways:(a) through the specifications/technical data for each raw material, based on knowledge of the process for manufacturing the raw material (origin of substance, production process, synthesis route, extraction process, solvent used, etc.);(b) through a physico-chemical analysis of possible impurities in raw materials and, if necessary, in the finalproduct (e.g. nitrosamines which are potentially generated during or after the manufacturing process).Traces of prohibited substances are dealt with in paragraph 3.4.2 of these Guidelines.Some traces have regulatory concentration limits. For the presence of traces of substances that are not prohibited, and for which there are no regulatory concentration limits, but which could be expected to impact consumer safety, the safety assessment needs to be carried out by the safety assessor.3.4.2. Evidence of the technical unavoidability of traces of prohibited substancesWhile the procedure outlined in paragraph 3.4.1 should be followed for all known impurities and traces to evaluate their toxicological impact, further investigation is required for prohibited substances present as traces in the finished product (1).When such presence is technically unavoidable, the cosmetics manufacturers are required to provide evidence of the technical unavoidability. That means they have to justify the presence of those traces by all necessary means.The presence of traces of prohibited substances should be kept as low as is reasonably achievable under good manufacturing practices. In addition, the safety assessor has to decide whether their levels are toxicologically acceptable and whether the product is still safe.Especially in the case of non-threshold genotoxic and carcinogenic substances (2), the cosmetic industry should keep improving its best practices in order to eliminate these substances (ALARA principle (3)) in the finished cosmetic product. The main concern is to ensure the protection of human health, as required by Article 3 of Regulation (EC) No 1223/2009.Traces generated by the degradation of substances within the final product (stability issues), by preservation or transport problems, or by the interaction of raw materials should be avoided through good manufacturing practices, or possibly through re-formulation of the product.3.4.3. The relevant characteristics of packaging materialPackaging material means the container (or primary packaging) that is in direct contact with the formulation. The relevant characteristics of packaging materials in direct contact with the final product are important for the safety of the cosmetic product. Reference to Regulation (EC) No 1935/2004 of the European Parliament and of the Council (4) could be useful.Experience with similar formulation/packaging combinations already on the market provides useful indications.Materials that have been developed for food packaging have often already been tested, so relevant information on stability and migration may be available. Additional testing may not be required. However, more evaluation may be needed for new or novel packaging.The combination of packaging material, formulation of the cosmetic product and contact with the external environment may have an impact on the safety of the finished product, due to the following factors:(a) interaction between the product and the packaging material;(b) barrier properties of the packaging material;(c) substance migration from/to the packaging material.The information on relevant characteristics of the packaging materials in direct contact with the product should allow an estimation of potential risks. Relevant characteristics could include, for example, the following:(a) composition of the packaging material, including technical substances such as additives;(b) technically unavoidable impurities;(c) possible migration from the packaging.(1) Article 17 of Regulation (EC) No 1223/2009 establishes that traces of prohibited substances are only permitted if they are technicallyunavoidable and if they have no impact on the safety of the cosmetic products.(2) The ‘non-threshold genotoxic and carcinogenic substances’ are the genotoxic and carcinogenic substances without a threshold for thecarcinogenic-genotoxic effects.(3) Opinion of the Scientific Committee on a request from EFSA related to A Harmonised Approach for Risk Assessment of SubstancesWhich are both Genotoxic and Carcinogenic, the EFSA Journal (2005) 282, pp. 1-31.(4) OJ L 338, 13.11.2004, p. 4.。

欧盟:临床阶段的GMP要求和QP放行欧盟临床阶段的D Si f i d S h itt PAREXELDr Siegfried Schmitt, PAREXELEU GMP REQUIREMENTS AT THE CLINICAL DEVELOPMENT STAGE & QP RELEASEAGENDAGMPs for Investigational Medicinal Products (IMP) and their Qualified Person (QP) releasetheir Qualified Person(QP)releaseNew developments and requirements in the EUN d l t d i t i th EUINVESTIGATIONAL MEDICINAL PRODUCTS (IMP)()Definition of Investigational Medicinal Products(IMPs)“a pharmaceutical form of an active substance or placebo beingftested or used as a reference in a clinical trial,including products already with a marketing authorization but used or assembled (formulated or packaged)in a way different from the authorised form,or when used for an unauthorised indication,or when used to gain further information about the authorised form.”An IMP must be registered in the EudraCT database/3rya6w7NON INVESTIGATIONAL MEDICINAL PRODUCTS (NIMP)Definition of Non Investigational Medicinal Products(NIMPs) Products which are not the object of investigation(i.e.other than the tested product,placebo or active comparator)may be supplied)to subjects participating in a trial and used in accordance with the protocol.For instance,some clinical trial protocols require the use of medicinal products such as support or rescue/escape medication for preventive,diagnostic or therapeutic reasons and/or to ensure q p j y y that adequate medical care is provided for the subject.They may also be used in accordance with the protocol to induce a physiological response./3rya6w7()THE QUALIFIED PERSON (QP)The person defined in Article 48 of Directive 2001/83/EC and Article 52 of Directive 2001/82/ECThe QP is the only person that can release product to be placed on the market in the EUplaced on the market in the EUThe QP is personally liable for the release decisionThe regulations apply also to IMPsTHE EUROPEAN UNIONEU -LEGAL INSTRUMENTSPrimary EU Law:Treaty of the Function of the European Union(TFEU)Treaty of the Function of the European Union (TFEU) Secondary EU Law:Medicinal(Drug) Secondary EU Law:a) Legislative Acts:Regulation, Directive, Decisionb)Non Legislati e ActsMedicinal (Drug)Products:E d L V l1b) Non-Legislative Acts:Recommendation, Opinionc) New Legal Acts (created byEudraLex Vol 1(human) &EudraLex Vol 5the Treaty of Lisbon):Delegated Act, ImplementingAct(veterinary)http://europa.eu/legislation_summaries/institutional_affairs/treaties/lisbon_treaty/ai0032_en.htm_y_EU REGULATIONSA regulation is a legal instrument that is immediately enforceableExample:Council Regulation(EEC)No2309/93on Community procedures for the authorization and supervision of medicinal products for human and veterinary use and establishing a European Agency for the Evaluation of Medicinal ProductsEU DIRECTIVES•Require Member States to achieve a certain result•Do not dictate the means of achieving the result•The contents of the Directive must be transposed into national law by Member States-and carried out by Member State National Competent Authorities“National Authorities”Example:Commission Directive2003/94/EC on the principles E l C i i Di ti th i i l and guidelines of good manufacturing practice in respect of medicinal products for human use and investigational medicinal products for human useEU GUIDELINESDo not have the force of law,but represent the agreed views of regulators on a certain topic.It is possible not to follow them if scientifically justifiedAre complemented by Questions and Answers to clarify specific points in guidelines•EMA scientific guidelines-/d2qvbjx•The GMP guide-/crj4qb3g p y j q•The guideline publication process-/c6dpjrxEU LEGISLATION ON THE WEBEU Legislationhttp://eur-lex.europa.eu/en/index.htmLegislation for Medicinal Products in the European Union http://ec.europa.eu/health/index_en.htmGXP FOR IMPS -APPLICABLE REGULATIONSThe regulations cover the lifecycle from development to discontinuationEU GXP FOR IMPS -APPLICABLE REGULATIONS EudraLex Volume4Annex13Manufacture of InvestigationalMedicinal ProductsEudraLex Volume4Parts I,II and IIIEudraLex Volume4Annex16Certification by a Qualified person and Batch Release July2001Draft revision of Annex162013http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm htt///h lth/d t/d l/l4/i d htEU GXP FOR IMPS -PHASE APPROPRIATEPDA Technical Report 56 GXP FOR IMPS VERSUS GXP FOR COMMERCIAL PRODUCTyParticularly for Clinical Phases I and II•Changes need to be documented,but do not need to go through a formal change approval process•Deviations will be the exceptiony y •Analytical methods need to be documented and suitability established,but do not need to be validated •Documentation has to be controlled,but laboratory journals may be used instead of batch recordsb d i t d f b t h d•Operational and analytical personnel may be the same •Critical parameters and quality attributes not fully established(e.g.yield not a critical parameter)GXP FOR IMPS VERSUS GXP FOR COMMERCIAL PRODUCTWhat must be in place for IMPs and Commercial product:•A formal quality system•Job descriptions and training•Calibration program•Appropriate environmental conditions(minar Air Fumehood(LAF)or HVAC)•Process controls•Rationales,e.g.for changes,analytical method selection, process and process controlsGXP FOR IMPS -SOME SPECIFIC ISSUESSterilisation steps in IMP manufacture•Must be fully validated•Associated analytical methods must be fully validatedBiotech Products•Production scale for Clinical Phase III material must be equivalent to commercial scale(preferably the same equipment)GXP FOR IMPS -SOME SPECIFIC ISSUESClinical Trial LogisticsPharmaceutical Engineering Nov/Dec 2014 GXP FOR IMPS -SOME SPECIFIC ISSUESClinical Trial LogisticsPAREXEL InternationalGXP FOR IMPS -SOME SPECIFIC ISSUESOptimising IMP UtilisationPAREXEL InternationalTHE ROLE OF THE QP IN IMP RELEASEThe QP is responsible for certifying that each batch of IMP has been produced and tested/checked in accordance with:•EU GMPG•The Product Specification File•The IMPD(Investigational Medicinal Product Dossier)or the CTA(Clinical Trial Authorisation)Th P d t S ifi ti Fil(di A t th EU •The Product Specification File(according Annex13to the EU-GMP Guide)THE ROLE OF THE QP IN IMP RELEASEThe IMP QP is accountable from manufacture all the way to the patient by:•assessing GMP issuesG•participating in inspections and audits at sites involved in the manufacturing and distribution of IMPs•being a reliable contact for the health authorities•knowing applicable legislation and processes including k i li bl l i l ti d i l di exceptions which may impact the quality and safety of the IMP •being involved in complaint handling and recall processes 2014QP RELEASE -ABOUT THE QPThere is very little harmonisation in Europe regarding the prerequisites for becoming a QPE.g.in the UK a biologist,a chemist or a pharmacist will be eligible to become a QP if they have chartered status with the respective Royal Societyg y Q p y yE.g.in Italy the QP must be employed by the MAIHE.g.in Germany the QP must have police clearance certificationQP RELEASE -THERE IS A PRICE TO PAYWentworth Pharmaceutical QP Survey2015THE BASIS OF THE QP DECLARATIONFor human and veterinary medicinal products,the QP declaration should be based upon an audit of the active substance manufacturers.It is established good practice that the audit should be conducted at the manufacturing site i.e.an on-site auditAudits should be by or on behalf of the Manufacturing Importation Authorisation Holder(MIAH),by suitably trained p p(),y p y and experienced person(s),who may be a third party contractorThe audit cannot be replaced by GMP certificates from a relevant competent authorityQP RELEASE -TEMPLATES FOR API AND IMPEMA Guidance for the template for the qualified person’s declaration concerning GMP compliance of active substance manufacture The QP declaration template2014“The template”-/pmnkfggThe template-/o63p7ogp q p g Template for the qualified person’s declaration concerning GMP compliance of investigational medicinal products manufactured in non-EU countries-/pnh4m68QP RELEASE -TEMPLATES FOR BATCH RELEASE Internationally harmonised requirements for batch certification -/posc88wThis certificate may also be used for active pharmaceutical ingredients and investigational medicinal products used in clinical trial authorisations.t i l th i tiIMP RELEASE -TWO-STEP PROCESSStep1Certification by the QPStep2Release following fulfilment of the requirements of Article9(Commencement of a clinical trial)of Directive(C f)f2001/20/EC-i.e.the clinical trial must be authorisedQP RELEASE -SOME SPECIFIC ISSUESQ:How do you perform batch record review of batches produced in China,when they are not bilingual or translated? How about the following options:f•Not required•Only in audits•Translation of one example•Full translation of every batch•Only summaryQP RELEASE -SOME SPECIFIC ISSUESQ:How do you perform batch record review of batches produced in China,when they are not bilingual or translated?fA:A translation of one batch record as an example and a summary of each batch would be required together with a CoA.It is important that the QP can understand the process and whether there were any excursions,CAPAs,changes,etc., and what they were and how they were concludedPlease note:The QP has to release each individual batchQP RELEASE -SOME SPECIFIC ISSUESQ:Can a member state can invalidate a procedure because a GMP Certificate was not provided for an API for which the QP declaration was provided?A:No.A satisfactory QP Declaration is always necessary and is normally sufficient to confirm that the manufacture of active pharmaceutical ingredients(APIs)comply with Good Manufacturing Practice(GMP),as required by Article8g p()Paragraph3(ha)of Directive2001/83/EC-http://tinyurl com/p7uecnqCMDh/268/2012/p7uecnqGXP FOR IMPS -INDUSTRY EXPERIENCEThere will be a shortage of QPs as many QPs are due to retire in the coming yearsf Q For logistical reasons it is often practical to use the QP services of a Contract Research Organisation(CRO)often rely on third party audit reports,provided they are QPs reportsprepared by suitably qualified auditorsy Q p It may be advisable to have a second QP named as a back-up optionSUMMARY AND OUTLOOK¾The level of GMP applied to IMP manufacture has to increase depending on the clinical phase.Rationales for (necessary)changes need to be documented¾IMPs require a QP release in Europe¾specific requirements for QPs differ in each EU Themember state¾IMP manufacture,IMP supply and clinical trial management,pp y g are intricately linked-working with a suitably qualified and experienced third party may be advisableYOUR PRESENTERSiegfried Schmitt, PhD FRSC CChem CSciPrincipal ConsultantPAREXEL International+44 7824 592401siegfried.schmitt@。

Guidelines related to the Pressure Equipment Directive2014/68/EU (PED)In order to ensure a coherent application of the Pressure Equipment Directive 2014/68/EU (replacing the Directive 97/23/EC (PED) as of 19 July 2016), Guidelines are developed and agreed by the Commission's Working Group "Pressure" (WGP).This working group is composed of representatives of Member States, European federations, the Notified Bodies Forum and CEN and chaired by a representative of the Commission services. The PED Guidelines developed for Directive 97/23/EC will systematically be reviewed and possibly issued as a PED Guideline under the new Directive 2014/68/EU. Also new Guidelines may be issued to support the implementation of the Directive. This work is in progress and the new or updated Guidelines will be made available as soon as they are endorsed by the Working Group "Pressure" (WGP).Remarks or questions concerning this document should be addressed via the email to the unit in the European Commission dealing with the Pressure Equipment Directive:GROW-PRESSURE-EQUIPMENT@ec.europa.euStatus of the guidelinesThe PED Guidelines are not a legally binding interpretation of the Directive. The legally binding text remains that of the Directive. However, the PED Guidelines represent a reference for ensuring consistent application of the Directive. They represent, unless indicated differently in the respective guideline text, the unanimous opinion of the Member States.Classification of the guidelinesThe guidelines carry a x/yy type identification- (x) relates to the subject (A, B, C etc…),- the second (yy) is a sequential numbering.Remark: To facilitate the transition to the new Guidelines the sequential number is maintained as far as possible (e.g. Guideline A-24 under the new PED 2014/68/EU corresponds to Guideline 1-24 under PED 97/23/EC)The letter x refers to one of the following subjects:A.S COPE AND EXCLUSIONS OF THE DIRECTIVEB.C LASSIFICATION AND CATEGORIESC.A SSEMBLIESD.E VALUATION ASSESSMENT PROCEDURESE.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON DESIGNF.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ONMANUFACTURINGG.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON MATERIALSH.I NTERPRETATION OF OTHER ESSENTIAL SAFETY REQUIREMENTSI.M ISCELLANEOUSJ.G ENERAL-HORIZONTAL QUESTIONSDocument historyVersion Date Comment1.0 31/3/2015 Includes PED 2014-68-EU Guidelines from the WGP meeting of 11/03/2015A.S COPE AND EXCLUSIONS OF THE DIRECTIVEGuideline A-24Guideline related to: Article 2, point (12)Question According to the definition of Article 2 point 12 fluids may contain a suspension of solids.Is a system of solid pieces or liquid drops distributed in a gas still a fluidin the sense of the PED?Answer YesReasonNote 1 A gas containing pieces of solids or drops of liquid is also to be considered a fluid.suspensionKeywords fluid,Accepted by Working Party Guidelines (WPG) on: 28/11/2014Accepted by Working Group Pressure (WGP) on: 11/03/2015Guideline A–54Guideline related to: Article 1 paragraph 2(s)Question How to understand the exclusion in PED Article 1 paragraph 2 (s) related to equipment covered by the regulations on transport of dangerousgoods?Answer This exclusion shall be read in the context of the scope of PED which applies to the design, manufacture and conformity assessment of pressure equipment.The exclusion only applies when the listed regulations on transport ofdangerous goods includes construction and conformity assessmentrequirements for the equipment concerned.ReasonNote In the context of the listed regulations on transport of dangerous goods, the term construction traditionally refers to design and manufacture.KeywordsAccepted by Working Party Guidelines (WPG) on: 28/11/2014Accepted by Working Group Pressure (WGP) on: 11/03/2015B.C LASSIFICATION AND CATEGORIESGuideline B-21Guideline related to: Annex I sections 2.2.1 and 2.3,Annex II Table 1 Annex IITable 6Question How does one define an unstable gas as referred to in Tables 1 & 6 of Annex II of PED?Answer An unstable gas in this context is a gas liable to transform itselfspontaneously, producing a sudden pressure increase.Such transformation as an example can result from a relatively small variationof an operating parameter (e.g. pressure, temperature, presence of catalysingmaterial) in a confined volume.This includes gases that are classified as chemically unstable gases accordingto CLP Regulation (EC) No 1272/2008 as amended.Typical examples of unstable gases: acetylene (UN 1001), methyl acetylene(UN 1060), vinylfluoride (UN 1860), ozone and dinitrogen oxide (UN 1067).For further examples, see Table 35.1 of the UN Manual of Tests and Criteria. NotegasKeywords UnstableAccepted by Working Party Guidelines (WPG) on: 12/02/2015Accepted by Working Group Pressure (WGP) on: 11/03/2015Guideline B-41Guideline related to: Article 13 QuestionWhere to find additional information on classification of fluids based on PED Article 13 as of 1 June 2015?AnswerAs of 1 June 2015 classification of fluids is based on article 13 of PED 2014/68/EU. Article 13 paragraph 1 (a) lists the physical and health hazard classes and categories for substances and mixtures included in Group 1. The classification is based on the CLP Regulation (EC) No 1272/2008. The table below provides an overview of the hazard classes and categories and the corresponding hazard statements according the CLP Regulation including references to the criteria and label elements in the CLP Regulation.CLP hazard classes and categories (as listed in article 13 of PED) Criteria according to Annex I to CLP Hazard statementaccording toCLP Label elementsaccording to Annex I to CLP(i) unstable explosives or explosives of Divisions 1.1, 1.2, 1.3, 1.4 and 1.5; Section 2.1.2 H200, H201, H202, H203, H204, H205 Table 2.1.2 (ii) flammable gases, category 1 and 2; Section 2.2.2 H220, H221 Table 2.2.3 (iii) oxidising gases, category 1;Section 2.4.2H270 Table 2.4.2 (iv) flammable liquids, category 1 and 2;Section 2.6.2 H224, H225 Table 2.6.2 (v) flammable liquids, category 3 where the maximum allowable temperature is above the flashpoint;Section 2.6.2 H226Table 2.6.2(vi) flammable solids, category 1 and 2; Section 2.7.2 H228 Table 2.7.2(vii) self-reactive substances and mixtures, type A to F;Section 2.8.2 H240, H241, H242 Table 2.8.1 (viii) pyrophoric liquids, category 1; Section 2.9.2 H250 Table 2.9.2 (ix) pyrophoric solids, category 1;Section 2.10.2H250 Table 2.10.2 (x) substances and mixtures which in contact with water emit flammable gases, category 1,2 and 3;Section 2.12.2 H260, H261Table 2.12.2(xi) oxidising liquids, category 1, 2 andSection 2.13.2 H271, H272 Table 2.13.2 3;(xii) oxidising solids, category 1, 2 andSection 2.14.2 H271, H272 Table 2.14.2 3;(xiii) organic peroxides types A to F; Section 2.15.2 H240, H241,Table 2.15.1H242Table 3.1.1 H300 Table 3.1.3 (xiv) acute oral toxicity, category 1 and2;(xv) acute dermal toxicity, category 1Table 3.1.1 H310 Table 3.1.3 and 2;Table 3.1.1 H330, H331 Table 3.1.3 (xvi) acute inhalation toxicity, category1, 2 and 3;(xvii) specific target organ toxicity –Table 3.8.1 H370 Table 3.8.4 single exposure, category 1.Note 1 Article 13 paragraph 1 (a) also states that "Group 1 comprises alsosubstances and mixtures contained in pressure equipment with amaximum allowable temperature which exceeds the flash point of thefluid". The purpose of this provision is to ensure that the flammabilityhazard is properly addressed for those substances and mixtures which arenot classified as flammable under the CLP Regulation (based on thetemperature criteria of the CLP Regulation) but which are presenting thishazard due to the maximum allowable temperature (TS).For example, Heat transfer oils are not classified as flammable liquidsaccording to the CLP Regulation because their flashpoint is above 60 °C(see CLP Regulation Annex I, Table 2.6.1 in Section 2.6 FlammableLiquids, 2.6.2 Classification criteria). However, if the maximumallowable temperature (TS) is above the flashpoint, the hazard of heattransfer oil corresponds to a Group 1 fluid.Note 2 Please note that the CLP Regulation is subject to adaptations to technical progress and therefore the information in the table above should bechecked with the version of the CLP Regulation in force at the time theequipment is placed on the market.Note 3 For questions related to the CLP Regulation please consult your national CLP-helpdesks. Further information on the CLP Regulation can be foundon the European Chemicals Agency (ECHA) website:www.echa.europa.eu. On the ECHA website there is also a list with thecontact details of all national CLP-helpdesks.fluids, classificationKeywords CLPRegulation,Accepted by Working Party Guidelines (WPG) on: 12/02/2015Accepted by Working Group Pressure (WGP) on: 11/03/2015C.A SSEMBLIESD.E VALUATION ASSESSMENT PROCEDURESE.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON DESIGNF.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ONMANUFACTURINGG.I NTERPRETATION OF THE ESSENTIAL SAFETY REQUIREMENTS ON MATERIALSH.I NTERPRETATION OF OTHER ESSENTIAL SAFETY REQUIREMENTSI.M ISCELLANEOUSJ.G ENERAL-HORIZONTAL QUESTIONS。

EUROPEAN COMMISSIONHEALTH AND CONSUMERS DIRECTORATE-GENERALPublic Health and Risk AssessmentPharmaceuticalsBrussels,SANCO/C8/AM/sl/ares(2010)1064599EudraLexThe Rules Governing Medicinal Products in the European UnionVolume 4Good Manufacturing PracticeMedicinal Products for Human and Veterinary UseAnnex 11: Computerised SystemsLegal basis for publishing the detailed guidelines: Article 47 of Directive 2001/83/EC on the Community code relating to medicinal products for human use and Article 51 of Directive 2001/82/EC on the Community code relating to veterinary medicinal products. This document provides guidance for the interpretation of the principles and guidelines of good manufacturing practice (GMP) for medicinal products as laid down in Directive 2003/94/EC for medicinal products for human use and Directive 91/412/EEC for veterinary use.Status of the document: revision 1Reasons for changes: t he Annex has been revised in response to the increased use of computerised systems and the increased complexity of these systems. Consequential amendments are also proposed for Chapter 4 of the GMP Guide.Deadline for coming into operation: 30 June 2011Commission Européenne, B-1049 Bruxelles / Europese Commissie,B-1049 Brussel - BelgiumTelephone: (32-2) 299 11 11PrincipleThis annex applies to all forms of computerised systems used as part of a GMP regulated activities. A computerised system is a set of software and hardware components which together fulfill certain functionalities.The application should be validated; IT infrastructure should be qualified.Where a computerised system replaces a manual operation, there should be no resultant decrease in product quality, process control or quality assurance. There should be no increase in the overall risk of the process.General1. Risk ManagementRisk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerised system.2. PersonnelThere should be close cooperation between all relevant personnel such as Process Owner, System Owner, Qualified Persons and IT. All personnel should have appropriate qualifications, level of access and defined responsibilities to carry out their assigned duties. 3. Suppliers and Service Providers3.1 When third parties (e.g. suppliers, service providers) are used e.g. to provide, install, configure, integrate, validate, maintain (e.g. via remote access), modify or retain a computerised system or related service or for data processing, formal agreements must exist between the manufacturer and any third parties, and these agreements should include clear statements of the responsibilities of the third party. IT-departments should be considered analogous.3.2 The competence and reliability of a supplier are key factors when selecting a product or service provider. The need for an audit should be based on a risk assessment.3.3 Documentation supplied with commercial off-the-shelf products should be reviewed by regulated users to check that user requirements are fulfilled.3.4 Quality system and audit information relating to suppliers or developers of software and implemented systems should be made available to inspectors on request.Project Phase4. Validation4.1 The validation documentation and reports should cover the relevant steps of the life cycle. Manufacturers should be able to justify their standards, protocols, acceptance criteria, procedures and records based on their risk assessment.4.2 Validation documentation should include change control records (if applicable) and reports on any deviations observed during the validation process.4.3 An up to date listing of all relevant systems and their GMP functionality (inventory) should be available.For critical systems an up to date system description detailing the physical and logical arrangements, data flows and interfaces with other systems or processes, any hardware and software pre-requisites, and security measures should be available.4.4 User Requirements Specifications should describe the required functions of the computerised system and be based on documented risk assessment and GMP impact. User requirements should be traceable throughout the life-cycle.4.5 The regulated user should take all reasonable steps, to ensure that the system has been developed in accordance with an appropriate quality management system. The supplier should be assessed appropriately.4.6 For the validation of bespoke or customised computerised systems there should be a process in place that ensures the formal assessment and reporting of quality and performance measures for all the life-cycle stages of the system.4.7 Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered. Automated testing tools and test environments should have documented assessments for their adequacy.4.8 If data are transferred to another data format or system, validation should include checks that data are not altered in value and/or meaning during this migration process.Operational Phase5. DataComputerised systems exchanging data electronically with other systems should include appropriate built-in checks for the correct and secure entry and processing of data, in order to minimize the risks.6. Accuracy ChecksFor critical data entered manually, there should be an additional check on the accuracy of the data. This check may be done by a second operator or by validated electronic means. The criticality and the potential consequences of erroneous or incorrectly entered data to a system should be covered by risk management.7. Data Storage7.1 Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability and accuracy. Access to data should be ensured throughout the retention period.7.2 Regular back-ups of all relevant data should be done. Integrity and accuracy of back-up data and the ability to restore the data should be checked during validation and monitored periodically.8. Printouts8.1 It should be possible to obtain clear printed copies of electronically stored data.8.2 For records supporting batch release it should be possible to generate printouts indicating if any of the data has been changed since the original entry.9. Audit TrailsConsideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated "audit trail"). For change or deletion of GMP-relevant data the reason should be documented. Audit trails need to be available and convertible to a generally intelligible form and regularly reviewed.10. Change and Configuration ManagementAny changes to a computerised system including system configurations should only be made in a controlled manner in accordance with a defined procedure.11. Periodic evaluationComputerised systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security and validation status reports.12. Security12.1 Physical and/or logical controls should be in place to restrict access to computerised system to authorised persons. Suitable methods of preventing unauthorised entry to the system may include the use of keys, pass cards, personal codes with passwords, biometrics, restricted access to computer equipment and data storage areas.12.2 The extent of security controls depends on the criticality of the computerised system. 12.3 Creation, change, and cancellation of access authorisations should be recorded.12.4 Management systems for data and for documents should be designed to record the identity of operators entering, changing, confirming or deleting data including date and time.13. Incident ManagementAll incidents, not only system failures and data errors, should be reported and assessed. The root cause of a critical incident should be identified and should form the basis of corrective and preventive actions.14. Electronic SignatureElectronic records may be signed electronically. Electronic signatures are expected to:a. have the same impact as hand-written signatures within the boundaries of thecompany,b. be permanently linked to their respective record,c. include the time and date that they were applied.15. Batch releaseWhen a computerised system is used for recording certification and batch release, the system should allow only Qualified Persons to certify the release of the batches and it should clearly identify and record the person releasing or certifying the batches. This should be performed using an electronic signature.16. Business ContinuityFor the availability of computerised systems supporting critical processes, provisions should be made to ensure continuity of support for those processes in the event of a system breakdown (e.g. a manual or alternative system). The time required to bring the alternative arrangements into use should be based on risk and appropriate for a particular system and the business process it supports. These arrangements should be adequately documented and tested.17. ArchivingData may be archived. This data should be checked for accessibility, readability and integrity. If relevant changes are to be made to the system (e.g. computer equipment or programs), then the ability to retrieve the data should be ensured and tested.GlossaryApplication: Software installed on a defined platform/hardware providing specific functionalityBespoke/Customized computerised system: A computerised system individually designed to suit a specific business processCommercial of the shelf software: Software commercially available, whose fitness for use is demonstrated by a broad spectrum of users.IT Infrastructure: The hardware and software such as networking software and operation systems, which makes it possible for the application to function.Life cycle: All phases in the life of the system from initial requirements until retirement including design, specification, programming, testing, installation, operation, and maintenance.Process owner: The person responsible for the business process.System owner: The person responsible for the availability, and maintenance of a computerised system and for the security of the data residing on that system.Third Party: Parties not directly managed by the holder of the manufacturing and/or import authorisation.。

最新IEC标准--EN IEC62776双端LED灯管安全要求.pdf的翻译®注册商标国际电工委员会34A/1501/NP新工作项目提案投保人IEC SC 34A日期的建议2011-07-05TC / SC34A秘书处BSI英国流通日期2011-07-22截止日期为投票2011-10-28一个新的工作项目范围内现有的技术委员会或小组委员会的建议应提交中央办公室。
建议将被分配到P-成员的技术委员会或小组委员会的表决它变成了工作方案,并介绍O-成员的信息。
投保人可能是一个全国委员会IEC,秘书处本身,另一个技术委员会或小组委员会会议上,联络,组织,标准化管理委员会或咨询委员会,或总书记。
为提出和证明一个新的准则工作项目在ISO / IEC导则第1部分,附录C(见提取物背页),这种形式是不被用于修订或修改现有的出版物。
“建议(投保人可完成)标题的建议普通照明用双端LED灯-安全规范。
标准技术规格范围(ISO / IEC导则第2部分,第6.2.1节中定义的)本标准规定了的安全性和互换性的要求,交换操作的测试方法和必要条件,以表明符合双端帽G5和G13的LED灯,用于更换荧光灯同样的帽子,其额定功率高达60 W,额定电压高达250 V。
目的和理由,包括市场的相关性,它是否是一个建议的水平标准(指南108)1)和关系到安全(指南104),EMC(指南107),环境因素(指南109)和质量保证(指南102)。
(作为附件附加一个单独的页面,如果有必要)1)其他TC / SC的要求表明他们的兴趣,如果有的话,这NP TC / SC书记的双端荧光灯在办公照明,路灯照明,安装在大容量工业照明等等。
双端LED灯的目的是作为一个可能的G5-G13-皑皑的荧光灯的替代品。
本标准的保障措施的变化从荧光灯,LED灯和落后的变化,从LED灯,荧光灯进行了安全LED灯,并在指定的交换条件。
目标日期第一张CD-IS / TS2012年10月会议的估计数2会议次数:2每年第一次会议的日期和地点:2011年10月,代尔夫特/ NL建议工作方法电子邮件协作工具要考虑的相关文件IEC 62560,IEC 61199其他国际机构的活动项目之间的关系联络组织需要在ISO或IEC的协调®版权所有©2011,IEC国际电工委员会。

Annex 4: Registration Items and Application Information Requirements of Supplemental Application of Drug RegistrationI Registration ItemsA Supplemental applications to be approved by SFDA1) Application for Drug Approval Number of a new drug by the New Drug Certificate holder of the drug.2) Use of the name of the Trade Name of drugs.3) Additional indications or functions of TCM, or the indication approved in China for chemical drug or biological products.4) Change in the strengths of the drugs.5) Change of strength of drugs6) Change of the supplementive in the formula of the drugs, where there is a medial requirement for it.7) A change in the drug manufacture technology and process affecting drug quality.8) Amendment of drug registration standards.9) Substitute or removal of the drug material listed in formula of National Drug Standards as toxic or endangered..10) Change of the immediate packing material or container of the drugs.11) Application for combined packing of drug.12) The transfer of new drug technology.13) Transfer of temporary standards into formal standards.14) A change in items within the import drug registration certificate, such as name of the drug, drug enterprise name, registered location, packing specification.15) Change of the location where the import drug is manufactured.16) Change of the location where the import drug is packed overseas.17) Repacking of import drugs in China.18) Change of the location where the raw material for import preparation is manufactured.B Supplemental applications to be filed for record at SFDA19) Change of the name of a domestic drug manufacturer.20) Internal change of the manufacture workshop of a domestic drug manufacturer.21) Amendment of insert sheet of the drugs according to national drug standards or required by SFDA.22) Supplementing and perfecting of the drug safety part of the insert sheet.23) Modification of design of packing and label of the drugs according to the regulation.24) Change of the packing specification of drugs manufactured at domestic.25) Change of valid period of drugs manufactured at domestic26) Change of the location where the raw material for domestic preparation is manufactured.27) Change of appearance of the drug without change of drug standards.28) Change of the agent for import drug registration.II Application Information Items1) Copies of drug approval certificate and appendixes.Include all the approval documents related to the application, such as drug registration approval, approval for supplemental application, approval for trade name, publishing document of drug standards, approval for amendment of drug standards and uniformly renewed drug approval number, New Drug Certificate, Import Drug License or Drug Product License. Annex should include all the annexes to the above approval, such as drug standards, insert sheet, design of packing and label and other annex.2) Certified Documents:i) If the Applicant is a drug manufacture enterprise, copies of the drug manufacture certificate, business licenses, and GMP Certificate should be provided. If the Applicant is not a drug manufacture enterprise, copies of the certified document of legal registration of the Applicant should be provided.If the drug registration is conducted by the resident office of the foreign manufacturer, copy of Registration Certificate Of Resident Office Of Foreign Enterprise should be submitted. If drug registration is conducted by a Chinese agent with authorization from the foreign manufacturer, the certified documents of the authorizations, notarized documents and the Chinese translation should be submitted together with the copy of Business License of the domestic agent.ii) For different application items, the certified document should be provided according to the Table of Application Information Items respectively for each item.iii) For import drug, the certified documents, notarized documents of the approval of the changes of the drugs issued by the competent drug administration authorities at the local country or region and the Chinese translation should by provided. The form of the documents should comply to the requirements for certified document set out in the application information items of TCM, natural drugs, chemical drugs, biological products.3) Draft of the amended design of the insert sheet of the drugs, attached with detailed notes for the amendments.4) Draft of the amended design of the packing and label of the drugs, attached with detailed notes for the amendments.5) Pharmaceutical study information.Part or all the experiment information of Pharmaceutical study and necessary global literature should be provided respectively for the different registration category, information items should be provided according to the corresponding application information items of Annexes 1-3. 6) Pharmacology and toxicology study information.Part or all the experiment information of Pharmacology and toxicology study and necessary global literature should be provided respectively for the different registration category,information items should be provided according to the corresponding application information items of Annexes 1-3.7) Clinical study information:If there is a need for a clinical study, the information items should be provided according to the corresponding application information items of Annexes 1-3. Information should be provided respectively before and after clinical trail. If there is no need for clinical study, the literature of the clinical study may be provided instead.8) Sample product of the drugs.III Table of Application Information ItemsRegistration Items Application Information Items1 2 3 4 5 6 7 8①②③Supplemental Applications Items to Approved by SFDA 1.Application for Drug Approval Number of a new drug by the New Drug Certificate holder of the drug + + --- + *1 ---2.Use of the name of the Trade Name of drugs + + *2 + + + ----3.Additional indications or functions of TCM, or indication already approved in China for chemical drug or biological products + + - + + + - # # -4.Change in dosage form or group of people of using of the drugs + + - + + + - # # -5.Change in the strengths of the drugs + + - + + + + - *3 +6.Change of the supplementive in the formula of the drugs, where there is a medial requirement for + + - + *4 *4 + -- +7.A change in the drug manufacture technology and process that affecting drug quality + + - + *4 *4 + # # +8.Amendment of drug registration standards + + - + *4 *4 *5 ---9.Substitute or removal of the drug material listed in formula of National Drug Standards as toxic or endangered. + + *6 + + + # # # +10.Change of the immediate packing material or container of the drugs + + - + *4 *4 *7 -- +11.Application for combined drug packing + + - + + + - *8 *8 +12.The transfer of new drug technology *9 + *10 - + + *1 - *11 +13.Transfer of temporary standards into formal standards *12 + -- *4 *4 # -- +14.A change in items within the import drug registration certificate, such as name of the drug, drug enterprise name, registered location, valid period and/ or packing specification + + - + + + *4 ---15.Change of the location where the import drug is manufactured + + - + + + + -- +16.Change of the overseas location where the import drug is packed + + *13 + + + *14 -- +17.Repacking of import drugs in China + + *15 - + + *16 -- +18.Change of the location where the raw material for import preparation is manufactured + + - + -- + -- +Supplemental applications items to be filed for record at SFDA 19.Change of the name of a domestic drug manufacturer + + *17 - + + ----20.Internal change of workshop of domestic drug manufacturer + + *18 - *4 *4 *1 -- +21.Amendment of insert sheet of the drugs according to national drug standards or required by SFDA + + *19 - + + ----22.Supplementing and perfecting of the drug safety part of the insert sheet + + - + + + - *20 *21 -23.Modification of design of packing and label of the drugs according to the regulations + + *22 + - + ----24.Change of the packing specification of drug manufactured at domestic + + -- + + *4 - *3 +25.Change of valid period of domestic drug + + - + + + *23 ---26.Change of the location where the raw material for domestic preparation is manufactured + + --- *4 *24 -- +27.Change of appearance of the drug without change of drug standards + + - + + *4 + -- +28.Change of the agent for import drug registration + + *25 -------Notes:*1 Only the test reports for the 3 consecutive batches of sample product are needed.*2 Certified documents for trademark retrieval should be provided.*3 Report of literature of clinical use should be provided*4 Should be provided if there is any amendment.*5 Only the experiment information and literature during quality study, draft for drug standards and notes to the draft, and the test reports for the 3 consecutive batches of sample product should be provided.*6 Certified document of toxic drug material, or endangered drug material, or certified document from authorities to request substitute or removal.*7 Only the experiment information during drug stability study and the test reports for the 3 consecutive batches of sample products should be provided.*8 Information should be provide according to the corresponding requirement for the preparation marketed ex-China under the registration category of TCM, Chemical Drug, or biological products. For the section of pharmaceutical study, only the test reports for the 3 consecutive batches of sample products, the experiment information during drug stability study, the basis for selection of the immediate packing and container of the drugs and quality standards should be provided.*9 original of new drug certificate should also be provided.*10 Transfer contract signed by the parties of the technology transfer should be provided; the relevant documents should be provided if the original drug manufacturer abandoned the drug manufacture.*11 Required separately be SFDA during conducting evaluation and when there is a need.*12 The summary and pharmaceutical study section of the original new drug application information that already passed its examination, and its examination recommendation should be provided at the same time.*13 The certified document of the GMP certificate of the packing enterprise issued by the competent drug administration authorities in the local country or region where the packing enterpriser is located should be provided.*14 Only the re-packing technology and process, experimental information of the drug stability, and basis for selection of the immediate packing and container of the drug and quality standard, as well as the test reports for the 3 consecutive batches of sample product should be provided.*15 Re-packing contract for import drug (including the authorization of use of the Trade Mark) should be provided.*16 Only the repacking technology and process, basis for the selection of immediate packing material for the drug and container as well as quality standards should be provided.*17 Approvals from the relevant administration of the change of the name, business licenses, Drug Manufacture Certificate, and the GMP Certificate before and after the name is change should be provided.*18 Certified document from the competent administration to approve the manufacture of the drug in a different workshop should be provided.*19 New national drug standards or the document from SFDA to request the amendment of the insert sheet should be provided.*20 Experimental information or literature of toxicology study may be provided.*21 Literatures may be provided.*22 relevant document should be provided for change of drug packing and label as required by regulation.*23 only experiment data of drug stability and test report of drug sample from 3 consecutive batches of drug should be provided.*24 only the approval of the drug substance and certified document to evidence the legal source of the drug substance as well as the test report of 1 batch of the preparation should be provided.*25 Authorization document, notarization document of the foreign drug manufacture to authorize the domestic agent to conduct its drug registration, and the Chinese translation, business license of the domestic agent, the agreement document or other certified document that the original agent has waive its agency right should be provided.“#”see the corresponding numbers in “VI Notes and relevant requirement of the registration items and application information items”.IV Notes and relevant requirement of the registration items and application information items.1) Registration item 1, Application for Drug Approval Number of a drug by the New Drug Certificate holder of the drug shall refer to the application for the manufacture of new drug when the manufacture conditions to manufacture the new drug are ready, but the manufacture conditions to manufacture the new drug was not ready when the research institution of the new drug obtained New Drug Certificate, and the new drug has not transferred to other enterprise for manufacture.2) Registration items 2, trade name of drugs only apply to new chemical drug or new biological products.3) Registration item 3. For the additional indications or functions of TCM, or for chemical drug or biological drug with indication of domestic drug of the same drug, pharmacology and toxicology study and the clinical study of the drug should be conducted according to the following,i) If a prolonged treatment period or increased dosage is needed for the additional indications or functions of the drugs, experiment information and literature on main pharmacodynamic, experiment information and literature on primary pharmacology, experiment information and literature on acute toxicity, as well as experiment information and literature on long term toxicity should be provided. Relevant experiment information should be provided for topical use of the drugs. After approval, clinical study should be conducted for with cases at trial group of not less than 300 cases.ii) For the additional indications or functions of the drugs where the same indications of the same kind of drug has been approved overseas, experiment information and literature on main pharmacodynamic should be provided and randomized controlled trials should be conducted for clinical study with at lease 60 pair of cases.iii) For the additional indications or functions of the drugs where the same indications of the same kind of drug has been approved at domestic, randomized controlled trial should be conducted for clinical study with at lease 60 pair of cases, or a bioequivalence trials should be conducted with use of the same kind of drug with the approved indication as Control.4) Registration item 4. For change in drug usage, drug dosage or group of people to use the drug, the experiment information and literature of safety study to support the changes should be provided, a clinical study should be conducted if necessary.5) Registration item 5, Change of strength of drugs should meet the following requirements:i). Strength of the drug in the application should be the same with that of the marketed drug. The principle of being scientific, reasonable and necessary should be followed for any inconsistency.ii). Strength of the drug in the application should be reasonably decided based on the drug usage and dosage where strength should be not less than signal minimal use, or greater than signal maximal use.iii). If there are change both in usage or dosage and group of people to use the drug, information required in registration category 4 should also be provided, and clinical trail should be conducted when necessary.6) Registration item 7, a change in the drug manufacture technology and process should not cause the change of the basis of the medical use of material. If there is a change of the basis ofthe medical use for TCM drugs, comparison test information on Pharmacy and toxicity test should be provided, and clinical trial with at lease 100 pair of cases should be conducted at specific objective based characteristic.7) Registration item 9, Substitute or removal of the drug material listed in formula of National Drug Standards as toxic or endangered, refers to the Substitute or removal of the drug material applied by applicant but not uniformly required by the State.i). For application for use of approved substitute of TCM material to replace the TCM material of any preparation, experiment information of new preparation process, drug standards and stability. Pharmacology and toxicology and clinical information may be exempted.ii). For application for use of TCM material collected in statutory standards as replacement, if the drug material acts as supplemental, experiment information of new preparation process, drug standards and stability, pharmacology and toxicology and clinical information may be exempted. In the substitute of drug is toxic drug, information on drug safety should also be provided, and including toxicology comparison test and clinical test should also be conducted. If the drug material acts as primary substance, in addition to the above information, clinical equivalence test should be conducted with the preparation of the drug, if necessary, pharmacology and toxicology comparison test should be conducted. In the substitute of drug is toxic drug, pharmacology and toxicology comparison test should be conducted.iii). For application for removal of toxic drug, experiment information of new preparation process, drug standards and stability, pharmacology and toxicology should be provided and clinical trail should be conducted.iv). Requirements of Pharmacy, pharmacology and toxicology information and clinical trails are as follows:Pharmacy: (1), production process: production process should be the same with original process before and after the substitute and removal of the drug. (2) Drug standards, specificity and content determination should be established to the substitute of drug. If it is not possible to establish specificity and content determination, experiment information should be provided. (3) stability, if stability will be affected due to the substitute of drug, stability test should be conducted.Pharmacology and toxicology: after the drug material is substituted, comparison study on pharmacodynamic and acute toxicity should be conducted on main indication between the new drug and the original drug. After the toxic drug material is removed, comparison study on pharmacodynamic should be conducted on main indication between the new drug and the original drug.8) Registration item 11, combined packing of drug refers to packing of 2 or more drugs of independent indication and / or dosage and usage, excluding the following:i). Marketing of preparation made of component of same active substance.ii). Lack of internationally recognized fully developed treatment protocol as supporting basis. iii). Drug with different route of administration.iv). Others not meeting the requirements.No separate drug approval number will be issued to combined packing of drug with no monitoring period, and no trade name is allowed to be used.Combined packing of drug should also meet the following requirementsi). Enterprise apply for combined packing of drug should be enterprise holding GMP Certificate, combined packing of drug should be manufactured by the enterprise itself with drug approval number available.ii). Insert sheet and packing label should be decided based on the pre-clinical study and clinical trails results, but not simple addition of all insert sheets, where regulation on insert sheet and packing label should be followed.iii). Immediate packing material should be compatible to the drug packed.iv). Specified validity period should be the validity period of drug of shortest validity period. v). Storage condition should be compatible to the drug packed.vi). Name of combined packing should read X/Y/Z combined packing, where X, Y and Z denote the INN of each drugs.9) Registration item 7, transfer of temporary standards into formal standards, should be conducted according to the procedure for the transfer of temporary standards into formal standards. The following information (with the exception of the those regulated otherwise by the State) should be provided for the pharmaceutical study part of the application information item:i) Drug standard under the application of transfer into formal standard and the notes to its amendments(including a comparison table with the drug standard overseas). Additional and revised test items for some material should be added based on the characteristic of the production process.ii) Improvement to follow up the examination recommendation of original drug registration approval certificate and the notes.iii) Total numbers of batches manufactured, and a compete test data of the partial products (normally annual statistic should include the results of not less than 10 consecutive batches of products).iv) Quality stability of the products and the determining of the valid period of the products within the last 2 years of the temporary standards.10) Registration item 19, change of the name of a domestic drug manufacturer, refers to the case where the application for the corresponding change of the enterprise name, after the name change of Drug Manufacturing License is approved.11) Registration item 20, Internal change of the manufacture workshop of a domestic drug manufacturer, include renovation of the existing location, expansion and new construction at different location.12) Registration item 21, refers to the amendment of some item of insert sheet of the drugs according to uniform requirements by SFDA, such as adverse reaction, counter indication, caution, except for specified otherwise, excluding change of indication, usage and dosage, or strength and others.13) Registration item 22, Supplementing and perfecting of the drug safety part of the insert sheet.14) Registration item 23, modification of design of packing and label of the drugs according to the regulation refers to modification of design of packing and label based on the relevant drug regulation, national standards, or the content of approved insert sheet of drug.15) Registration item 24, change of the packing specification of drugs manufactured at domestic should meet the following requirements:i). Packing specification of drug should be economic and convenient, if there is a treatment period for the drug, Packing specification of drug should be decided based on the treatment period.ii). If disposable syringe is used for injection, or special solvent is used for intravenous infusion and injection, no separate name is allowed. Registration of syringe and solvent should have been approved. Valid period of syringe and solvent should not be shorter than that of drug packed16) Registration item 26, change of the location where the raw material for domestic preparation is manufactured, refer to the case where a domestic drug manufacture enterprise changed its drug manufacturer of the raw material used for the manufacture of its preparation. There must be a drug approval number or an import drug registration certificate available for the raw material, and the certified document to evidence the legal sourcing of the raw material must be provided.17) Registration item 1, 5-10, 12, 15, 20, registration inspection should be conducted to the 3 batches of drugs. Registration item 26, registration inspection should be conducted to the 1 batches of drugsAnnex 5: Application Information Items of Drug Re-RegistrationI Drug manufactured in China1) Certified documentsi. Certified documents of drug approval as approval of sequential changes issued from food and drug administration.ii. Copy of Drug Manufacturing License.iii. Copy of business license.iv. Copy of GMP certificate.2) Summary of the production, marketing and random inspection during the past five years, with explanation of the cases where product failed to pass quality certification.3) Necessary document or explanation should be provided for any of the following cases:i. If there is any work to be completed as required in the certified document of drug approval or the re-registration approval, a summary report of completion of work should be providedattached with relevant information.ii. During the first time application for re-registration, if there is a need for Phase IV clinical trial, the summary report of Phase IV clinical trial should be provided.iii. During the first time application for re-registration, if there is a need for monitoring period for new drug, the summary report of monitoring period for new drug should be provided.4) Formula, production process and drug standards should be provided. If there is any change in Formula, production process and drug standards, the specific change should be provided with certified approval documents.5) Source of the raw material used for the production of the drug preparation should be provided, if any change in the source, certified approval documents should be provided.6) The sample of the current packing, label and insert sheet of smallest retail package.II Import Drug1) Certified documentsi. Copies of certified documents of Import Drug License and Drug Product License and the approval of supplemental application issued from SFDA.ii. Certified Documents, notarized documents for the approval of the marketing of the drugs issued from the competent drug authorities at the local country or region where the manufactured is located, and the GMP Certificate of the manufacturer, and the Chinese translation.iii. Certified Documents, notarized documents for the approval of sequential changes issued from the competent drug authorities at the local country or region where the manufactured is located, and the Chinese translation.iv. If the drug registration is conducted by the resident office of the foreign manufacturer, copy of Registration Certificate Of Resident Office Of Foreign Enterprise should be submitted.v. If drug registration is conducted by a Chinese agent with authorization from the foreign manufacturer, the certified documents of the authorizations, notarized documents and the Chinese translation should be submitted together with the copy of Business License of the domestic agent.2) Summary of the drug importation and marketing in China during the past five years, with explanation of the cases where product failed to pass quality certification.3) Summary of the clinical use, adverse reaction during the past three years of marketing since importation.4) Necessary document or explanation should be provided for any of the following cases:i. During the first time application for re-registration, if there is a need for Phase IV clinical trial, the summary report of Phase IV clinical trial should be provided.ii. If there is any work to be completed as required in the certified document of drug approval or the re-registration approval, a summary report of completion of work should be provided attached with relevant information.5) Formula, production process, drug standards and validation methods should be provided. If there is any change in Formula, production process drug standards and validation methods, the specific change should be provided with certified approval documents.6) Source of the raw material used for the production of the drug preparation should be provided, if any change in the source, certified approval documents should be provided.7) The sample of the current packing, label and insert sheet of smallest retail package used in China.8) The current version of the insert sheet in the original language from the original manufacturer approved by the competent drug authorities at the local country or region where the manufactured is located and the translation~~~~~~~~~~~~~~~~~~~~THE END~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~。
PRODUCT USER GUIDETo view the full MadgeTech product line, visitour website at .2Product Overview 2Installation Guide 2Device Operation 3Device Maintenance 4Appendix AProduct OverviewThe Temp1000Ex carries hazardous location, intrinsically safe certification in accordance with the latest issue of:IEC 60079-0, IEC 60079-11Directive 2014/34/EU (known as ATEX)Certified Intrinsically Safe for:• Electrical Protection Concepts: IEC: 60079-11 Ex ia – Ex ic, Intrinsic Safety Zones 0-2• Equipment Protection Level: Ga – Gc, Zones 0-2• Gas Groups: IIC• Temperature Class: T4Operational Warnings• When used in hazardous locations, the Temp1000Ex is to be installed prior to the location becoming hazardous and removed only after the area is no longer hazardous.• The maximum allowed ambient temperature for the Temp1000Ex (under any circumstances) is 80 °C. The minimum rated operating temperature is -40 °C.• The Temp1000Ex is approved for use only with the Tadiran TL-2150/S battery. Replacement with any other battery will void the safety rating.• Batteries are user replaceable, but are to be removed or replaced only in locations known to be non-hazardous.• Tampering or replacement of non-factory components may adversely affect the safe use of the product, and is prohibited. Except for replacement of the battery, the user may not service the Temp1000Ex. MadgeTech, Inc. or an authorized representative must perform all other service to the product.Ordering Information• 902153-00 — Temp1000Ex-2 (2 inch Probe)• 902155-00 — Temp1000Ex-1 (1 inch Probe)• 902156-00 — Temp1000Ex-5.25 (5.25 inch Probe)• 902157-00 — Temp1000Ex-7 (7 inch Probe)Key Ring End Cap• 902209-00 — Temp1000Ex-1-KR (1 inch Probe)• 902210-00 — Temp1000Ex-2-KR (2 inch Probe)• 902211-00 — Temp1000Ex-5.25-KR (5.25 inch Probe)• 902212-00 — Temp1000Ex-7-KR (7 inch Probe) Accessories• 900319-00 — IFC400• 900325-00 — IFC406• 901745-00 — Battery Tadiran TL-2150/S Installation GuideInstalling the SoftwareThe Software can be downloaded from the MadgeTech website at . Follow the instructions provided in the Installation Wizard.Installing the USB Interface DriversIFC400 or IFC406 — Follow the instructions provided in the Installation Wizard to install the USB Interface Drivers. Drivers can also be downloaded from the MadgeTech website at .Device OperationConnecting and Starting the Data Logger1. Once the software is installed and running, plug theinterface cable into the docking station (IFC400 orIFC406).2. Connect the USB end of the interface cable into anopen USB port on the computer.3. Place the data logger into the docking station (IFC400or IFC406).4. The data logger will automatically appear under Connected Devices within the software.5. For most applications, select Custom Start fromthe menu bar and choose the desired start method,reading rate and other parameters appropriate for the data logging application and click Start. (Quick Start applies the most recent custom start options, BatchStart is used for managing multiple loggers at once,Real Time Start stores the dataset as it records whileconnected to the logger.)6. The status of the device will change to Running or Waiting to Start, depending upon your start method.7. Disconnect the data logger from the interface cableand place it in the environment to measure.Note: The device will stop recording data when the end of memory is reached or the device is stopped, unless user selectable memory wrap is enabled. At this point the device cannot be restarted until it has been re-armed by the computer.Product User Guide | 2Device Operation (cont’d)Downloading Data from a Data Logger1. Place the logger into the docking station (IFC400 or IFC406).2. Highlight the data logger in the Connected Device s list. Click Stop on the menu bar.3. Once the data logger is stopped, with the logger highlighted, click Download .4. Downloading will offload and save all the recorded data to the PC.Device MaintenanceBattery ReplacementMaterials: Replacement Battery (Tadiran TL-2150/S)1. Move device to a non-hazardous location before replacing battery.2. Observe Operational Warnings when removing and replacing the battery.3. Unscrew the bottom of the data logger and remove the battery.4. Place the new battery into the logger. Caution: Observe correct battery polarity when installing.5. Screw the cover onto the data logger.O-RingsO-ring maintenance is a key factor when properly caring for the Temp1000Ex. The O-rings ensure a tight seal and prevent liquid from entering the inside of the device. Please refer to the application note “O-Rings 101: Protecting Your Data”, found at , for information on how to prevent O-ring failure.RecalibrationRecalibration is recommended annually. To send devices back for calibration, visit .Additional Services:Custom calibration and verification point options available, please call for pricing.Call for custom calibration options to accommodate specific application needs.Prices and specifications subject to change. See MadgeTech’s terms and conditions at .To send devices to MadgeTech for calibration, service or repair, please use the MadgeTech RMA Process by visiting .CommunicationTo ensure desired operation of the TEMP1000Ex, please keep the surface clear of any foreign objects orsubstances . The TEMP1000Ex’s data is downloaded through external contact with the IFC400 or IFC406 docking station. Covering the surface with foreign objects (i.e. Calibration Labels) can prevent the communication and/or downloading process.DOC-1367036-00 REV 5 2020.11.306 Warner Road, Warner, NH 03278(603) 456-2011******************EU Declaration of ConformityNo. DC-2020-250According to EN ISO/IEC 17050-1:2004The name and address of the manufacturer:MadgeTech, Inc.6 Warner RoadWarner, NH 03278 USAProduct model and description:902153-00 - Temp1000EX-2 902154-00 - RHTemp1000EX 902155-00 - Temp1000EX-1 902156-00 - Temp1000EX-5.25 902157-00 - Temp1000EX-7902208-00 - RHTemp1000EX-KR 902209-00 - Temp1000EX-1-KR 902210-00 - Temp1000EX-2-KR 902211-00 - Temp1000EX-5.25-KR 902212-00 - Temp1000EX-7-KRThis product conforms with the following Union harmonization legislation:2014/34/EU – ATEX DirectiveThe following harmonized standards and other technical specification were used in support of the declaration: Harmonized Standards: EN 60079 - 0 Edition 2018EN60079-11 Edition2012Notified body SGS Fimko Oy, number 0598 performed EU-Type examination in accordance with Annex III of the directive and issued the certificate: Baseefa19ATEX0126Notified body SGS Fimko Oy, number 0598 performed Conformity to type based on quality assurance of the production process in accordance with Annex IV of the directive and issued the QA Notification document: Baseefa19ATEX0126 Additionally, product complies with the essential requirements, and carries the CE marking accordingly with:2014/30/EU – EMC Directive2015/863/EU – RoHS3 Directive1907/2006/EU – REACH DirectiveAnd conforms with the following product standards and/or normative documents:IEC 61326-1 Edition 2013Emission RequirementsCISPR 11, Radiated Emissions, 30 MHz to 1 GHzLimit: CISPR 11, Group 1, Class ALimit: FCC Class AEnclosure PortIEC 61000-4-2, Electrostatic DischargeLevel: 4 kV Contact, 8 kV Air DischargesIEC 61000-4-3, Radiated Immunity (EM Field)Level: 10 V/M, 80 to 1000 MHz3 V/M, 1.4 to 2.0 GHz1 V/M, 2.0 to 2.7 GHzSupplementary Information:The products were tested in a typical usage configuration.RoHS Exemptions applied 6(a)), 7(c)-IIDeclared on behalf of MadgeTech, Inc.Dianne Moulton, Quality ManagerIssued from MadgeTech, Inc. Warner, NH USA November 25, 2020BAS-CERT-084SGS Baseefa Limited is an associate of SGS Fimko OYIssue 21 EU - TYPE EXAMINATION CERTIFICATE2 Equipment or Protective System Intended for use in Potentially Explosive AtmospheresDirective 2014/34/EU3 EU - Type Examination Certificate Number:Baseefa19ATEX0126 – Issue 24 Product:RHTEMP1000EX and TEMP1000EX5 Manufacturer:MadgeTech, Inc6 Address:6 Warner Road, Warner, NH 03278, USA7This re-issued certificate extends EU Type Examination Certificate No. Baseefa19ATEX0126 to apply to product designed and constructed in accordance with the specification set out in the Schedule of the said certificate but having any variations specified in the Schedule attached to this certificate and the documents therein referred to.8SGS Fimko Oy, Notified Body number 0598, in accordance with Article 17 of Directive 2014/34/EU of the European Parliament and of the Council, dated 26 February 2014, certifies that this product has been found to comply with the Essential Health and Safety Requirements relating to the design and construction of products intended for use in potentially explosive atmospheres given in Annex II to the Directive.8.1The original certificate was issued by SGS Baseefa Ltd (UK Notified Body 1180). It, and any supplements previously issued by SGS Baseefa Ltd have been transferred to the supervision of SGS Fimko Oy (EU Notified Body 0598). The original certificate number is retained.The examination and test results are recorded in confidential Report No. See Certificate History 9Compliance with the Essential Health and Safety Requirements has been assured by compliance with:EN IEC 60079-0: 2018 EN 60079-11: 2012except in respect of those requirements listed at item 18 of the Schedule.10 If the sign “X” is placed after the certificate number, it indicates that the product is subject to the Specific Conditions of Use specified in the schedule to this certificate.11This EU - TYPE EXAMINATION CERTIFICATE relates only to the design and construction of the specified product. Further requirements of the Directive apply to the manufacturing process and supply of this product. These are not covered by this certificate.12The marking of the product shall include the following : II 1 G Ex ia IIC T4 Ga (-40°C ≤ T a ≤ +80°C)SGS Fimko Oy Customer Reference No. 8001Project File No. 20/0489This document is issued by the Company subject to their General Conditions for Certification Services accessible at/en/Terms-and-Conditions.aspx . Attention is drawn to the limitation of liability, indemnification and jurisdiction issues defined therein. Any holder of this document is advised that information contained herein reflects the Company’s findings at the time of their intervention only and within the limits of Client’s instructions, if any. It does not necessarily indicate that the equipment may be used in particular industries or circumstances. The Company’s sole responsibility is to its Client and this document does not exonerate parties to a transaction from exercising all their rights and obligations under the transaction documents. This document cannot be reproduced except in full, schedule included, without prior written approval of the Company. Any unauthorized alteration, forgery or falsification of the content or appearance of this document is unlawful and offenders may be prosecuted to the fullest extent of the law.SGS Fimko OyTakomotie 8FI-00380 Helsinki, Finland Telephone +358 (0)9 696 361 e-mail *****************web site www.sgs.fiBusiness ID 0978538-5 Member of the SGS Group (SGA SA)R S SINCLAIRAuthorised Signatory for SGS Fimko OyBAS-CERT-084 SGS Baseefa Limited is an associate of SGS Fimko OYIssue 213 Schedule14 Certificate Number Baseefa19ATEX0126 – Issue 215Description of ProductThe product is a temperature sensor TEMP1000EX and temperature with humidity sensor RHTEMP1000EX with additional model variants. These dataloggers are to monitor temperature and humidity for intended area of deployment. The data logging stops once the maximum memory capacity is reached. This data is extracted by removing the product from the area of deployment and putting it onto a docking station, through serial communication. Data recording will re-start only once reset by the computer. The data is then collected. These sensors are designed to be intrinsically safe for temperature range of -40°C < Tamb < 80°C. Equipment is designed for Zone 0, under EPL Ga for gas group IIC, classified under temperature code T4. Humidity range for the product is from 0% to 100% on RH scale. Equipment is powered by one Tadiran TL 2150 1/2AA cell. Product dimensions for TEMP Model Series is 2.65 in. X .97 in. dia., with various lengths of probe attachments based on utilization. And, RHTEMP Model Series is 1.7 in. X 0.97 in. X 0.97 in. They appear alike and are cylindrical portable sensors, with probe attachments, which is the difference in the Model Series. In addition, there is mechanical key ring feature provided on enclosure for aesthetic looks only. This design does not relate to safety or protection type of the product. 16Report NumberSee Certificate History 17 Specific Conditions of UseNone 18Essential Health and Safety Requirements In addition to the Essential Health and Safety Requirements (EHSRs) covered by the standards listed at item 9, the following are considered relevant to this product, and conformity is demonstrated in the report: Clause Subject 1.4.1External effects1.4.2Aggressive substances, etc.19Drawings and DocumentsNew drawings submitted for this issue of certificate: Number Sheet Issue Date DescriptionDOC-902154-00 1 of 1 1.3 11/4/2020 RHTEMP100EX, TEMPERATURE AND HUMIDITY DATA LOGGERDOC-902155-00 1 of 1 1.3 11/4/2020 TEMP100EX TEMPERATURE DATA LOGGER SUB-1367007-XX 1 of 3 1.1 9/29/2020 TEMP1000EX-XX ELECTRICAL ASSEMBLY SUB-1367007-01 1 of 3 1.1 11/5/2020 TEMP1000EX-1 ELECTRICAL ASSEMBLY SUB-1368007-001 of 31.111/5/2020RHTEMP1000EX ELECTRICAL ASSEMBLYThe above drawings are associated and held with IECEx Certificate No. IECEx BAS 19.0109 Iss. 2BAS-CERT-084 SGS Baseefa Limited is an associate of SGS Fimko OYIssue 2Current drawings which remain unaffected by this issue: Number Sheet Issue Date DescriptionSUB-2106007-02 1 of 1 1.1 11/22/2019 HITEMP140 & 1000Ex 2" PROBE BOARD SUB ASSEMBLYSVC-2104007-00 1 of 1 1.1 11/22/2019 BOARD HT & 1000Ex 2-PIN BATTERY CONTACT PCB ASSEMBLYSVC-2105007-01 1 to 2 1.1 11/22/2019 1000EX PCB ASSEMBLYSVC-2107007-01 1 of 1 1.1 11/22/2019PCB ASSEMBLY, HITEMP140 & 1000EX RH SENSOR ADAPTER ASSEMBLYSUB-2106007-03 1 of 1 1.1 11/22/2019 HITEMP140 & 1000Ex 5.25" PROBE BOARD SUBASSEMBLYSUB-2106007-04 1 of 1 1.1 11/22/2019 HITEMP140 & 1000Ex 7" PROBE BOARD SUBASSEMBLYDOC-2104002-00 1 of 1 1.0 01/21/2019 HITEMP140 AND 1000EX SCHEMATICDRAWING, 2-PIN BATTERY CONNECTIONDOC-2104003-00 1 of 1 1.0 01/21/2019 PCB ASSEMBLY, HITEMP140 AND 1000EX 2-PINBATTERY CONNECTIONDOC-2105002-01 1 of 1 1.0 03/25/2019 SCHEMATIC DRAWING 1000EX PCB DOC-2105003-01 1 of 1 1.0 05/30/2019PCB ASSEMBLY, 1000EXDOC-2106002-XX 1 of 1 1.0 05/31/2019 HITEMP140 AMD 1000EX SCHEMATICDRAWING, RIGID AND FLEXIBLE ROBE ADAPTERDOC-2106003-XX 1 of 1 1.0 05/31/2019 PCB ASSEMBLY, HITEMP140 AND 1000EX RIGIDAND FLEXIBLE PROBE ADAPTERDOC-2107002-01 1 of 1 1.0 05/31/2019 HITEMP140 AND 1000EX RELATIVE HUMIDITYSCHEMATIC DRAWING, SENSOR ADAPTER (RHI)DOC-2107003-011 of 11.005/31/2019 PCB ASSEMBLY, HITEMP140 AND 1000EXRELATIVE HUMIDITY SENSOR ADAPTER (RHi)The above drawings are associated and held with IECEx Certificate No. IECEx BAS 19.0109. 20Certificate History Certificate No. Date CommentsBaseefa19ATEX012619 December 2019The release of the prime certificate. The associated test and assessment against the requirements of EN IEC 60079-0: 2018 & EN 60079-11: 2012 is documented in Certification Report No. GB/BAS/ExTR19.0287/00 (held with IECEx BAS 19.0109 Iss. 0), Project File No. 19/0594.Baseefa19ATEX0126 Issue 127 July 2020This issue of the certificate permits the addition of variants of the equipment fitted with a key ring and other minor drawing changes not affecting the original assessment.The equipment description on page 2 of the certificate was updated to include reference to these key ring variants. No additional changes performed under this revision.The associated test and assessment of the above is documented in Certification Report No. GB/BAS/ExTR20.0079/00 (held with IECEx BAS 19.0109 Iss. 1), Project File No. 20/0143.BAS-CERT-084 SGS Baseefa Limited is an associate of SGS Fimko OYIssue 2Certificate No. Date CommentsBaseefa19ATEX0126 Issue 211 November 2020 This issue of the certificate permit minor drawings changes notaffecting the previous assessment. As a result of these drawing changes, drawing No’s 902153-00, 902154-00, 902155-00, 902156-00, 902157-00, SUB-1367006-01, SUB-1367006-02, SUB-1367006-03, SUB-1367006-04, SUB-1367006-K1, SUB-1367006-K2, SUB-1367006-K3, SUB-1367006-K4, SUB-1368006-00, SUB-1368006-K0, SUB-2105007-01 & SUB-2107007-00 now no longer require listing as controlled drawings and were removed from the above drawing list.The associated test and assessment of the above is documented in Certification Report No. GB/BAS/ExTR20.0167/00 (held with IECEx BAS 19.0109 Iss. 2), Project File No. 20/0489.For drawings applicable to each issue, see original of that issue.。
1.1List1.1Module模块定义,类似于class,模块不能包含子模块,但是可以从其他模块引入定义。
定义部分:定义测试成分、通信端口、数据类型、常数、测试数据模板、函数、测试端口上调用的过程特征(signatures)、测试例等等。
控制部分:分调用测试例并控制它们的执行。
控制部分也可以声明(局部)变量等,程序语句(如ifelse和do-while)可以用于各个测试例的选择和执行顺序。
TTCN-3不支持全局变量的概念。
1.2Type用户自定义数据类型type float pi (3.1415926);type set MySetType{integer field1,charstring field2}1.3Template模板(Templates)用于传送一个特定值的集合或是测试接收的值的集合是否与模板说明匹配。
a) 模板提供了一种组织和重复使用测试数据的方法b) 模板能够被参数化;c) 模板允许匹配机制;d) 模板既能够用于基于消息的通信,也能用于基于过程的通信1.4ComponentThe component type defines which ports are associated with a component.•It is also possible to declare constants, variables and timers local to a particular component type.•These declarations are visible to all testcases, functions and altsteps that run on an instance of the given component type. This shall be explicitly stated using the runs on keyword (see clause 16) in the testcase, function or altstep header.1.5Port && messagePort:端口可以基于消息(message标识),和基于过程(procedure)的,甚至是二者混合(mixed)的。
Declaration of Compliancefor articles made of plastic and metal intended to come into contact with food02.03.2020Hereby we confirm that our productItem name: Insulation Mug Motion 3.35l and 0.5lItem No.: 06.96xx.xxxx / CMMF No.:complies with the legal regulations laid down in the German Commodity Ordinance (or the respective stipulations laid down in the European Regulations on Plastic materials)as well as Regulation (EU)No 10/2011,the Regulation (EC)No 1935/2004,in particular Article 3,11(5),15and 17,and the Regulation (EC)No 2023/2006,in their relevant versions.When used as specified,the overall migration as well as the specific migration do not exceed the legal limits;the transition of metal ions does not exceed the SRLs (Specific Release Limits)according to Council of Europe Resolution CM/Res(2013)9on metals and alloys used in food contact materials and articles (effective 2013–1st Edition).The tests are performed according to articles 17and 18of Regulation (EU)No 10/2011in conjunction with Annex III and V resp.according to the …Technical guide on metals and alloys used in food contact materials and articles“.The materials and raw materials used comply with the German Commodity Ordinance (or the respective stipulations laid down in European Regulations on Plastic materials)as well as the Regulation (EC)No 1935/2004and the Regulation (EU)No 10/2011,respectively according to the latest version.In the context of the quality assurance measures there was not found any indication of an incorrect utilization of substances evaluated.The following substances,subject to limitations and/or specification are used in the above mentioned product:Specification of the intended use or limitations:- Type(s) of food intended to come into contact with the material:- Intended duration and temperature of treatment and storage while in contact with the food:-Ratio of the area of the food contact surface to the volume used to determine the compliance of the food contact material or article:Insulation Mug Motion Cromargan 18/10 stainless steel, with black coating and copper coating. Lid made of polypropylene (PP) and silicone black. Mug 0.35l: 06.9616.6040, 06.9616.6600, 06.9616.6610Mug 0.5l: 06.9617.6040, 06.9617.6600, 06.9617.6610Description (material/optics)/ analogue items :repeated contact, short-time also above 100°Cliquid food (aqueous, fatty, acidic)------material:name of substance/ CAS-No. (limitation):The following dual-use-additives are enclosed:There is no functional barrier used in the plastic material.This declaration is valid for the product delivered by us as specified above.Under consideration of the food contact conditions stated,the product complies with the stipulations of these Directives regarding the specified foods.The user shall verify himself that the product is suitable for the intended food beyond the stipulations of the Directives.The validity of the declaration is ending if the requirements are changed.This document was created electronically and is valid without signature.Name of the document: A01_06.96xx.xxxx_EN_20200302_Insulation Mug Motion 3.35l and 0.5l_MS.pdfnoneDrückknopf/push button: 160ml/0.96dm²; Deckel/lid: 169ml/1.01dm²; Verschluss/closing:144ml/0.86dm²; Dichtung/sealing: 130ml/0.78dm² for the plastic piece334ml/2.24dm² for the metal piece WMF Group - a Groupe SEB company WMF Group GmbHVORSITZENDER DES AUFSICHTSRATES SITZ DER GESELLSCHAFT BANKVERBINDUNG Eberhardstraße 35Thierry de La Tour d'Artaise Geislingen | Steige Commerzbank AG Göppingen 73312 Geislingen | SteigeRECHTSFORM IBAN DE06 6104 0014 0160 300000GermanyGESCHÄFTSFÜHRER Gesellschaft mit beschränkter Haftung BIC COBADEFFXXX Oliver Kastalio | VORSITZENDER REGISTERGERICHT telefon+49 7331 25 1Bernd Stoeppel Amtsgericht Ulm HRB 732253fax +49 7331 45 387 WEEE-Reg.-Nr. DE 78426351USt.-ID.-Nr. DE 298 927 552。
Alpha锡膏OM340EN规格书英文版FINE __, ZERO __1, LOW HEAD IN PILLOW DEFECT, HIGHLY PIN __E__TION__-340 - NO-CLEAN, LEAD-FREE SOLDER PASTEALPHA OM-340 is a lead-free, no-clean solder paste designed for a broad range of applications. ALPHA OM-340 provides best in class low defect rate for Head in Pillow defects combined with excellent first pass yield on ICT/pin testing. ALPHA OM-340 also yields excellent print capability performance across various board designs and, particularly, with ultra fine feature repeatability and high “through-put” applications.Outstanding reflow process window delivers superior soldering on CuOSP with excellent coalescence on a broad range of deposit sizes, excellent random solder ball resistance and mid-chip solder ball performance. ALPHA OM-340 is formulated to deliver excellent visual joint cosmetics and best in class in circuit pin test yields. Additionally, ALPHA OM-340’s capability of IPC Class III for voiding and ROL0 IPC classifications ensures maximum long-term product reliability.1Zero halogen is defined as no halogen is intentionally added to the formulation__S __SMaximizes reflow yield for lead-free processing, allowing full alloy coalescence at circular dimensions as small as 200μm (8 mil) with 100μm (4 mil) thick stencilsExcellent print consistency with high process capability index across all board designs.Print sp eeds of up to 150mm/sec (6”/sec), enabling a fast print cycle time and a high throughput. Wide reflow profile window with good solderability on various board / component finishes. Excellent solder and flux cosmetics after reflow soldering Best in class low defect rate for Head in Pillow Best in class in circuit pin test yieldReduction in random solderballing levels, minimizing rework and increasing first time yield Meets highest IPC 7095 voiding performance classification of Class III Excellent reliability properties, halide-free material Compatible with either nitrogen or air reflowZero halogen (No halogen intentionally added to theformulation)__ __TIONSAC305 (96.5%Sn/3.0%Ag/0.5%Cu) SAC405 (95.5%Sn/4.0%Ag/0.5%Cu)SACX Plus 0307 (99%Sn/0.3%Ag/0.7%Cu) SACX Plus 0807 (98.5%Sn/0.8%Ag/0.7%Cu)InnoLot (90.95%Sn/3.8%Ag/0.7%Cu/1.4%Sb/0.15%Ni/3%Bi)For other alloys, please contact your local Cookson Electronics Sales Office. Type 3 (25 - 45μm per IPC J-STD-005) Type 4 (20 - 38μm per IPC J-STD-005)Type 4.5 (Proprietary Powder Size Distribution) C available upon request Type 5 (15 - 25μm per IPC J-STD-005) C available upon request Approximately 5% by (w/w) Complies with RoHS Directive 2022年/95/EC.__TIONFormulated for both standard and fine pitch stencil printing, at print speeds of between 25mm/sec (1”/sec) and 150mm/sec (6”/sec), with stencil thickness of 100μm (4 mil) to 150μm (6 mil), particularly when used with ALPHA Stencils. Blade pressures should be 0.18-0.27 kg/cm of blade (1.0 -1.5 Ibs/inch), depending upon the print speed. The higher the print speedemployed, the higher the blade pressure that is required. The reflow process window will give high soldering yield with good cosmetics and minimized rework.__ STATUSALPHA OM-340 is a Zero Halogen product and passes the standards listed in the Table below:Halogen StandardsStandardJEITA ET-7304Definition of Halogen Free Soldering Materials IEC __-2-21 JEDECA Guideline for Definin g “Low Halogen" ElectronicsRequirement1000 ppm Br, Cl, F in solder material solids Post Soldering Residues contain900 ppm each or total of 1500 ppm Br or Cl from flame retardant sourcePost soldering residues contain1000 ppm Br or Cl from flame retardant sourceTest Method StatusPassTM EN __PassPassZero Halogen: No halogenated compounds have been intentionally added to this productSAFETYWhile the ALPHA OM-340 flux system is not considered toxic, its use in typical reflow will generate a small amount of reaction and decomposition vapors. These vapors should be adequately exhausted from the work area. Consult the most recent MSDS (available at ) for additional safety information.____RES/__ALPHA OM-340 Technical Data__Y__L __IESActivity Level Halide Content Halogen Content Copper MirrorCopper Corrosion Test____CAL __IES SIR(IPC 7 days @ 85° C/85% RH) SIR(Bellcore 96 hours @ 35°C/85%RH) Electro migrationROL0 IPC J-STD-004 Halide free (by titration). Passes Ag IPC J-STD-004 Chromate TestPass, Zero Halogen - No halogen EN__, by oxygen bomb combustion, intentionally added Non-detectable (ND) at 50 ppm Pass IPC J-STD-004PassPass, 8.6 x 109 ohms Pass, 2.1 x 1011 ohms Pass, Initial = 3.9 x 10 ohmsClear, Colorless Flux Residue(Bellcore 96 hours @ 65°C/85%RH 10V 500 hours)__L __IES (Using 88.0% Metal, IPC Type 3 Powder, unless otherwise noted) ColorTack Force vs. Humidity(t=8 hours)Final = 1.9 x 109 ohmsIPC J-STD-0048(Pass ≥ 1 x 10ohm) Bellcore GR78-CORE11(Pass ≥ 1 x 10 ohm) Bellcore GR78-CORE (Pass=final initial/10)Pass, Change of 1 g/mm2 over 24 hoursat 25% and 75 % Relative HumidityIPC J-STD-005 TM-650 2.4.44 JIS Z3284 Annex 9Pass, Change of 10% when stored at25±2C and 50±10% relative humidity. Type 3 powder, 88.0% metal load designated M16 for printingViscosity (Typical) 1600 poise at 10 RPM MalcomType 4 powder, 88.3% metal load designated M18 for printingViscosity (Typical) 1800 poise at 10 RPM MalcomType 4 powder, 84% metal load, designated M06 for dispensingType 4 powder, 85% metal load, designated M08 for dispensingType 5 powder, 88.3% metal load designated M19 for printingType 5 powder, 78% metal load, designated M04 for dispensingType 6 powder, 78% metal load, designated M04 for dispensingoViscosity Malcom Spiral Viscometer; J-STD-005Solderball Stencil Life SpreadAcceptable (SAC 305 and SAC405 alloys) Pass, Class I - 1 hour and 72 hour8 hoursIPC J-STD-005DIN Standard 32 513, 4.4@ 50%RH, 25C (74°F) JIS-Z-3197: 1999 8.3.1.1 IPC J-STD-005 (10 min 150C) TM-650 2.4.35JIS-Z-3284-1994 Annex 7oPass PassSlumpNo bridging 0.2 mm gap above No bridging 0.3 mm gap aboveJIS-Z-3284-1994 Annex 8ALPHA OM-340 Processing Guidelines__ and __GRefrigerate to guarantee stability @ 0-10°C (32-50°F)__GRecommend CooksonElectronics ALPHA CUT ,ALPHA NICKEL-CUT , ALPHA__ND , or __RM stencils @ 0.100mm - 0.150 mm (4-6 mil) thick for 0.4 - 0.5 mm (0.016” or 0.020”) pitch. Stencil design is subject to many process variables. Contact your local Cookson Electronics stencil site for advice.Metal (recommended)PASTE ROLL: 1.5-2.0 cmdiameter and make additions when roll reaches 1-cm (0.4”) diameter (min). Max roll size will depend upon blade__E: 0.45 to 0.7 kg/inch 25 to 150mm per second (1 to 6 inches per second). : 3-10mm/sec.PRINT PUMP HEAD: Passes DEK ProFlow compatibility test REFLOW (See Figure 1)Clean-dry air or nitrogen atmosphere.Acceptable reflow / coalescence for feature size down to 8 mil (200 μm). IPC Class IIIvoiding obtained for both straight ramp and soak profiles.__GALPHA OM-340 residue isdesigned to remain on the board after reflow.If reflowed residue cleaning is required, the following aqueous cleaners are recommended: In-line or Batch Cleaners - ALPHA BC-2200 - Zestron Vigon A201 - Zestron Vigon A250 - Zestron Vigon USManual or solvent cleaning: - ALPHA SM-110 and SM-110E - BioactTM SC-10 and SC-10EMisprints and stencil cleaning may be done with the following cleaners:ALPHA SM-110E ALPHA SM-440Zestron Vigon SC200TMBioact SC-10EShelf life of refrigerated paste is 6 months.Paste can be stored for 2 weeks at roomotemperatures up to 25C (77°F) prior to use.When refrigerated, warm-up of paste container to room temperature for up to 4 hours. Paste must beoo≥19C (66F) before processing. Verify pastetemperature with a thermometer to ensure paste isooat 19C (66F) or greater before setup. Printing canoobe performed at temperatures up to 32C (89F).Paste can be manually stirred before use. A rotating, centrifugal force mixing operation is not required. If a rotating/centrifugal force mixing is used, 30 - 60 seconds at 300 RPM is adequate.Do not remove worked paste from stencil and mix with unused paste in jar. This will alter rheology of unused paste.These are starting recommendations and allprocess settings should be reviewed independently.Note 1: Refer to component and board supplier data for thermal properties at elevated temperatures. Lower peak temperatures require longer TAL for improved joint cosmetics. Keeping the peaktemperature below 240°C will lower the amount of voiding.Figure 1: ALPHA OM-340 SAC305 Typical Reflow ProfileParameter Atmosphere SAC305, SAC405, SACX Plus 0807 SACX Plus 0307 Setting Zone* o 40°C to 225 C 170°C to 225°C 120°C to 225°C TAL (217 - 225°C) Peak temperatureALPHA OM-340 - General Reflow Profile Guidelines Guideline Additional Information Air or N2 217 - 225°C Melting Range 217 - 227°C Melting Range Optimal Dwell Period Extended window 2:30 to 4:30 min. 5:00 min. 0:30 to 2:00 min 2:30 min. 1:25 to 3:00 min. 3:30 min. 45 - 90 sec. Not Recommended Compatible with most common surface finishes. (Entek HT, Entek OM, Alpha Star, ENIG, SACX 235 - 245°C HASL) 1 - 6°C/second Recommended to prevent surface cracking issues.Joint cool down rate from 170°C* Above recommendations are for SAC305. For alternative alloys, please follow the liquidus temperature of the respective alloy.109 Corporate Blvd., South Plainfield, NJ 07080, 1-800-367-5460,。
EUROPEAN COMMISSIONENTERPRISE AND INDUSTRY DIRECTORATE-GENERALConsumer goodsPharmaceuticalsBrussels, 01 September 2008EudraLexThe Rules Governing Medicinal Products in the European UnionVolume 4EU Guidelines toGood Manufacturing PracticeMedicinal Products for Human and Veterinary UseAnnex 7Manufacture of Herbal Medicinal Products Document HistoryRevision to specify application of GMP provisions for active substances used as starting materials (Part II) for the manufacture of herbal medicinal products. Additional changes are in particular related to the new Directive 2004/24/EC on traditional herbal medicinal products. Collaboration between GMDP Inspectors Working Group (formerly Ad Hoc GMP Inspection Services Working Group) and Committee of Herbal Medicinal Products (HMPC). May 2005 – March2006Public consultation May - July 2006Date of revised version coming into operation 01 September 2009 Commission Européenne, B-1049 Bruxelles / Europese Commissie, B-1049 Brussel – Belgium. Telephone: (32-2) 299 11 11PrincipleBecause of their often complex and variable nature, control of starting materials, storage and processing assume particular importance in the manufacture of herbal medicinal products.The “starting material” in the manufacture of a herbal medicinal product1 can be a medicinal plant, a herbal substance2 or a herbal preparation1. The herbal substance shall be of suitable quality and supporting data should be provided to the manufacturer of the herbal preparation/herbal medicinal product. Ensuring consistent quality of the herbal substance may require more detailed information on its agricultural production. The selection of seeds, cultivation and harvesting conditions represent important aspects of the quality of the herbal substance and can influence the consistency of the finished product. Recommendations on an appropriate quality assurance system for good agricultural and collection practice are provided in the HMPC guidance document: “Guideline on Good Agricultural and Collection Practice for starting materials of herbal origin”.This Annex applies to all herbal starting materials: medicinal plants, herbal substances or herbal preparations.1 Throughout the annex and unless otherwise specified, the term “herbal medicinal product/ preparation” includes “traditional herbal medicinal product/ preparation”.2 The terms herbal substance and herbal preparation as defined in Directive 2004/24/EC are considered to be equivalent to the Ph. Eur. terms herbal drug and herbal drug preparation respectively.Table illustrating the application of Good Practices to the manufacture of herbal medicinal products 3.Activity Good Agricultural and Collection Practice (GACP)4 Part II of the GMP Guide † Part I ofthe GMP Guide †Cultivation, collection and harvesting of plants, algae, fungi and lichens, andcollection of exudatesCutting, and drying of plants, algae, fungi, lichens and exudates *Expression from plants and distillation **Comminution, processing of exudates, extraction from plants, fractionation,purification, concentration or fermentationof herbal substancesFurther processing into a dosage form including packaging as a medicinal product†Explanatory Note.The GMP classification of the herbal material is dependent upon the use made of it by the manufacturing authorisation holder. The material may be classified as an active substance, an intermediate or a finished product. It is the responsibility of the manufacturer of the medicinal product to ensure that the appropriate GMP classification is applied.* Manufacturers should ensure that these steps are carried out in accordance with the marketing authorisation/registration. For those initial steps that take place in the field, as justified in the marketing authorisation/registration, the standards of Good Agricultural and Collection Practice for starting materials of herbal origin (GACP) is applicable. GMP is applicable to further cutting and drying steps.** Regarding the expression from plants and distillation, if it is necessary for these activities to be an integral part of harvesting to maintain the quality of the product within the approved specifications, it is acceptable that they are performed in the field, provided that the cultivation is in compliance with GACP. These circumstances should be regarded as exceptional and justified in the relevant marketing authorisation/ registration documentation. For activities carried out in the field, appropriate documentation, control, and validation according to the GMP principles should be assured. Regulatory authorities may carry out GMP inspections of these activities in order to assess compliance.Premises & EquipmentStorage areas1. Herbal substances should be stored in separate areas. The storage area should be equipped in such a way as to give protection against the entry of insects or other animals, especially rodents. Effective measures should be taken to prevent the spread of any such animals and 3This table expands in detail the herbal section of Table 1 in part II of the GMP Guide. 4 as published by the European Medicines Agency EMEAmicro-organisms brought in with the herbal substance, to prevent fermentation or mould growth and to prevent cross-contamination. Different enclosed areas should be used to quarantine incoming herbal substances and for the approved herbal substances.2. The storage area should be well aerated and the containers should be located in such a way so as to allow free circulation of air.3. Special attention should be paid to the cleanliness and maintenance of the storage areas particularly when dust is generated.4. Storage of herbal substances and herbal preparations may require special conditions of humidity, temperature or light protection; these conditions should be provided and monitored. Production area5. Specific provisions should be made during sampling, weighing, mixing and processing operations of herbal substances and herbal preparations whenever dust is generated, to facilitate cleaning and to avoid cross-contamination, as for example, dust extraction, dedicated premises, etc.Equipment6. The equipment, filtering materials etc. used in the manufacturing process must be compatible with the extraction solvent, in order to prevent any release or undesirable absorption of substance that could affect the product.DocumentationSpecifications for starting materials7. Herbal medicinal product manufacturers must ensure that they use only herbal starting materials manufactured in accordance with GMP and the Marketing Authorisation dossier. Comprehensive documentation on audits of the herbal starting material suppliers carried out by, or on behalf of the herbal medicinal product manufacturer should be made available. Audit trails for the active substance are fundamental to the quality of the starting material. The manufacturer should ensure that the suppliers of the herbal substance/preparation are in compliance with Good Agricultural and Collection Practice.8. To fulfil the specification requirements described in the basic requirements of the Guide (chapter 4), documentation for herbal substances/preparations should include: —the binomial scientific name of plant (genus, species, subspecies/variety and author(e.g. Linnaeus); other relevant information such as the cultivar name and thechemotype should also be provided, as appropriate;—details of the source of the plant (country or region of origin, and where applicable, cultivation, time of harvesting, collection procedures, possible pesticides used, possible radioactive contamination etc.);—which part(s) of the plant is/are used;—when a dried plant is used, the drying system should be specified;—a description of the herbal substance and its macro and microscopic examination;—suitable identification tests including, where appropriate, identification tests for constituents with known therapeutic activity, or markers. Specific distinctive tests are required where an herbal substance is liable to be adulterated/ substituted. A reference authentic specimen should be available for identification purposes;—the water content for herbal substances, determined in accordance with the European Pharmacopoeia;—assay of constituents of known therapeutic activity or, where appropriate, of markers;the methods suitable to determine possible pesticide contamination and limits accepted, in accordance with European Pharmacopoeia methods or, in absence thereof, with an appropriate validated method, unless otherwise justified;—tests to determine fungal and/or microbial contamination, including aflatoxins, other mycotoxins, pest-infestations and limits accepted, as appropriate;—tests for toxic metals and for likely contaminants and adulterants, as appropriate;—tests for foreign materials, as appropriate;—any other additional test according to the European Pharmacopoeia general monograph on herbal substances or to the specific monograph of the herbal substance, as appropriate.Any treatment used to reduce fungal/microbial contamination or other infestation should be documented. Specifications and procedures should be available and should include details of process, tests and limits for residues.Processing instructions9. The processing instructions should describe the different operations carried out upon the herbal substance such as cleaning, drying, crushing and sifting, and include drying time and temperatures, and methods used to control cut size or particle size.10. In particular, there should be written instructions and records, which ensure that each container of herbal substance is carefully examined to detect any adulteration/substitution or presence of foreign matter, such as metal or glass pieces, animal parts or excrement, stones, sand, etc., or rot and signs of decay.11. The processing instructions should also describe security sieving or other methods of removing foreign materials and appropriate procedures for cleaning/selection of plant material before the storage of the approved herbal substance or before the start of manufacturing.12. For the production of an herbal preparation, instructions should include details of solvent, time and temperature of extraction, details of any concentration stages and methods used. Quality controlSampling13. Due to the fact that medicinal plant/herbal substances are heterogeneous in nature, their sampling should be carried out with special care by personnel with particular expertise. Each batch should be identified by its own documentation.14. A reference sample of the plant material is necessary, especially in those cases where the herbal substance is not described in the European Pharmacopoeia or in anotherPharmacopoeia of a Member State. Samples of unmilled plant material are required if powders are used.15. Quality Control personnel should have particular expertise and experience in herbal substances, herbal preparations and/or herbal medicinal products in order to be able to carry out identification tests and recognise adulteration, the presence of fungal growth, infestations, non-uniformity within a delivery of crude material, etc.16. The identity and quality of herbal substances, herbal preparations and of herbal medicinal products should be determined in accordance with the relevant current European guidance on quality and specifications of herbal medicinal products and traditional herbal medicinal products and, where relevant, to the specific Ph. Eur. Monographs.。
Notified Bodies Recommendation GroupConsensus Paperfor the Interpretation and Application of Annexes Z inEN ISO 14971: 2012Draft V 1.1 from the NBRG WG RMJune, 25th, 2014!! Not yet adopted from NBRG !!Content1.) Introduction (32)2.) Terminology (3)3.) General Considerations (4)4.) Recommendations for Industry (5)5.) Recommendations for the NB audit process (9)Annex 1: Recommendations for Standardization Bodies (11)Members of the workgroup:Michael Bothe, MBA, VDE Testing & Certification Institute, ChairMarina Belonogova, St. Jude Medical (Eucomed)Oliver Bisazza, (COCIR)Gert Bos, BSICaterina Brusasco, IBA (COCIR)Uwe Herrmann, Roche Diagnostics (EDMA)Hans-Heiner Junker, TÜV SüdPeter Linders, Philips Healthcare (COCIR)Katalin Mate, (EDMA)Peter Meeremans, Alcon (Eucomed)Gerd Neumann, Siemens Healthcare (COCIR)Martin Penver, LRQARydin, Carrie, Terumo BCTThecla Sterk, (Eucomed)Martin Schraag, Philips Healthcare, (ZVEI)Andre Schmitz, TÜV Rheinland LGA Products GmbHSue Spencer, BSIJos van Vroonhoven, Philips Healthcare (COCIR)1.) IntroductionIn October 2010, the regular review of ISO 14971:2007 which is the basis of EN ISO 14971:2009 was closed by a broad majority of votes confirming the existing status and the wide-spread acceptance of this standard in the medical devices community, including competent authorities. In November 2010, the European Commission raised a formal objection against the use of several harmonized standards, including EN ISO 14971, followed by an in-depth assessment of the coverage of the Essential Requirements of the Medical Device Directives (90/385/EEC, 93/42/EEC and 98/79/EC) by these standards.As a result of these objections, the Annexes Z to EN ISO 14971 were modified, resulting in EN ISO 14971:2012. This amendment of the EN ISO 14971 standard did not modify the normative parts of ISO 14971:20071. The Annexes Z describe the extent of presumption of conformity that can be based on application of the normative requirements of ISO 14971 alone. The “content deviations”, expressed in the revised Annexes Z, between ISO 14971:2007 and the Medical Device Directives have been commented by many experts in the field of risk management and resulted in diverging interpretations from different stakeholders (e.g. manufacturers, notified bodies, competent authorities).This document has been prepared as a Notified Body Consensus Paper by a working group headed by the NBRG Vice Chair, with representatives from several European Notified Bodies and industry associations COCIR, Eucomed, EDMA and ZVEI. The paper aims to provide a practical interpretation of these “content deviations” to the Medical Device Directives and give guidance as to how to implement the risk management requirements. The work consolidates prior publications of various sources and is intended to facilitate common understanding between industry and Notified Bodies.2.) TerminologyThe three medical devices directives (93/42/EEC, 90/385/EEC and 98/79/EC) refer to “risk” and “safety” in a general sense. Since the time of writing these directives about 20 years ago, the knowledge of and experience in risk management have evolved considerably. This is reflected by the successive publication of EN1441 in 1994 and ISO14971-1 in 1998 as well as first (2000) and second (2007) editions of ISO 14971 which all have been recognized globally as the state of the art for risk management at the moment of their publication.The wording of the risk management aspects in the essential requirements of the Medical Devices Directives has not been modified over time, however.The international standard ISO 14971:2007 and its European equivalent EN ISO 14971:2012 contain specific defined terms with a clear and precisely described 1EN ISO 14971:2012 Annexes Z apply to manufacturers placing devices on the market in the European Union; for the rest of the world, ISO 14971:2007 remains the applicable standard.meaning. See Table 1 for an overview of the most relevant terms used in this document. Note that these defined terms are more precise than the general terms as used in the Medical Device Directive. For example, “risk” in the medical devices directives can bear the meaning of “risk”, “hazard” or “hazardous situation” depending on the context. This document is compiled on the assumption that “risk” in the Medical Device Directives is equivalent to “unacceptable risk” in ISO 14971:2007.Table 1: Relevant terms from (a) ISO 14971:2007 and (b) ISO/TR 24971:2013 3.) General ConsiderationsThis Consensus Paper intends to bridge the gap between the interpretation of the relevant Essential Requirements of the Medical Devices Directives, as given in the Annexes ZA, ZB, and ZC of EN ISO 14971:2012, and the practice of placing safe medical devices on the market in the EU and in other countries where the above-mentioned directives apply. This chapter provides some considerations about risk management and how this is to be interpreted in the context of the European Medical Devices Directives.The practical approach of this Consensus Paper safeguards the principle that only medical devices that are “compatible with a high level of protection of health and safety”2can be placed on the EU market. With this in mind, two aspects of the European Commission’s interpretation of EN I SO 14971:2012, as reflected in the Annexes Z, deserve further consideration:1.Reduce risk “as far as possible”, and2.Economical considerations in Risk Management1. Reducing risk “as far as possible”2 COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 concerning medical devices.The phrase “as far as possible” has led to significant confusion for those involved in placing medical devices on the market. Strict interpretation would create practical problems such as where to stop in reducing risk before a product can be placed on the market. This may restrict patient access to safe and affordable devices.In line with Clause 1.1 of the 2013 edition of the European Commission, Parliament and Council’s Joint Practical Guide of the European Parliament, the Council and the Commission for persons involved in the drafting of European Union legislation3, this consensus paper offers Notified Bodies and manufacturers an interpretation of “as far as possible” that is “clear, easy to understand and unambiguous.”2. On economical considerations in Risk Management“C ontent deviation” 3 in the Annexes Z of EN ISO 14971:2012 states:a) Annex D.8 to ISO 14971, referred to in 3.4, contains the concept of reducing risks "as lowas reasonably practicable" (ALARP concept). The ALARP concept contains an element ofeconomic consideration.b) However, the [Essential Requirements require risks to be reduced "as far as possible"without there being room for economic considerations.c) Accordingly, manufacturers and Notified Bodies may not apply the ALARP concept withregard to economic considerations.This disregard of economic considerations when reducing risk is not coherent with the Medical Device D irectives’ objective as stated in, for example, the following recital4 of Directive 93/42/EEC:It should be noted that this specific recital on the relevance of economic considerations exists since the first publication of the original Directive.4.) Recommendations for IndustryThe Annexes Z of EN ISO 14971:2012 list seven “content deviations”, i.e., differences between the wording of the Essential Requirements (ERs) in the Medical Device Directives and the wording of the requirements of the standard. Nevertheless, the Medical Device Directives and the standard share the same objectives of achieving a high level of product safety and ensuring continuous improvement. Parts of the content deviations are quoted and given here in italics. Specific 3http://eur-lex.europa.eu/content/pdf/techleg/joint-practical-guide-2013-en.pdf4Recitals are part of the legal text, as stated in Clause 10.1 of the Joint Practical Guide mentioned above and confirmed in the ECJ ruling C-219/11.recommendations for medical device manufacturers in relation to those “content deviations” are given below.Content deviation 1:Treatment of negligible risks“..the manufacturer must take all risks into account..”Recommendation:The manufacturer must identify known and foreseeable hazards and estimate the risk for each hazardous situation identified (Clause 4 of EN ISO 14971:2012). The risk control measures and the results of the risk evaluation must be recorded in the risk management file (Clause 5 and Clause 6.2 of EN ISO 14971:2012). This process ensures that all risks are given sufficient attention.The manufacturer shall document all identified hazards and hazardous situations, their associated risks and the risk control measures for each individual risk, in the risk management file.Compliance may be demonstrated by review of the risk management file.Content deviation 2:Discretionary power of manufacturers as to acceptability of risks“...all risks combined, regardless of any “acceptability assessment,” need to be balanced, together with all other risks, against the benefit of the device.”“Accordingly, the manufacturer may not apply any criteria of risk acceptability prior to applying Sections 1 and 2 of Annex I to Directive 93/42/EEC (Sections 1 and 6 of Annex I to Directive 90/385/EEC respectively Sections A.1 and A.2 of Annex I to Directive 98/79/EC).”Recommendation:When determining the criteria for risk acceptability, the manufacturer shall consider whether death or serious deterioration of health is unlikely to occur in normal operation or due to device malfunctions or deterioration of characteristics or performance, or any inadequacy in the labeling or instructions for use.5If unlikely to occur the risk shall be considered acceptable.Otherwise, the risk must be reduced. In doing so, the manufacturer may choose an end-point for risk reduction, for example:1.) The risk acceptability is preferably based on harmonized standards specifyingstate of the art risk control measures for particular categories of medical devices.Basing the risk reduction end-point on harmonized standards ensures that the risk is reduced to an acceptable level.5 coherent with criteria from Article 10 of Directive 93/42/EEC and corresponding requirements in Annexes (e.g. Annex II, 3.1) when deciding about Field Corrective Actions2.) If no harmonized standards are available, other national or internationalrecognized standards or publications should be considered. Basing the risk reduction end-point on recognized international standards ensures that the risk is reduced to an acceptable level.3.) Where those publications are not available, the manufacturer must assess thebest risk reduction means and shall include in the description of the risk management process what criteria were used to determine the acceptability of risks. The criteria for risk acceptability are then based among others on historical data, best medical practice and state of the art.4.) Further risk control measures do not improve the safety.If a reduction to an acceptable level cannot be achieved, a risk-benefit analysis must demonstrate that the residual risk is outweighed by the medical benefit as explained in content deviation 4.Compliance may be demonstrated by reflecting such end-points in the criteria for risk acceptability as part of the risk management file. Where safety cannot be demonstrated as such, existing clinical data is used to demonstrate that the medical benefit outweighs the risk.Content deviation 3: Risk reduction “as far as possible” versus “as low as reasonably practicable”“Essential requirements require risks to be reduced as far as possible without there being room for economic considerations...”With this deviation the European Commission raises the concern that economic considerations might surmount safety considerations. On the other hand the reduction of a risk “as far as possible” could be without limits and the resulting devices might no longer be affordable for a larger group of patients.Recommendation:Although economic considerations will always be relevant in decision-making processes, the safety of the product must not be traded off against business perspectives. For transparency the manufacturer must document the end-point criteria of risk reduction based on his risk policy.Compliance is checked by inspection of the documentationContent deviation 4: Discretion as to whether a risk-benefit analysis needs to take place“the manufacturer must undertake a risk-benefit analysis for the individual risk and the overall risk-benefit…”Recommendation:At the end of the risk management process the manufacturer shall perform a risk‐benefit analysis for individual risks that are not acceptable according to the criteria explained in content deviation 2 and for which further risk reduction is not possible. In any case the manufacturer shall perform an overall risk-benefit analysis considering all individual risks to provide a rationale for overall risk acceptance. Compliance is checked by inspection of the individual and overall risk-benefit analyses.Content deviation 5: Discretion as to the risk control options“…the manufacturer must apply all the control options and may not stop his endeavors if the first or second control option has reduced the risk to an “acceptable” level (unless the additional control options do not improve the safety).”Recommendation:As stated above for content deviation 2, the manufacturer can justify ceasing further risk reduction where it is determined that the risk is acceptable, i.e. that risk reduction has progressed to a level as described above in the section of content deviation 3 . The manufacturer shall consider all risk control measures in Essential Requirement 2 that are appropriate to reduce the risk to an acceptable level. In so doing, the manufacturer shall document the control options in the priority order, as part of the risk management process.Compliance is checked by reviewing the documented risk management process. Content deviation 6: Deviation as to the first risk control option“inherent safety by design” vs.“inherently safe design and construction”Recommendation:The manufacturer shall ensure that, whenever possible, the first risk control option includes both safe design and safe construction.Not relevant for compliance verification.Content deviation 7: Information of the users influencing the residual risk“..manufacturers shall not attribute additional risk reduction to the information given to the users...”ISO 14971 (Annex J) and ISO/TR 24971 describe ‘information for safety’ as instructions for use, warnings, required maintenance, etc.. ‘Information for safety’ comprises instructions of what actions the user can take or avoid in order to prevent a hazardous situation from occurring. On the other hand, ‘d isclosure of residual risk’has the objective to inform users of remaining risk inherent to the use of the medical device, and concerns the risks remaining after all risk control measures have been taken.Recommendation:Any information for safety comprising instructions of what actions the user can take or avoid in order to prevent a hazardous situation from occurring may be considered a risk control measure. As required by Essential Requirement 13.1 of Directive 93/42/EEC (respectively ER B.8 of 98/79/EC) it may be considered as a risk control measure. The information includes the instructions for use, labels, etc.. Since ‘safe use’ is related to risk control measures, the Medical Device Directives do not deviate in that regard from EN ISO 14971. Any effects on risk reduction are to be documented by the manufacturer in the risk management file.‘Disclosure of residual risk’ should be conducted in compliance with EN ISO 14971 Clause 6.4, 6.5 and 7. The manufacturer shall not claim a reduction to the probability of harm when disclosing residual risk.Compliance is checked by inspection of the risk management file.5.) Recommendations for the NB audit processThe role of the Notified Body is to assess compliance to the Directives, including the implementation of the risk management process and whether clinical benefits outweigh the risks to patients and users.Manufacturers placing devices on the European market should be aware that gaps between the requirements of the Directives and the Risk Management Standard (documented in the EN ISO 14971: 2012 edition) have to be addressed, if applicable. It is the discretion of the manufacturer to use this standard in conjunction with other means to demonstrate conformity with the Essential Requirements of the Directive. When EN ISO 14971 is used in upcoming audits and risk management file reviews, assessors and technical experts from Notified Bodies will focus on objective evidence on how manufacturers addressed those gaps and modified their Risk Management Process accordingly. More specifically they will evaluate:1. Are all design solutions in conformity with the safety principles given in theEssential Requirements and EN ISO 14971 (inherent safe design andconstruction > protection measures > information for safety)?2. Has the manufacturer demonstrated that all risks have been reduced to anacceptable level in the sense of this guidance paper?3. Has the manufacturer conducted a risk benefit analysis for all individualresidual risks that are not acceptable according to the risk acceptabilitycriteria?4. Has the manufacturer conducted an overall risk benefit analysis consideringall individual risks combined?5. Has the manufacturer demonstrated that information for safety is effective?6. Has the manufacturer included information on residual risks, if needed, in theaccompanying documents?Annex 1: Recommendations for Standardization Bodies Modification of IEC 60601-1 (Ed. 3.1)Amendment A1:2012 to IEC 60601-1:2005 (together Edition 3.1) has improved the consistency of risk management terminology. Nevertheless, this edition has room for further improvement. The primary objective of the standard (and its collateral and particular standards) is to provide state-of-the-art requirements and solutions for basic safety and essential performance. The standard should be restrictive in referring to risk management.Modification of EN ISO 14971As has been argued, the Annexes Z to EN ISO 14971:2012 contain errors and occasionally confusing phrases. It is therefore important that these Annexes are amended through a revision of EN ISO 14971. This is within the remit of the European Standards Organizations and within CEN/CENELEC TC3 in particular. The joint Notified Bodies do not have an explicit role in this Technical Committee but, upon acceptance and implementation of this consensus paper, the NBRG is willing to contribute to amendment suggestions that subsequently may be taken forward by individual members of CEN/CENELEC TC3.11。
ANNEX I‘A NNEX14INTRODUCTORY NOTES TO THE LIST IN ANNEX15Note1:The list sets out the conditions required for all products to be considered as sufficiently worked or processed within the meaning of Articles69and100.Note2:2.1.The first two columns in the list describe the product obtained.The first column gives the heading number orchapter number used in the Harmonised System and the second column gives the description of goods used in that system for that heading or chapter.For each entry in the first two columns,a rule is specified in column3 or4.Where,in some cases,the entry in the first column is preceded by an“ex”,this signifies that the rules in column3or4apply only to the part of that heading as described in column2.2.2.Where several heading numbers are grouped together in column1or a chapter number is given and thedescription of products in column2is therefore given in general terms,the adjacent rules in column3or4 apply to all products which,under the Harmonised System,are classified in headings of the chapter or in any of the headings grouped together in column1.2.3.Where there are different rules in the list applying to different products within a heading,each indent containsthe description of that part of the heading covered by the adjacent rules in column3or4.2.4.Where,for an entry in the first two columns,a rule is specified in both columns3and4,the exporter may opt,as an alternative,to apply either the rule set out in column3or that set out in column4.If no origin rule is given in column4,the rule set out in column3is to be applied.Note3:3.1.The provisions of Articles69and100,concerning products having acquired originating status which are used inthe manufacture of other products,shall apply,regardless of whether this status has been acquired inside the factory where these products are used or in another factory in the beneficiary country or republic or in the Community.Example:An engine of heading8407,for which the rule states that the value of the non originating materials which may be incorporated may not exceed40%of the ex works price,is made from“other alloy steel roughly shaped by forging”of heading ex7224.If this forging has been forged in the beneficiary country or republic from a non originating ingot,it has already acquired originating status by virtue of the rule for heading ex7224in the list.The forging can then count as originating in the value-calculation for the engine,regardless of whether it was produced in the same factory or in another factory in the beneficiary country or republic.The value of the non-originating ingot is thus not taken into account when adding up the value of the non-originating materials used.3.2.The rule in the list represents the minimum amount of working or processing required,and the carrying-out ofmore working or processing also confers originating status;conversely,the carrying-out of less working or processing cannot confer originating status.Thus,if a rule provides that non-originating material,at a certain level of manufacture,may be used,the use of such material at an earlier stage of manufacture is allowed,and the3.3.Without prejudice to Note3.2,where a rule uses the expression“Manufacture from materials of any heading”,then materials of any heading(s)(even materials of the same description and heading as the product)may be used,subject,however,to any specific limitations which may also be contained in the rule.However,the expression“Manufacture from materials of any heading,including other materials of heading...”or “Manufacture from materials of any heading,including other materials of the same heading as the product”means that materials of any heading(s)may be used,except those of the same description as the product as given in column2of the list.3.4.When a rule in the list specifies that a product may be manufactured from more than one material,this meansthat one or more materials may be used.It does not require that all be used.Example:The rule for fabrics of headings5208to5212provides that natural fibres may be used and that chemical materials,among other materials,may also be used.This does not mean that both have to be used;it is possible to use one or the other,or both.3.5.Where a rule in the list specifies that a product must be manufactured from a particular material,the conditionobviously does not prevent the use of other materials which,because of their inherent nature,cannot satisfy the rule.(See also Note6.2below in relation to textiles.)Example:The rule for prepared foods of heading1904,which specifically excludes the use of cereals and their derivatives, does not prevent the use of mineral salts,chemicals and other additives which are not products from cereals.However,this does not apply to products which,although they cannot be manufactured from the particular materials specified in the list,can be produced from a material of the same nature at an earlier stage of manufacture.Example:In the case of an article of apparel of ex Chapter62made from non-woven materials,if the use of only non-originating yarn is allowed for this class of article,it is not possible to start from non-woven cloth–even if non-woven cloths cannot normally be made from yarn.In such cases,the starting material would normally be at the stage before yarn–that is,the fibre stage.3.6.Where,in a rule in the list,two percentages are given for the maximum value of non originating materials thatcan be used,then these percentages may not be added together.In other words,the maximum value of all the non-originating materials used may never exceed the higher of the percentages given.Furthermore,the individual percentages must not be exceeded,in relation to the particular materials to which they apply.Note4:4.1.The term“natural fibres”is used in the list to refer to fibres other than artificial or synthetic fibres.It is restrictedto the stages before spinning takes place,including waste,and,unless otherwise specified,includes fibres which have been carded,combed or otherwise processed,but not spun.4.2.The term“natural fibres”includes horsehair of heading0503,silk of headings5002and5003,as well aswool-fibres and fine or coarse animal hair of headings5101to5105,cotton fibres of headings5201to5203,4.3.The terms“textile pulp”,“chemical materials”and“paper-making materials”are used in the list to describe thematerials,not classified in Chapters50to63,which can be used to manufacture artificial,synthetic or paper fibres or yarns.4.4.The term“man-made staple fibres”is used in the list to refer to synthetic or artificial filament tow,staple fibresor waste,of headings5501to5507.Note5:5.1.Where,for a given product in the list,reference is made to this Note,the conditions set out in column3shallnot be applied to any basic textile materials used in the manufacture of this product and which,taken together, represent10%or less of the total weight of all the basic textile materials used.(See also Notes5.3and5.4below.)5.2.However,the tolerance mentioned in Note5.1may be applied only to mixed products which have been madefrom two or more basic textile materials.The following are the basic textile materials:—silk;—wool;—coarse animal hair;—fine animal hair;—horsehair;—cotton;—paper-making materials and paper;—flax;—true hemp;—jute and other textile bast fibres;—sisal and other textile fibres of the genus Agave;—coconut,abaca,ramie and other vegetable textile fibres;—synthetic man-made filaments;—artificial man-made filaments;—current-conducting filaments;—synthetic man-made staple fibres of polypropylene;—synthetic man-made staple fibres of polyester;—synthetic man-made staple fibres of polyacrylonitrile;—synthetic man-made staple fibres of polyimide;—synthetic man-made staple fibres of polytetrafluoroethylene;—synthetic man-made staple fibres of poly(phenylene sulphide);—synthetic man-made staple fibres of poly(vinyl chloride);—other synthetic man-made staple fibres;—artificial man-made staple fibres of viscose;—other artificial man-made staple fibres;—yarn made of polyurethane segmented with flexible segments of polyether,whether or not gimped;—yarn made of polyurethane segmented with flexible segments of polyester,whether or not gimped;—products of heading5605(metallised yarn)incorporating strip consisting of a core of aluminium foil or of a core of plastic film whether or not coated with aluminium powder,of a width not exceeding5mm, sandwiched by means of a transparent or coloured adhesive between two layers of plastic film;—other products of heading5605.Example:A yarn,of heading5205,made from cotton fibres of heading5203and synthetic staple fibres of heading5506,is a mixed yarn.Therefore,non-originating synthetic staple fibres which do not satisfy the origin-rules(which require manufacture from chemical materials or textile pulp)may be used,provided that their total weight does not exceed10%of the weight of the yarn.Example:A woollen fabric,of heading5112,made from woollen yarn of heading5107and synthetic yarn of staple fibresof heading5509,is a mixed fabric.Therefore,synthetic yarn which does not satisfy the origin-rules(which require manufacture from chemical materials or textile pulp),or woollen yarn which does not satisfy the origin-rules(which require manufacture from natural fibres,not carded or combed or otherwise prepared for spinning),or a combination of the two,may be used,provided that their total weight does not exceed10%of the weight of the fabric.Example:Tufted textile fabric,of heading5802,made from cotton yarn of heading5205and cotton fabric of heading5210,is a only mixed product if the cotton fabric is itself a mixed fabric made from yarns classified in two separate headings,or if the cotton yarns used are themselves mixtures.Example:If the tufted textile fabric concerned had been made from cotton yarn of heading5205and synthetic fabric of heading5407,then,obviously,the yarns used are two separate basic textile materials and the tufted textile fabric is,accordingly,a mixed product.5.3.In the case of products incorporating“yarn made of polyurethane segmented with flexible segments of polyether,5.4.In the case of products incorporating“strip consisting of a core of aluminium foil or of a core of plastic filmwhether or not coated with aluminium powder,of a width not exceeding5mm,sandwiched by means of a transparent or coloured adhesive between two layers of plastic film”,this tolerance is30%in respect of this strip. Note6:6.1.Where,in the list,reference is made to this Note,textile materials(with the exception of linings and interlinings),which do not satisfy the rule set out in the list in column3for the made-up product concerned,may be used, provided that they are classified in a heading other than that of the product and that their value does not exceed 8%of the ex-works price of the product.6.2.Without prejudice to Note6.3,materials,which are not classified within Chapters50to63,may be used freelyin the manufacture of textile products,whether or not they contain textiles.Example:If a rule in the list provides that,for a particular textile item(such as trousers),yarn must be used,this does not prevent the use of metal items,such as buttons,because buttons are not classified within Chapters50to63.For the same reason,it does not prevent the use of slide-fasteners,even though slide-fasteners normally contain textiles.6.3.Where a percentage-rule applies,the value of materials which are not classified within Chapters50to63must betaken into account when calculating the value of the non-originating materials incorporated.Note7:7.1.For the purposes of headings ex2707,2713to2715,ex2901,ex2902and ex3403,the“specific processes”arethe following:(a)vacuum-distillation;(b)redistillation by a very thorough fractionation-process(1);(c)cracking;(d)reforming;(e)extraction by means of selective solvents;(f)the process comprising all of the following operations:processing with concentrated sulphuric acid,oleumor sulphuric anhydride;neutralisation with alkaline agents;decolourisation and purification with naturally-active earth,activated earth,activated charcoal or bauxite;(g)polymerisation;(h)alkylation;(i)isomerisation.7.2.For the purposes of headings2710,2711and2712,the“specific processes”are the following:(a)vacuum-distillation;(b)redistillation by a very thorough fractionation-process(1);(c)cracking;(d)reforming;(e)extraction by means of selective solvents;(f)the process comprising all of the following operations:processing with concentrated sulphuric acid,oleumor sulphuric anhydride;neutralisation with alkaline agents;decolourisation and purification with naturally-active earth,activated earth,activated charcoal or bauxite;(g)polymerisation;(h)alkylation;(ij)isomerisation;(k)in respect of heavy oils of heading ex2710only,desulphurisation with hydrogen,resulting in a reduction of at least85%of the sulphur-content of the products processed(ASTM D1266-59T method);(l)in respect of products of heading2710only,deparaffining by a process other than filtering;(m)in respect of heavy oils of heading ex2710only,treatment with hydrogen,at a pressure of more than20bar and a temperature of more than250°C,with the use of a catalyst,other than to effect desulphurisation, when the hydrogen constitutes an active element in a chemical reaction.The further treatment,with hydrogen,of lubricating oils of heading ex2710(e.g.hydrofinishing or decolourisation),in order,more especially,to improve colour or stability shall not,however,be deemed to be a specific process;(n)in respect of fuel oils of heading ex2710only,atmospheric distillation,on condition that less than30%of these products distils,by volume,including losses,at300°C,by the ASTM D86method;(o)in respect of heavy oils other than gas oils and fuel oils of heading ex2710only,treatment by means of a high-frequency electrical brush-discharge.(p)in respect of crude products(other than petroleum jelly,ozokerite,lignite wax or peat wax,paraffin wax containing by weight less than0,75%of oil)of heading ex2712only,de-oiling by fractional crystallisation.7.3.For the purposes of headings ex2707,2713to2715,ex2901,ex2902and ex3403,simple operations,such ascleaning,decanting,desalting,water-separation,filtering,colouring,marking,obtaining a sulphur-content as a result of mixing products with different sulphur-contents,or any combination of these operations or like operations,do not confer origin.'ANNEX II‘A NNEX15LIST OF WORKING OR PROCESSING REQUIRED TO BE CARRIED OUT ON NON-ORIGINATING MATERIALS IN ORDER THAT THE PRODUCT MANUFACTURED CAN OBTAIN ORIGINATING STATUSHS heading Description of product Working or processing,carried out on non-originating materials,which confers originating status(1)(2)(3)or(4)Chapter1Live animals All the animals of Chapter1shall bewholly obtainedChapter2Meat and edible meat offal Manufacture in which all the materialsof Chapters1and2used are whollyobtainedChapter3Fish and crustaceans,molluscs and otheraquatic invertebrates Manufacture in which all the materials of Chapter3used are wholly obtainedex Chapter4Dairy produce;birds'eggs;naturalhoney;edible products of animal origin,not elsewhere specified or included;except for:Manufacture in which all the materials of Chapter4used are wholly obtained0403Buttermilk,curdled milk and cream,yoghurt,kephir and other fermented oracidified milk and cream,whether or notconcentrated or containing added sugaror other sweetening matter or flavouredor containing added fruit,nuts or cocoa Manufacture in which:–all the materials of Chapter4used are wholly obtained,–all the fruit juice(except that of pineapple,lime or grapefruit)of heading2009used is originating,and –the value of all the materials of Chapter17used does not exceed30% of the ex-works price of the productex Chapter5Products of animal origin,not elsewherespecified or included;except for:Manufacture in which all the materials of Chapter5used are wholly obtainedex0502Prepared pigs',hogs'or boars'bristlesand hair Cleaning,disinfecting,sorting and straightening of bristles and hairChapter6Live trees and other plants;bulbs,rootsand the like;cut flowers and ornamentalfoliage Manufacture in which:–all the materials of Chapter6used are wholly obtained,and–the value of all the materials used does not exceed50%of the ex-works price of the productChapter7Edible vegetables and certain roots andtubers Manufacture in which all the materials of Chapter7used are wholly obtainedChapter8Edible fruit and nuts;peel of citrus fruitsor melonsManufacture in which:–all the fruit and nuts used are whollyobtained,and–the value of all the materials ofChapter17used does not exceed30%of the value of the ex-works price ofthe productex Chapter9Coffee,tea,matéand spices;except for:Manufacture in which all the materialsof Chapter9used are wholly obtained0901Coffee,whether or not roasted ordecaffeinated;coffee husks and skins;coffee substitutes containing coffee inany proportion Manufacture from materials of any heading0902Tea,whether or not flavoured Manufacture from materials of anyheadingex0910Mixtures of spices Manufacture from materials of anyheadingChapter10Cereals Manufacture in which all the materialsof Chapter10used are wholly obtainedex Chapter11Products of the milling industry;malt;starches;inulin;wheat gluten;except for:Manufacture in which all the cereals, edible vegetables,roots and tubers of heading0714or fruit used are wholly obtainedex1106Flour,meal and powder of the dried,shelled leguminous vegetables ofheading0713Drying and milling of leguminous vegetables of heading0708Chapter12Oil seeds and oleaginous fruits;miscellaneous grains,seeds and fruit;industrial or medicinal plants;straw andfodder Manufacture in which all the materials of Chapter12used are wholly obtained1301Lac;natural gums,resins,gum-resins andoleoresins(for example;balsams)Manufacture in which the value of all the materials of heading1301used does not exceed50%of the ex-works price of the product1302Vegetable saps and extracts;pecticsubstances,pectinates and pectates;agar-agar and other mucilages andthickeners,whether or not modified,derived from vegetable products:–Mucilages and thickeners,modified, derived from vegetable products Manufacture from non-modified mucilages and thickeners–Other Manufacture in which the value of allthe materials used does not exceed50%of the ex-works price of the productChapter14Vegetable plaiting materials;vegetableproducts not elsewhere specified orincluded Manufacture in which all the materials of Chapter14used are wholly obtainedex Chapter15Animal or vegetable fats and oils andtheir cleavage products;prepared ediblefats;animal or vegetable waxes;exceptfor:Manufacture from materials of any heading,except that of the product1501Pig fat(including lard)and poultry fat,other than that of heading0209or1503:–Fats from bones or waste Manufacture from materials of anyheading,except those of heading0203,0206or0207or bones of heading0506–Other Manufacture from meat or edible offal ofswine of heading0203or0206or ofmeat and edible offal of poultry ofheading02071502Fats of bovine animals,sheep or goats,other than those of heading1503–Fats from bones or waste Manufacture from materials of anyheading,except those of heading0201,0202,0204or0206or bones ofheading0506–Other Manufacture in which all the materialsof Chapter2used are wholly obtained1504Fats and oils and their fractions,of fishor marine mammals,whether or notrefined,but not chemically modified:–Solid fractions Manufacture from materials of anyheading,including other materials ofheading1504–Other Manufacture in which all the materialsof Chapters2and3used are whollyobtainedex1505Refined lanolin Manufacture from crude wool grease ofheading15051506Other animal fats and oils and theirfractions,whether or not refined,butnot chemically modified:–Solid fractions Manufacture from materials of anyheading,including other materials ofheading1506–Other Manufacture in which all the materialsof Chapter2used are wholly obtained 1507to1515Vegetable oils and their fractions:–Soya,ground nut,palm,copra,palm kernel,babassu,tung and oiticica oil, myrtle wax and Japan wax,fractions of jojoba oil and oils for technical or industrial uses other than the manufacture of foodstuffs for human consumption Manufacture from materials of any heading,except that of the product–Solid fractions,except for that of jojoba oil Manufacture from other materials of headings1507to1515–Other Manufacture in which all the vegetablematerials used are wholly obtained1516Animal or vegetable fats and oils andtheir fractions,partly or whollyhydrogenated,inter-esterified,re-esterifiedor elaidinised,whether or not refined,but not further prepared Manufacture in which:–all the materials of Chapter2used are wholly obtained,and–all the vegetable materials used are wholly obtained.However,materials of headings1507,1508,1511 and1513may be used1517Margarine;edible mixtures orpreparations of animal or vegetable fatsor oils or of fractions of different fats oroils of this Chapter,other than ediblefats or oils or their fractions ofheading1516Manufacture in which:–all the materials of Chapters2and4 used are wholly obtained,and–all the vegetable materials used are wholly obtained.However,materials of headings1507,1508,1511 and1513may be usedChapter16Preparations of meat,of fish or ofcrustaceans,molluscs or other aquaticinvertebrates Manufacture:–from animals of Chapter1,and/or–in which all the materials of Chapter3 used are wholly obtainedex Chapter17Sugars and sugar confectionery;exceptfor:Manufacture from materials of any heading,except that of the productex1701Cane or beet sugar and chemically puresucrose,in solid form,containing addedflavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price of the product1702Other sugars,including chemically purelactose,maltose,glucose and fructose,insolid form;sugar syrups not containingadded flavouring or colouring matter;artificial honey,whether or not mixedwith natural honey;caramel:–Chemically-pure maltose and fructose Manufacture from materials of anyheading,including other materials ofheading1702–Other sugars in solid form,containing added flavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price of the product–Other Manufacture in which all the materialsused are originatingex1703Molasses resulting from the extraction orrefining of sugar,containing addedflavouring or colouring matter Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex works price1704Sugar confectionery(including whitechocolate),not containing cocoaManufacture:–from materials of any heading,exceptthat of the product,and–in which the value of all the materialsof Chapter17used does not exceed30%of the ex-works price of theproductChapter18Cocoa and cocoa preparations Manufacture:–from materials of any heading,exceptthat of the product,and–in which the value of all the materialsof Chapter17used does not exceed30%of the ex-works price of theproduct1901Malt extract;food preparations of flour,groats,meal,starch or malt extract,notcontaining cocoa or containing less than40%by weight of cocoa calculated on atotally defatted basis,not elsewherespecified or included;food preparationsof goods of headings0401to0404,notcontaining cocoa or containing less than5%by weight of cocoa calculated on atotally defatted basis,not elsewherespecified or included:–Malt extract Manufacture from cereals of Chapter10–Other Manufacture:–from materials of any heading,exceptthat of the product,and–in which the value of the materials ofeach of Chapters4and17used doesnot exceed30%of the ex-works priceof the product1902Pasta,whether or not cooked or stuffed(with meat or other substances)orotherwise prepared,such as spaghetti,macaroni,noodles,lasagne,gnocchi,ravioli,cannelloni;couscous,whether ornot prepared:–Containing20%or less by weight of meat,meat offal,fish,crustaceans or Manufacture in which all the cereals and derivatives(except durum wheat and its1902 (cont'd)–Containing more than20%by weightof meat,meat offal,fish,crustaceans ormolluscsManufacture in which:–all the cereals and their derivatives(except durum wheat and itsderivatives)used are wholly obtained,andall the materials of Chapters2and3used are wholly obtained–all the materials of Chapters2and3used are wholly obtained1903Tapioca and substitutes therefor preparedfrom starch,in the form of flakes,grains,pearls,siftings or similar forms Manufacture from materials of any heading,except potato starch of heading11081904Prepared foods obtained by the swellingor roasting of cereals or cereal products(for example,corn flakes);cereals(otherthan maize(corn))in grain form or inthe form of flakes or other workedgrains(except flour,groats and meal),pre-cooked or otherwise prepared,notelsewhere specified or included Manufacture:–from materials of any heading,except those of heading1806,–in which all the cereals and flour (except durum wheat and Zea indurata maize,and their derivatives)used are wholly obtained,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the product1905Bread,pastry,cakes,biscuits and otherbakers'wares,whether or not containingcocoa;communion wafers,emptycachets of a kind suitable forpharmaceutical use,sealing wafers,ricepaper and similar products Manufacture from materials of any heading,except those of Chapter11ex Chapter20Preparations of vegetables,fruit,nuts orother parts of plants;except for:Manufacture in which all the fruit,nuts or vegetables used are wholly obtainedex2001Yams,sweet potatoes and similar edibleparts of plants containing5%or moreby weight of starch,prepared orpreserved by vinegar or acetic acid Manufacture from materials of any heading,except that of the productex2004and ex2005Potatoes in the form of flour,meal orflakes,prepared or preserved otherwisethan by vinegar or acetic acidManufacture from materials of anyheading,except that of the product2006Vegetables,fruit,nuts,fruit-peel andother parts of plants,preserved by sugar(drained,glacéor crystallised)Manufacture in which the value of all the materials of Chapter17used does not exceed30%of the ex-works price2007Jams,fruit jellies,marmalades,fruit ornut purée and fruit or nut pastes,obtained by cooking,whether or notcontaining added sugar or othersweetening matter Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the productex2008–Nuts,not containing added sugar orspirits Manufacture in which the value of all the originating nuts and oil seeds of headings0801,0802and1202to1207 used exceeds60%of the ex-works price of the product–Peanut butter;mixtures based on cereals;palm hearts;maize(corn)Manufacture from materials of any heading,except that of the product–Other except for fruit and nuts cooked otherwise than by steaming or boiling in water,not containing added sugar, frozen Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the product2009Fruit juices(including grape must)andvegetable juices,unfermented and notcontaining added spirit,whether or notcontaining added sugar or othersweetening matter Manufacture:–from materials of any heading,except that of the product,and–in which the value of all the materials of Chapter17used does not exceed 30%of the ex-works price of the productex Chapter21Miscellaneous edible preparations,exceptfor:Manufacture from materials of any heading,except that of the product2101Extracts,essences and concentrates,ofcoffee,tea or matéand preparationswith a basis of these products or with abasis of coffee,tea or maté;roastedchicory and other roasted coffeesubstitutes,and extracts,essences and Manufacture:–from materials of any heading,except that of the product,and–in which all the chicory used is。