关联分析软件TASSEL使用教程
- 格式:pdf
- 大小:3.25 MB
- 文档页数:85


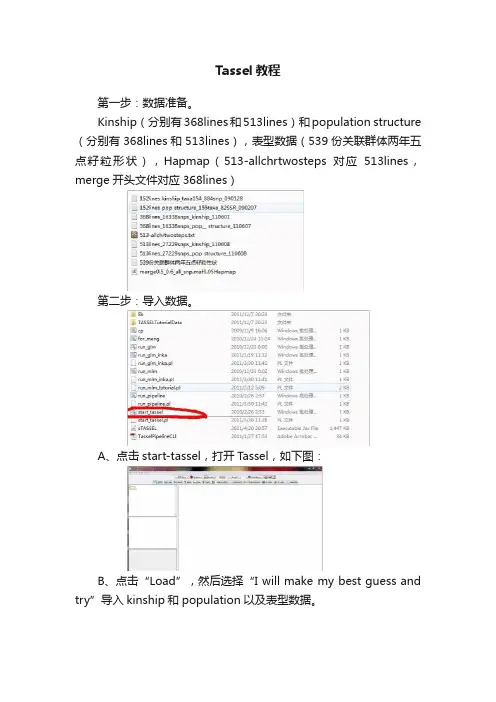
Tassel教程第一步:数据准备。
Kinship(分别有368lines和513lines)和population structure (分别有368lines和513lines),表型数据(539份关联群体两年五点籽粒形状),Hapmap(513-allchrtwosteps对应513lines,merge开头文件对应368lines)第二步:导入数据。
A、点击start-tassel,打开Tassel,如下图:B、点击“Load”,然后选择“I will make my best guess and try”导入kinship和population以及表型数据。
导入kinship和population structure以及表型数据的时候,可以按住“Ctrl”键和文件,同时选上这几个文件,一起导入。
C、导入成功的结果如下:D、再点击“Load”,选择“Load Hapmap”,导入基因型数据,如下图:注意:如果导入的是368lines的kinship和population structure,导入的基因型是merge开头的文件;如果导入的是513lines的kinship和population structure,导入的基因型是513-allchrtwosteps。
因为不同群体大小,对应的kinship和population structure以及基因型是不同的。
我们有368的群体大小和513的群体大小,368的kinship和population structure 以及基因型得对应起来。
E、导入基因型成功后的图如下:到这一步所有数据就导入完成了。
第三步:进行过滤。
A、过滤population structure,先点击population structure 文件,后点击“Traits”,如下图:然后再弹出的对话框中去掉“TST”前面的勾,点击“OK”,就过滤成功,会生成一个以“Filtered”开头的新文件,如下图:B、过滤基因型。

命令行tassel用法命令行TASSEL(Trait Analysis by aSSociation, Evolution and Linkage)是一个用于分析遗传多样性和关联的开源软件。
它提供了一套功能强大的命令行工具,可以帮助研究人员快速高效地进行遗传相关和群体遗传学分析。
以下是对命令行TASSEL的详细介绍和用法说明。
命令行TASSEL的安装和配置:在运行TASSEL之前,你需要设置一些系统环境变量,以便在任意路径下都可以方便地运行TASSEL。
命令行TASSEL的基本用法:TASSEL提供了多个命令行工具,可以处理各种不同类型的数据和分析。
下面是TASSEL的几个常用命令和用法的介绍:1. tassel-5.2.61. 这是TASSEL的主要命令行工具,用于执行各种不同的数据分析任务。
-用法:tassel-5.2.61 -h-解释:该命令将打印出所有可用的选项和子命令的详细帮助信息。
这是一个很好的起点,可以帮助你了解每个子命令的功能和使用方法。
2. hapmap2structure。
这个命令行工具用于将TASSEL生成的HapMap(.hmp.txt)文件转换成Structure(.str)文件格式,以便进行遗传群体结构分析。
-用法:tassel-5.2.61 -h hapmap2structure-解释:该命令将打印出hapmap2structure工具的帮助信息和使用方法。
你可以查看这些信息来了解如何使用该工具将HapMap文件转换成Structure文件。
3. kinship。
这个命令行工具用于计算群体内个体之间的亲缘关系水平,以帮助分析遗传相关性。
-用法:tassel-5.2.61 -h kinship-解释:该命令将打印出kinship工具的帮助信息和使用方法。
你可以通过查看这些信息来了解如何计算个体之间的亲缘关系。
4. svtool。
这个命令行工具用于通过诸如SNP Variation等方式进行群体遗传变异分析。

关联分析的一般方法关联分析的般方法杨小红中国农业大学国家玉米改良中心2011.5.25一、候选基因关联分析(TASSEL V2.1)二、全基因组关联分析(TASSEL V3.0)二全基因组关联分析(V30数据输入123SNP抽提3124SNP抽提结果位点序号与实际序号差1InDel 抽提3124InDel抽提结果SNP InDel与的整合13 2SNP与InDel的导出1324LD分析1324LD plot132LD decay的绘制多态性位点、群体结构、表型的整合多态性位点群体结构表型的整合312整合数据的核对31241212Manhanttan图单个位点所解释的表型变异R2──ANOVA (Excel)R2=SS intergroup/SS overall单因素方数据数据分析差分析单倍型分析134 25数据的输入12数据的导出12基因型数据的抽提3124基因型数据抽提结果群体结构的设置2413分析表型的设置1243基因型表型群体结构的整合基因型、表型、群体结构的整合12ctrl整合数据的核对运行——GLM1234GLM1结果GLM结果2运行——MLM_P3D&Compression 312MLM——Compression1p结果MLM——Compression2p结果MLM——Compression3p结果最优Compression 的选择2760278027202740L k266026802700‐2L n 262026401.0 1.52.33.4 5.2 7.8 11.9 22.7 250.0Compression运行——MLM_P3D&No Compression123MLM_P3D&No Compression结果QQ plot_TASSEL13 2Manhattan plot TASSELp_132数据输入Obp1Obp2观察值p预测值定义数据标记定义坐标轴格式绘图程序QQ plot_SAS结果。

tassel格式转化-回复tassel格式转化是一种数据处理工具,它的主要功能是将不同格式的数据转化成tassel可识别的格式,以便进行进一步的分析和处理。
本文将分步骤详细介绍如何使用tassel格式转化工具,并以“基因组学研究中的tassel 格式转化”为主题展开讨论。
第一步:理解tassel格式转化工具的作用和意义在基因组学研究中,研究人员通常面临的一个重要任务是处理和分析大量的基因组数据。
这些数据来自于各种不同的来源和实验,以不同的格式存在。
为了能够对这些数据进行统一的分析和比较,研究人员需要将它们转化为统一的格式。
tassel格式转化工具就是一种帮助研究人员完成这一任务的工具。
第二步:下载和安装tassel格式转化工具tassel格式转化工具是一个开源的软件包,可以从其官方网站免费下载和安装。
在安装之前,需要确认计算机上已经安装了Java运行环境。
下载完成后,按照官方提供的指南进行安装。
第三步:准备数据文件在开始使用tassel格式转化工具之前,需要准备好待转化的数据文件。
这些文件可以是来自DNA测序仪的原始数据或已经处理过的数据,可以是各种不同格式的文件,比如文本文件、Excel文件或者FASTA文件。
第四步:了解tassel格式转化工具的命令和参数tassel格式转化工具提供了一系列命令和参数,用于指定转化的输入文件、输出文件、转化方式等。
在开始转化之前,需要对这些命令和参数进行初步了解,以便选择合适的转化方式。
第五步:使用tassel格式转化工具进行转化在已经准备好数据文件和熟悉了工具的命令和参数之后,就可以使用tassel格式转化工具进行转化了。
根据具体的数据格式和转化需求,选择合适的命令和参数,执行转化操作。
第六步:检查和验证转化结果转化完成后,需要对转化结果进行检查和验证。
可以对部分数据进行抽样检查,比较转化前后的差异,确保数据转化的正确性和完整性。
第七步:进一步分析和处理转化后的数据一旦确认转化结果无误,就可以进一步使用tassel格式转化后的数据进行后续的分析和处理。

如何用spss做相关性分析例:学生每天学习时间T与学习综合成绩G之间的相关性原始数据T G1.1 541.5 602.2 623 70.13.4 744 74.54.2 775.5 81.55.9 856 85.56.5 86.28 90G=f(T),其中T为自变量,G为因变量step1:建立数据文件 file——new——data;定义变量选中左下角菜单Variable view,输入变量名T,其他选项不变,令起一行,输入变量名G其他选项不变,切换到data view(在左下角),将数据复制进去。
Step2:进行数据分析:在spss最上面菜单里面选中Analyze——correlate——bivariate(双变量)左边包含G,T的框为源变量框,后面的空白框为分析变量框,我们现在需要分析G和T的关系,因此将源变量框中的G和T选进分析变量框待分析。
(1)correlation coefficients(相关系数)包括三个选项:Pearson:皮尔逊相关,计算连续变量或是等间距测度的变量间的相关分析;Kendall:肯德尔相关,计算等级变量间的秩相关;Spearman:斯皮尔曼相关,计算斯皮尔曼秩相关。
注:Pearson可用来分析①分布不明,非等间距测度的连续变量Kendall可用来分析①分布不明,非等间距测度的连续变量,②完全等级的离散变量,③数据资料不服从双变量正态分布或总体分布型未知。
第②种情况只能用Kendall分析Spearman可用来分析数据资料不服从双变量正态分布或总体分布型未知(2)Test of significance选项Two-tailed:双尾检验,如果事先不知道相关方向(正相关还是负相关)则可以选择此项;One-tailed:单尾检验,如果事先知道相关方向可以选择此项。
(3)Flag significant correlations:表明显著水平,如果选择此项,输出结果中在相关系数值右上方使用*标示显著性水平为5%,用**标示其显著性水平为1%首先使用pearson,two-tailed(下图),点击右侧optionsstatistics为统计量,包括均值和标准差叉积离方差和协方差missing values 选择默认点击continue——ok输出结果(下图)相关系数为0.975,显著性p=0.000<0.01,有统计学意义选用Kendall 肯德尔,结果如下:选用spearman 斯皮尔曼,结果如下:画散点图:选中Graphs——Scatter/dot-----Simple scatter------define。

tassel格式转化-回复关于tassel格式转化的步骤与方法。
首先,我们需要了解什么是tassel格式以及它的用途。
Tassel是一个用于分析大规模遗传数据的软件套件,可用于研究多种植物和动物的基因组。
该软件提供了许多功能,包括基因型过滤、种群结构分析、关联分析等。
然而,Tassel软件在输入和输出数据的格式上有一些特定要求,因此我们需要将数据转换为符合Tassel的格式。
第一步是准备你的数据。
你可以使用任何喜欢的方法,例如基因芯片、基因测序等来获取你想要分析的数据。
确保你已经得到了一个数据集,包含了你感兴趣的个体的所有遗传信息,例如SNP、INDEL等。
第二步是将你的数据转化为Tassel可识别的格式。
Tassel软件对输入数据有特定的要求,通常需要将原始数据转化为VCF(Variant Call Format)格式。
VCF是一种常见的基因组数据存储格式,包含了个体的基因型信息以及可能的变异(例如SNP)。
有一些工具可以帮助你将数据转化为VCF 格式,例如BCFtools、GATK等。
根据你所使用的方法不同,可能需要使用特定的工具进行格式转化。
第三步是将VCF格式的数据转化为Tassel可以识别的格式。
Tassel通常需要使用一种称为HapMap格式的数据进行分析。
HapMap是一种用于存储基因组数据的格式,包含了个体的基因型信息、位点信息以及其他相关的注释。
你可以使用一些工具,例如TASSEL-Convert,将VCF文件转化为HapMap格式。
第四步是检查你的数据是否符合Tassel的要求。
Tassel对数据的格式有一些特定的要求,例如数据是否包含缺失值、是否有冗余数据等。
你可以使用Tassel软件自带的一些功能或者其他工具来检查你的数据是否符合要求。
第五步是导入数据到Tassel软件中进行分析。
一旦你的数据转换符合Tassel的要求,你就可以将数据导入到Tassel软件中进行进一步的分析了。

Tassel主要是研究基因型与表现型之间的关系。
有多种功能,包括:关联分析、评估进化关系、主成分分析、聚类分析、推断缺失数据、数据的可视化。
安装:1、下载Java Runtime Environment并安装;2、下载tassel_standalone.zip并解压;3、双击解压后文件里的jar文件(sTASSEL.jar),或者打开cmd命令窗口输入命令java-Xms256M –Xmx768M –jar sTASSEL.jar(要指定sTASSEL.jar文件路径),即可使用。
使用:1.data菜单栏里面的常用功能介绍:1.1、Load(下面介绍常见的几种格式)1.1.1、Hapmap 是一种储存序列数据的格式1.1.2、Plink数据储存在两个文件中(以.map和.ped结尾的文件中).ped:包含所有的SNP 值和六列(Family ID, Individual ID, PaternalID, Maternal ID, Sex and Phenotype ).map:每行都表示一个SNP 的信息(四列:Chromosome, rs#,Genetic distance and Position)1.1.3、Flapjack 数据储存在两个文件中(以.map和.geno结尾的文件中).geno:第一行的第一列是空的,后面的列是SNP ID,从第二行起的第一列都为种系名字.map:每行都表示一个SNP 的信息(三列:SNP ID,Chromosome,Position)1.1.4、Polymorphism1.1.5Numerical data协方差格式:1.1.6、Square Numerical Matrix1.1.7、Genetic Map1.2、Sites1.3、Site Names 从数据库中选择基因型数据1.4、Taxa 从数据库中选择基因型、表型、群体结构数据1.5、Traits用于numerical data sets中:可以改变性状类型、遗弃不需要的性状1.6、TransformWhen a genotype data set is selected, the data are transformed to numbers. When a numerical data set is selected, mathematical transformation, data imputation and principal component analysis (PCA) can be performed. The Transform columns tags will be displayed in a Data dialog box with three tabs: Trans, Impute and PCA.基因型转化为表型后出现的窗口:点击转化数字性数据后出现的窗口:1.7、Synonymize Taxa Names1.8、Union Join 取数据的并集1.9、Intersection Join 取数据的交集2、Analysis Mode2.1、Diversity2.2、Linkage Disequilibrium 针对SNP数据的首先要做一下过滤数据(apply Data Sites first);然后将进行LD分析(估计出D', r2 and P-values)2.3、Cladogram2.4、Kinship 针对SNP数据计算的2.5、GLM进行这项分析时,要有经过Trait Filter处理后的三个文件(gene sequence + population structure + phenotype),将这三个文件进行Intersection (∩) Join。
By 杂zá交jiāo粳jīng稻dào2012-1-12 根据目的不同,图示基因型有两种常见的制作方法。
一、使用软件GGT。
使用方法可以查看Menu,当然这里还是要简要介绍一下的。
这种方法通常用于查看作图群体,比如RIL,F2的图示基因型。
其中的染色体/连锁群的长度是根据你所使用的标记构建的连锁群确定的,与染色体实际的长度并不同。
下面以水稻为例介绍。
1,打开GGT软件,首先弹出下面这个窗口。
这时你可以打开已经做好的.ggt格式的文件,查看图示基因型。
也可以制作.ggt格式的文件。
一下内容主要介绍.ggt格式文件的制作。
2,打开“file”,选择其中的“Build GGT file…”。
弹出下面的窗口。
3,点击“Load marker maps”,输入map文件。
Map文件格式如下:Map文件实际就是染色体的信息,标记及其对应的遗传距离的信息。
在txt文档中保存,然后把后缀名改成map就行啦。
4,这时原来灰色显示的“Load Locus data”变的可选,点击它,输入.loc文件。
Loc文件格式如下:Loc文件其实就是你跑胶的结果。
5,点击“Merge data”,.ggt文件就做好了。
然后保存就可以啦。
输出的.ggt文件如下:6,用GGT软件打开就可以浏览这个群体的图示基因型了。
二,手工制作图示基因型下面介绍一种适用于NIL或其他类似用途的图示基因型做法。
这种方法做出的图示中染色体的长度可以由你自己定,通常我们根据染色体的实际大小来做。
下面仍然以水稻NIL为例。
假设这个NIL的8号染色体的50cM出有个目标基因。
我们需要做的就是把这个位点在12条染色体中表示出来。
1,确定染色体的长度。
水稻各条染色体的长度如下:(大致的长度)Chr 1 2 3 4 5 6 7 8 9 10 11 12cM 181.8 157.9 166.4 129.6 122.3 124.4 126.3 118.6 121.2 93.5 83.8 109.5 2,打开EXCEL,规定1个小格为10cM。

命令行tassel用法1. 简介Tassel是一个用于处理和分析遗传数据的开源软件。
它提供了一系列命令行工具,可以用来进行基因组关联分析、群体结构分析、遗传多样性分析等。
Tassel的命令行界面提供了丰富的功能和灵活性,使用户能够根据自己的需求选择合适的命令进行数据处理和分析。
本文将详细介绍Tassel命令行的使用方法,包括安装、配置环境、基本命令和常见应用案例。
2. 安装与配置2.1 安装Java运行环境Tassel是基于Java开发的,所以在使用之前需要安装Java运行环境。
可以从Oracle官网下载最新版本的Java Development Kit(JDK)并按照安装向导进行安装。
2.2 下载TasselTassel可以从官方网站上下载最新版本的压缩包。
解压后,可以看到如下文件和文件夹:- tassel.jar // Tassel程序主文件- lib/ // Tassel所需的依赖库文件夹- doc/ // Tassel文档文件夹- examples/ // Tassel示例数据文件夹2.3 配置环境变量为了方便使用Tassel命令行,可以将Tassel所在目录添加到系统的环境变量中。
以Linux系统为例,在终端中执行以下命令:export TASSEL_HOME=/path/to/tasselexport PATH=$PATH:$TASSEL_HOME其中,/path/to/tassel是Tassel所在的路径。
2.4 测试安装在终端中执行以下命令,如果能够正常输出版本信息,则表示安装成功:tassel -version3. 基本命令3.1 数据格式转换3.1.1 PED格式转换为VCF格式PED和VCF是两种常见的遗传数据格式。
使用Tassel可以方便地将PED格式转换为VCF格式。
tassel -convertFormat -inputFile input.ped -outputFile output.vcf -outputForma t VCF其中,input.ped是输入文件的路径,output.vcf是输出文件的路径。
如何使⽤Tassel做GWAS说明⽂档之前写的Tassel说明⽂档,虽然我都是使⽤命令⾏相关的软件,但是我发现,Linux,命令⾏对⼤多数⼈还是可望⽽不可即,分享⼀篇我做的说明⽂档,⽤⽰例数据,⼀步⼀步进⾏GWAS分析。
具体如下:⽬录1. 下载安装软件2. 导⼊数据3. 处理数据3.1 清洗数据3.2 主成分分析3.3 ⽤基因标记估计系谱3.4 ⽤⼀般线性模型分析GLM3.5 ⽤混合线性模型分析4. 欢迎关注我的微信公众号1. 下载安装软件这⾥下载的是win的64为系统,截图如下:安装成功后,打开菜单如下:2. 导⼊数据截图如下:打开data,load,选择Make Best Guess选择⼏个⽰例数据:打开后的数据如下⾥⾯包括系谱数据、性状数据和基因型数据(snp)。
3. 处理数据3.1 清洗数据选中mdp_trait,然后选择:Data中的TransformPhenotype,可以对数据进⾏转化、标准化等操作,注意,要先对数据进⾏选择,然后再进⾏操作:也可以对缺失值的数据进⾏删除,点击imput,Numerical impute,就会⽣成没有缺失值的数据,这只是缺失值的不同替换⽅法。
3.2 主成分分析主成分分析(PCA)是⼀种统计⽅法,它可以将相互关联的变量转化为独⽴的主成分(PC),第⼀种成分包含最多的组分,其它依次降低。
另⼀个主成分的作⽤可以⽤标记的主成分来代表群体结构。
这种⽅法⽐最⼤似然法节省时间。
因为⼤部分的分⼦标记都是字符,需要先将其转化为数值,然后再进⾏主成分分析,⼀般将纯合的标记⽤0代替,另⼀个纯合⼦⽤2代替,杂合的⽤1代替。
PCA要求变量不能有缺失值,因此,在进⾏主成分分析时,需要对数据进⾏清洗,去除缺失值。
去掉频率⼩于0.05的标记,可以选择Data,选择Site,然后在最⼩频率的框中键⼊0.05,然后选择Remove minor SNP status,然后点击Filter,进⾏过滤,模型如下:选择PCA,然后选择5个主成分(默认项),点击确定,就会⽣成结果,模型如下:结果如下:3.3 ⽤基因标记估计系谱利⽤主成分分析可以判断群体的结构特征,但是如果利⽤系谱信息,这种结果会更加准确。
tassel使⽤说明Kinship: The average relationship between individuals or lines can be estimated by kinship (K)calculated either from pedigrees or a suitable number of random markers across the entire genomeTassel运⾏使⽤:java –jar sTASSEL.jarhapmap格式: ⽤genotypetohapmap.py脚本转换成hapmap格式。
hapmap格式⼀定要有标头,并且个体名称要与structure results、phenotype的个体名称⼀致。
还有要有前11列,没有就表⽰为NA。
Tassel的使⽤说明:1.load -> hapmap(有许多格式,我的脚本genotypetohapmap.py是hapmap的格式) ->⽂件在Sequence中,点击就可看。
2.Sites -> Filter Alignment ⾥⾯有⼀些参数(minimum frequency、起始位置to结束位置(即可局部也可全基因组)),之后点击“Remove minor SNP states”(过滤0.05) -> Filter3.load -> I will make my best guess and try. ->导⼊表型数据:表型数据格式(如⽂件early.txt)是个体⾏数性状数⽬⾏数(标题⾏数)#此⾏显⽰的都是数⽬标题(开头不⽤空格)#此⾏显⽰的是性状名称,可放好⼏个性状,不需要个体标题(name之类)个体名字性状值(如:天数等)4.load -> I will make my best guess and try. ->导⼊structure数据。
Tassel 使用详细步骤1.转换导入文件格式(性状)注意文件格式要将excel文件为.txt 文件,注意文件开头格式2.打开tassel文件导入SNP数据库Data-Load-load hapmap导入性状txt文件Data-Load-3. 将‘SNP数据库’进行filter-----删除单态的和低覆盖度的位点:加亮mdp_genotype(即SNP数据文件),在菜单栏上单击F il t e r->S i t e s。
把“Minimum Frequency”设置为0.05、“Maximum Frequency”设置为1.0,“Minimum Count”设置为150。
单击Filter(过滤器)来产生一个过滤了的基因型数据集。
4.5.合并数据:通过按住Ctrl键同时选择过滤后的‘SNP数据集’和‘表型性状数据集’。
然后单击菜单项Data -> Intersect Join来产生一个合并的数据集,使表型与基因型一一对应。
6. 关联分析:加亮合并的数据集然后单击菜单项Analysis -> GLM来进行关联分析。
两个报告将被添加到数据树。
GLM-statsmarker_p: 对标记进行F检验的P值;单击“marker_p”将按照P值对表格排序。
最小的P 值是3.5963×10-6。
一个合理的显著性阈值是1.9×10-5,它是在Bonferroni多重检验矫正之后的5%(0.05/2559)。
Bonferroni矫正中的分母是检验的SNPs的总数。
该关联是显著的。
GLM-genotypesMLM-stats需要首先对snp数据库利用Analysis ->下的kinship进行亲缘关系计算,计算后右侧matrix 处会出现相应centered的结果,通过按住Ctrl键同时选择过滤后的‘SNP数据集’和‘表型性状数据集’然后单击菜单项Data -> Intersect Join来产生一个合并的数据集,使表型与基因型一一对应。
tassel格式转化-回复tassel格式转化,是一种数据处理工具,可用于将不同格式的数据转换为tassel软件所需的格式。
tassel是一款用于分析遗传多样性和关联分析的软件,广泛应用于植物和动物基因研究领域。
在进行tassel格式转化之前,首先要准备好需要转换的原始数据。
这些原始数据可以是各种格式,如文本文件、Excel表格或其他数据库格式。
接下来,我们将介绍如何使用tassel软件进行格式转换的一般步骤。
第一步是下载和安装tassel软件。
tassel是一个自由开放源代码的软件,可以从其官方网站上下载到最新版本的软件。
安装过程相对简单,只需按照提示进行操作即可。
第二步是准备原始数据。
通常,原始数据包含不同的变量和属性,如基因型、表型和环境因素等。
这些数据可以保存在不同的文件中,或者以表格的形式保存在同一个文件中。
确保原始数据的格式正确,并且每个变量和属性都有明确的标记。
第三步是打开tassel软件。
在tassel的主界面中,可以看到各种功能和选项。
在这里,我们关注的是数据转换功能,即将原始数据格式转换为tassel 所需的格式。
第四步是导入原始数据。
在tassel的主界面中,选择“导入数据”选项,然后选择原始数据文件所在的位置。
然后,根据数据的格式和结构,选择适当的导入选项。
例如,如果数据是以逗号分隔的文本文件,可以选择“导入文本文件”选项;如果数据是以Excel表格的形式保存的,可以选择“导入Excel文件”选项。
第五步是选择转换格式。
在tassel的主界面中,选择“转换格式”选项,然后选择需要转换的变量和属性。
根据需要进行选择,并设置合适的参数。
这些参数可以控制数据的处理方式和转换结果。
第六步是开始转换。
在tassel的主界面中,点击“开始转换”按钮,然后等待软件完成格式转换过程。
转换的时间可能取决于数据的大小和复杂性。
一旦转换完成,tassel将生成一个新的文件,其中包含转换后的数据。
tassel格式转化-回复转化tassel格式的步骤Tassel是一个常用的遗传学数据分析软件,可以用来执行各种高效而复杂的分析。
它支持多种数据格式,并可以进行多种统计分析和可视化。
本文将一步一步介绍如何使用tassel软件转化数据格式,帮助研究人员更好地应用这一工具。
第一步:安装和启动tassel软件在执行数据格式转化之前,首先需要下载和安装tassel软件。
它可以在其官方网站上免费获得。
一旦下载并完成安装,就可以启动tassel软件,并开始转化数据格式的操作。
第二步:准备输入文件在转化数据格式之前,需要准备输入文件。
tassel可以接受多种格式的文件作为输入,包括VCF、HapMap、PLINK等。
根据你的数据类型选择一个合适的输入文件格式,并确保文件准备就绪。
第三步:选择转化格式在tassel软件中,格式转化是一个常用的操作。
在“Convert”菜单下,可以找到“ToTASSELFormat”选项。
点击这个选项将打开一个对话框,让你选择要转化的文件和输出格式。
第四步:选择输入文件和输出格式在对话框中,点击“Open File”按钮选择你准备好的输入文件。
然后,在“Output Format”下拉菜单中选择你想要的输出格式。
tassel支持多种输出格式,包括HapMap、Numeric、Plink Whole、Simple等。
根据你的需求选择适当的输出格式。
第五步:设置输出文件路径和名称在对话框中,你还需要设置输出文件的路径和名称。
点击“Output File”按钮选择一个输出文件路径,并在“Output File Name”文本框中输入一个文件名。
确保文件路径和名称是正确的,并且你有相应的写入权限。
第六步:开始转化一旦输入文件和输出格式设置好,点击对话框中的“OK”按钮开始转化。
tassel将执行格式转化操作,并在转化过程完成后显示一个状态信息。
第七步:检查和保存输出文件转化过程完成后,tassel将生成一个转化后的输出文件。