
中国科学B辑:化学 2009年 第39卷 第10期: 1089~1101 https://www.doczj.com/doc/608704955.html, https://www.doczj.com/doc/608704955.html, 《中国科学》杂志社SCIENCE IN CHINA PRESS
化学反应过渡态的结构和动力学
戴东旭, 杨学明*
分子反应动力学国家重点实验室, 中国科学院大连化学物理研究所, 大连116023
* 通讯作者, E-mail: xmyang@https://www.doczj.com/doc/608704955.html,
收稿日期:2009-07-17; 接受日期:2009-08-02
摘要化学反应过渡态决定了包括反应速率和微观反应动力学在内的化学反应的基本特性, 而无论是从理论还是实验上研究和观测化学反应过渡态都是极具挑战性的课题. 近年来, 我国科学家们利用交叉分子束-里德堡氢原子飞行时间谱仪, 结合高精度的量子动力学计算, 对H + H2和F + H2这两个教科书式的典型反应体系进行了全量子态分辨的反应动力学研究, 从中得出了关于这两个反应体系的过渡态的结构和动力学性质的结论性的研究成果. 关键词
反应过渡态
化学反应动力学微分反应截面动力学共振态交叉分子束
1引言
化学反应过渡态是化学反应体系在反应过渡区域的量子态. 化学反应过渡态决定了包括反应速率和微观反应动力学在内的化学反应的基本特性. 现代重要的化学反应理论, 如双分子反应过渡态理论以及单分子分解的RRKM理论等, 都是建构在反应过渡区域的量子化过渡态的理论基础上. 了解量子化过渡态的结构以及它们对反应动力学行为的影响机制, 对于更深入地理解化学反应的本质至关重要. 诺贝尔化学奖获得者John C. Polanyi和Ahemed H. Zeweil于1995年在Accounts of Chemical Research的Pauling纪念专辑中指出, “直接观测化学反应过渡态”是化学学科的“圣杯”之一[1].
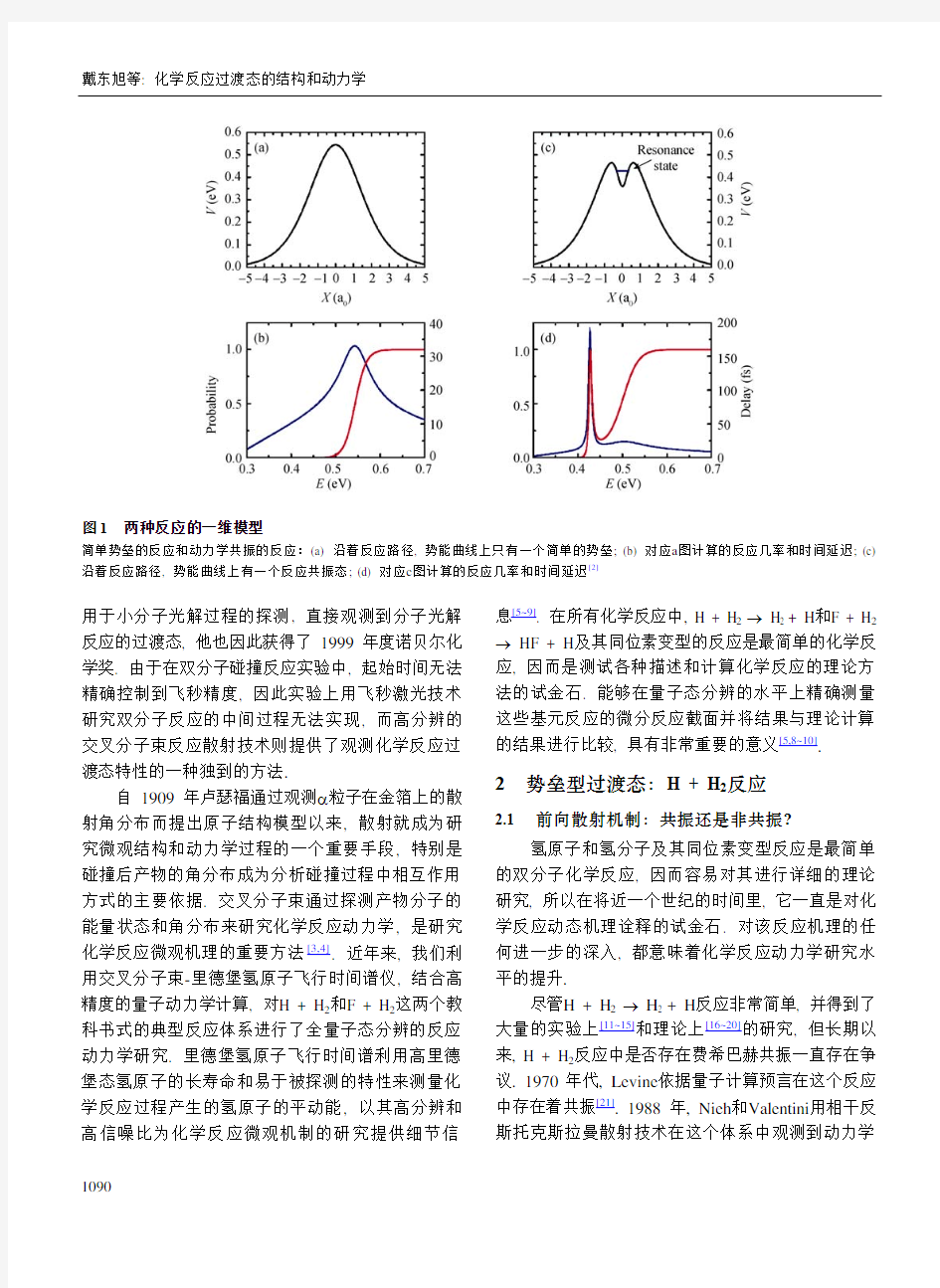
化学反应过渡态一般位于反应坐标中能量较高的区域. 对于势垒型的化学反应, 过渡态沿反应坐标方向有能量极大值, 因而不可能存在分立的量子化的过渡态结构(图1(a))[2]. 但是在垂直于反应坐标方向, 就会以最低能量路径为谷地, 形成分立的势垒型量子化过渡态, 或形象地称为量子化瓶颈态(quantized bottleneck state). 另一方面, 很多化学体系在反应过渡区域沿着反应坐标方向会存在能够容纳一定数量分立量子态的势阱, 因而可以存在非完全束缚的量子态结构, 这样的量子态结构被称为动力学共振态(dynamic resonance)或费希巴赫共振态(Feshbach resonance)(图1(c))[2].
化学反应过渡态的寿命很短, 无论从理论上还是实验上研究和观测化学反应过渡态都是极具挑战性的课题. 人们通常观测化学反应是在远长于过渡态寿命的条件下进行, 统计平均的效果往往掩盖了其中所包含的许多重要的关于过渡态的细节. 探测过渡态结构和性质的实验技术主要有两种途径, 一是超快探测技术, 使得时间分辨达到飞秒量级; 另外一种是分子束单次碰撞条件下的实验研究技术, 使分子只经历唯一的反应碰撞, 产物分子保持了单次碰撞后的状态, 因而可以通过动力学理论反推碰撞过程的细节. 1980年代中期, 李远哲等人发展了交叉分子束实验技术, 对分子碰撞反应的研究取得了一系列成就, 使人们能了解化学反应过程和细节, 他也因此获得了1986年度诺贝尔化学奖. 九十年代美国加州理工学院Zewail教授发展了飞秒激光技术, 并
1089
戴东旭等: 化学反应过渡态的结构和动力学
图1 两种反应的一维模型
简单势垒的反应和动力学共振的反应:(a) 沿着反应路径, 势能曲线上只有一个简单的势垒; (b) 对应a 图计算的反应几率和时间延迟; (c) 沿着反应路径, 势能曲线上有一个反应共振态; (d) 对应c 图计算的反应几率和时间延迟[2]
用于小分子光解过程的探测, 直接观测到分子光解反应的过渡态, 他也因此获得了1999年度诺贝尔化学奖. 由于在双分子碰撞反应实验中, 起始时间无法精确控制到飞秒精度, 因此实验上用飞秒激光技术研究双分子反应的中间过程无法实现, 而高分辨的交叉分子束反应散射技术则提供了观测化学反应过渡态特性的一种独到的方法.
自1909年卢瑟福通过观测α粒子在金箔上的散射角分布而提出原子结构模型以来, 散射就成为研究微观结构和动力学过程的一个重要手段, 特别是碰撞后产物的角分布成为分析碰撞过程中相互作用方式的主要依据. 交叉分子束通过探测产物分子的能量状态和角分布来研究化学反应动力学, 是研究化学反应微观机理的重要方法[3,4]. 近年来, 我们利用交叉分子束-里德堡氢原子飞行时间谱仪, 结合高精度的量子动力学计算, 对H + H 2和F + H 2这两个教科书式的典型反应体系进行了全量子态分辨的反应动力学研究. 里德堡氢原子飞行时间谱利用高里德堡态氢原子的长寿命和易于被探测的特性来测量化学反应过程产生的氢原子的平动能, 以其高分辨和高信噪比为化学反应微观机制的研究提供细节信
息[5~9]. 在所有化学反应中, H + H 2 → H 2 + H 和F + H 2 → HF + H 及其同位素变型的反应是最简单的化学反应, 因而是测试各种描述和计算化学反应的理论方法的试金石. 能够在量子态分辨的水平上精确测量这些基元反应的微分反应截面并将结果与理论计算的结果进行比较, 具有非常重要的意义[5,8~10].
2 势垒型过渡态:H + H 2反应
2.1 前向散射机制:共振还是非共振?
氢原子和氢分子及其同位素变型反应是最简单的双分子化学反应, 因而容易对其进行详细的理论研究, 所以在将近一个世纪的时间里, 它一直是对化学反应动态机理诠释的试金石. 对该反应机理的任何进一步的深入, 都意味着化学反应动力学研究水平的提升.
尽管H + H 2 → H 2 + H 反应非常简单, 并得到了大量的实验上[11~15]和理论上[16~20]的研究, 但长期以来, H + H 2反应中是否存在费希巴赫共振一直存在争议. 1970年代, Levine 依据量子计算预言在这个反应中存在着共振[21]. 1988年, Nieh 和Valentini 用相干反斯托克斯拉曼散射技术在这个体系中观测到动力学
1090
中国科学B辑: 化学 2009年第39卷第10期
共振[22]. 然而更精确的理论研究从来没有支持这个实验观测. 2000年Kendrik等通过一个理论和实验的合作研究给出了共振结构的证据, 但是后来发现这是实验中的系统误差所致[23,24].
由于H + H2→ H2 + H反应主要是在排斥的、倾向于共线的基态势能面上进行的, 所以产物分子H2会逆着H原子进来的方向“反弹”回去, 产物的分布主要是后向散射, 因此在这种类型的反应中如果观测到产物的前向散射分布, 会被认为是“奇异”的现象. 一般来讲, 前向散射分布来源于化学反应体系在经历过渡态区域时发生了“时间延迟”(图1(b)和(d))[2], 在这个“时间延迟”中反应体系恰好转过了一个合适的角度从而产生向前散射的产物. 这样的时间延迟常常与反应过程中的共振相联系. 2000年Fernánderz-Alonso和同事在H + D2反应的产物分布中观测到了前向散射峰, 并将其归属为散射共振[25]. 2002年Althorpe等用全量子动力学计算分析认为, 在H + D2反应微分截面中的前向散射是与反应碰撞过程中在过渡态区域的一个时间延迟相关联的[26].
几乎与此同时, Harich等人用交叉分子束-里德堡氢原子飞行时间谱方法对H + HD → H2(v, j)+ D反应在 1.2 eV下的微分反应截面进行了全量子态分辨的测量并且清楚地观测到了H2的前向散射峰[27]. 图2是该反应产物的量子态分辨的微分反应截面三维示意图, (a)和(b)分别是实验和理论结果. 图中不同层的同心圆代表产物不同的振转态, 层的高度代表产物分布的强度. B代表后向散射方向, F代表前向散射方向. 由于测量的是D原子, 所以前向散射方向与HD分子束入射方向一致.
基于全量子散射计算和理论分析表明, 这个前向散射峰也是来自于散射过程中的时间延迟机制. 这个时间延迟机制是当反应体系的能量恰好和某个特定的量子化瓶颈态的势垒顶部一致时, 由于大部分能量转化为势能所致, 体系沿着反应坐标方向的运动减速, 因而在垂直于反应坐标的方向上发生振动并向前散射. 很显然, 这个结论说明了这样的一个事实:前向散射不是一定和动力学共振相联系的, 观测到前向散射不等同于观测到费希巴赫共振. 迄今为止, 尚没有任何可靠的实验和理论证据说明在H + H2这个简单的反应中存在费希巴赫共振, H + H2反应
图2 碰撞能1.2 eV下H + HD (v=0, j=0)反应量子态分辨微分截面
(a) 和 (b) 分别是实验和理论结果[27]
过渡态是以量子化瓶颈态的形式存在的[8].
2.2量子瓶颈态结构观测: 化学反应中的量子
干涉
在确认H + H2的反应过渡态属于量子化瓶颈态后, 观测和分析量子化瓶颈态的结构及其对反应动力学的影响机制就成为人们关注的课题. 对量子化瓶颈态的实验观测一直是化学动力学研究的一个难点. 在90年代早期, Moore和合作者在实验中发现乙烯酮分子光解速率随光解能量的变化函数存在类似台阶的结构, 并将其归属为量子化瓶颈态结构[28,29]. 然而这一结论立刻引起了若干个理论研究者的质疑[30~32]. 在双分子反应中观测量子化瓶颈态就更为困难, 因为碰撞参数的统计效应会抹平反应过程中的量子效应.
若要彰显双分子碰撞反应中的量子效应, 消除碰撞参数的统计效应, 就需要精确测量特定散射方向上的微分反应截面. 2003年戴东旭等人用交叉分子束-氢原子飞行时间谱方法测量了实验室角度70°, 相
1091
戴东旭等: 化学反应过渡态的结构和动力学
当于质心坐标角度大约160°方向上, 碰撞能量从0.4eV到1.0eV之间的H + D2→ HD + D反应的转动态分辨的微分截面[33]. 图3显示的是H + D2(v = 0, j = 0) → HD(v′ = 0, j′ = 2) + D反应微分截面随能量变化的图形, 圆点是实验值, 曲线是全量子动力学计算的结果, 理论和实验符合得很好. 从图3可以看出, 该反应转动态分辨的微分截面随能量变化而振荡. 进一步的理论分析表明, 这些振荡不是量子化瓶颈态的共振峰, 而是反应体系在不同的量子化瓶颈态反应通道之间的干涉结果(图4). 该项研究为双分子反应中的量子化瓶颈态的存在提供了强有力的实验依据.
图3 实验室角度70o上H + D2(v=0, j=0) → D + HD (v′=0, j′=2)微分截面随能量变化[33]
图 4 不同的量子化瓶颈态反应通道之间的量子干涉, 中间势垒上方的短横线代表垂直于反应坐标方向的不同的量子化瓶颈态2.3 量子化瓶颈态的选控:态态态反应动力学
通过量子调控的办法控制化学反应的路径一直是科学家们追求的目标, 研究和探讨化学反应过渡态与反应物初始状态的关系对化学反应的调控显然具有十分重要的指导意义. 2006年张建阳等人报道了对D + H2→ HD + H反应中反应物的初始转动取向与中间过渡态之间的关联的研究[34]. 他们在实验中发现, 处于j = 0转动态的反应物H2分子所对应的产物在后向散射方向上的转动态分布是出乎意料的双模式分布, 与处于j = 1转动态的反应物H2分子对应的分布非常不同, 这种不同已经不能用两种转动态之间的能量差异来解释. 理论分析表明这种不同分布的来源是由于在反应过程中因角动量沿反应坐标方向分量守恒而导致的对量子化瓶颈态的选择性. 氢分子在和氘原子互相接近时, 可以有两种转动旋性:一种是车轮型(k = 0), 即行进方向和转轴垂直; 另一种是直升机型(k = 1), 即行进方向和转轴平行. 由于对称性选择, 这两种碰撞方式对应着不同的量子化瓶颈态(图5). 这个研究结果暗示量子化瓶颈态的反应路径可能通过立体动力学方式控制, 并将以往从反应物量子态到产物量子态的态态(state-to-state)化学反应动力学扩展到反应物量子态-过渡态-产物量子态的态态态(state-to-state-to-state)化学反应动力学.
3反应共振态:F + H2反应
3.1历史背景
相对于H + H2反应机制是势垒型过渡态, F + H2反应则是沿着反应坐标方向存在非完全束缚的动力学共振态的典型, 是研究费希巴赫共振的经典例子.
F + H2反应和H + H2反应一样是少数几个可以用全量子动力学方法研究的化学体系, 并且F + H2反应是化学激光主要体系之一, 因此关于F + H2及其同位素变型反应的研究得到了大量的实验上和理论上的关注.
20世纪70年代就有人利用量子动力学计算, 预测F + H2中存在共振现象[35~38], 但由于所使用的势能面是一维的并且是经验性的,所以不完全清楚
1092
中国科学 B 辑: 化学 2009年 第39卷 第10期
图5 D + H 2反应中, 不同的转动取向对应于不同的中间过渡态
所预言的共振到底是真实物理现象还是由于近似导致的理论假象. 例如, Connor 在几个势能面上进行了中间络合物几何构型为共线情况下的计算, 得到了完全不同的共振结构[39,40]. 80年代中期, 李远哲研究组对F + H 2反应首次进行了产物振动量子态分辨的交叉分子束实验[3,4,41], 这是一个化学反应动力学领域里的里程碑式的实验. 他们在实验中观测到, 在碰撞能为1.84 kal/mol 时, F + H 2反应的产物HF(v ′=3)的分布出现前向散射峰; 在碰撞能为3.32 kcal/mol 时, F + D 2反应的产物DF(v ′ = 4)也出现了前向散射峰. 他们认为观测到的前向散射峰可能来自于动力学共振. 因为以往的势能面都不能预言实验中的产物振动态和微分截面的分布, Stark 和Werner 在1996年从第一原理出发构建了高精度的SW 势能面[42], 但是在SW 势能面上所进行的经典散射计算和量子散射计算的对比得出的结论却是否定了HF(v ′=3)前向散射峰来自于反应共振[43,44].
2000年Liu 等人利用多普勒选择(Doppler Se-lected)离子飞行谱方法对 F + HD 反应进行了实验 研究
[45,46]
. 他们发现, 产物HF + D 通道的激发
函数σR (E )在碰撞能约为0.5 kcal/mol 处有一个台阶(图6)[45]; 而产物DF + H 通道的激发函数σR (E )却没有类似的台阶. 在SW 势能面上进行的量子动力学计算发现有类似的结果, 证实这个台阶是费希巴赫共振的结果. 然而理论计算的激发函数σR (E )的台阶出现在高于0.5 kcal/mol 处, 而且“台阶”的高度是实验结果的两倍. Liu 等认为这可能是由这个反应的自旋-轨道耦合的非绝热效应引起的. 他们认为F + HD →
H F (v ′) + D 反应的共振形成机制是:产 物HF(v ′=2)的产生是通过HF-D 的(003)准束缚态的 共振机理进行反应的. 但是在F + H 2反应中, Liu 等 人在实验上并没有发现类似于 F + HD 的反应共振 峰[47].
图6 F + HD → HF + D 积分截面随能量变化[45]
其他实验化学动力学家们也对这个体系进行了一系列新的研究, 但都没有探测到F + H 2反应共振的任何新迹象[48,49]. 同时, 理论上SW 势能面存在的一些问题也逐渐被发现, 如产物HF(v ′ = 3)的延迟出现、自旋-轨道耦合的影响等. 2005年Skodje 研究组在SW 势能面基础上构建了一个新的势能面(SW MHS- PES)[50]. 但是在低碰撞能区域, 该势能面上的反应图像与在SW 势能面上的有非常大的差异, 并与实验 不符.
1093
戴东旭等: 化学反应过渡态的结构和动力学
3.2 F + H2反应共振态的研究
为了澄清F + H2反应的共振图像, 2006年邱明辉等人利用里德堡氢原子飞行时间谱方法对F + H2反应进行了全量子态分辨的交叉分子束散射研究[51]. 图7是碰撞能为0.52 kcal/mol时F + H2(j=0)反应产物HF的态分辨的微分截面三维示意图. 可以看到在前向散射方向产物HF(v′=2)有一个显著的散射峰. 这是在实验上首次在F + H2反应中观测到前向散射峰(图7(a)), 之前的实验或者很难达到这样低的碰撞能, 或者缺乏足够的灵敏度测量出如此低碰撞能下的产物空间分布. 该实验同时观测到HF(v′=2)的前向散射分布随能量的变化在碰撞能为0.52 kcal/mol处显示一个峰值(图8中的圆点). 为了解释这一新的实验现象, 理论学家们从第一原理出发构建了一个新的精确的势能面(XXZ-PES)[52]. 基于该势能面的全量子动力学计算几乎完美地再现了实验的结果(图7(b)和图8中的
图7 碰撞能0.52 kcal/mole时F + H2反应产物HF态分辨微分截面
(a) 实验结果; (b) 理论结果
[51]图8 F + H2反应产物HF(v′=2)的前向散射分布随能量的变化
圆点为实验值, 曲线为量子动力学理论计算结果[51]
图9 F + H2反应势能面的沿反应坐标的一维图示[51]
曲线), 并清晰地阐述了有关该反应的动力学共振的机理. 在XXZ势能面上, HF(v′=3)-H的振动绝热势存在一个较深的、与反应势垒紧邻的势阱, 另外还存在一个浅的范德瓦尔斯(vdW)势阱. 两个费希巴赫共振, 分别为(003)和(103)共振态, 就被束缚在HF(v′=3)-H′的振动绝热势阱中(图9). 它类似于SW势能面的图像, 但不同于SWMHS势能面的图像. 从一维波函数的位置可知基共振态(003)主要是束缚在较深的HF(v′=3)-H势阱中, 而激发态(103)的波函数主要是范德瓦尔斯共振. 基共振态的主要特征类似于在F + HD反应中观测到的共振特征. 由于这两个共振态与
1094
中国科学B辑: 化学 2009年第39卷第10期
HF(v′=2)的通道之间存在强烈的耦合, 相当于多一个形成HF(v′=2)产物的通道. 但是产物HF(v′=2)是前向散射还是后向散射由中间络合物HHF驻留在两个共振态的时间决定. 从实验和理论结果上可以得到结论:处于这两个共振态的络合物大部分衰变为产物HF(v′=2), 而且是前向散射, 从而造成前向散射的信号比后向的信号强得多.而这两个共振态与HF(v′=0,1)的通道之间的耦合较弱, 很难通过这两个共振态打开产物HF(v′=0,1)的通道, 因此实验上观测到产物HF(v′=1)的前向散射很弱, 完全没有观测到产物HF(v′=0). 这正是长期以来人们一直在该反应中努力寻找的费希巴赫共振, 而且实验和理论完全一致.
3.3同位素取代效应:在光谱精度上研究反应
共振
Kopin Liu等人对F + HD反应的研究说明这一反应与F + H2反应有着非常明显的差别, 即同位素替代对反应共振态有着相当大的影响. 当用XXZ势能面对F + HD反应进行动力学计算时发现, 尽管相对于SW势能面来讲, 依据XXZ势能面计算的共振峰的高度与Liu的实验结果相符, 但在峰的位置上依然存在差别. 2008年, 任泽峰等利用里德堡氢原子飞行时间谱方法对F + HD反应进行了高精度的交叉分子束实验研究[53]. 他们发现当碰撞能仅仅变化0.28 kcal/mol(98 cm?1), 该反应的微分截面就发生了剧烈的变化(图10). 这项实验以前所未有的光谱精度(达到几个cm?1的精度)对这一重要反应的共振现象进行了动力学测量, 也对该反应体系的势能面构造和动力学计算提出了苛刻的要求.
量子动力学计算表明前述XXZ势能面尽管能精确地描述F+H2反应中的共振态动力学现象, 但对F +HD反应中的共振态描述不够理想. 图11 显示了实验测得的F + HD(j=0) → HF(v′, j′) + D反应产物HF(v′ = 2, j′ = 0~3)的后向信号之和随碰撞能的变化以及在XXZ势能面势能面上进行的量子散射动力学计算结果. 该曲线显示出一个明显的共振峰. 显然XXZ势能面的计算结果与实验结果有很大差别, 计算所得的共振峰位置比实验结果偏移了约0.2 kcal/mol. 为此张东辉等人用CCSD(T)方法重新构建
图10 F + HD(j=0) → HF(v′, j′) + D 反应在不同碰撞能下产物D原子的微分截面示意图
(a) 0.43; (b) 0.48; (c) 0.52; (d) 0.71 kcal/mol[53]
了该反应的一个高精度的势能面(FXZ-PES)[54], 从图11可以看出由此新势能面计算的结果与实验符合很好. FXZ在其他动力学性质方面的计算结果也与实验结果良好符合. 当把FXZ势能面应用于F+H2反应体系时, 理论计算得到的结果也比XXZ势能面更好地符合实验结果.
图12(a)显示的是FXZ势能面和XXZ势能面分别描述的F + H2体系的一维HF(v'=3)--H 绝热势阱和共振态. FXZ势能面的基态共振态(003)相对XXZ势
图11 F + HD(j = 0) → HF(v′, j′) + D反应产物HF(v′ =2, j′= 0-3)的后向信号之和随碰撞能的变化
实心圆为实验结果, 实线为XXZ势能面和FXZ势能面上进行的计算结果[53]
1095
戴东旭等: 化学反应过渡态的结构和动力学
能面向下移动了约0.12 kcal/mol, 而激发态共振态(103)位置仅仅向下移动了0.01 kcal/mol. 之前的研究说明, 0.52 kcal/mol 时产物HF(v′=2)的前向散射是(003)态和(103)态两个共振态干涉的结果, 而且主要由(103)态决定, 因此不难理解两个势能面上的计算均能与实验吻合. 图12(b)是这两个势能面分别描述的F + HD体系的一维HF(v′=3)--D绝热势阱和共振态. 在F + HD体系中, HF(v′=3)-D绝热势阱只能维持一个基共振态(003), 而且FXZ势能面的势阱比XXZ势能面的势阱深了约0.3 kal/mol, 相应地FXZ势能面的基共振态(003)也较XXZ势能面的低0.16 kal/mol, 所以只有FXZ势能面才能更好地描述F + HD反应体系的动力学. FXZ势能面与F + H2反应和F + HD反应的实验结果高度吻合说明我们对化学反应动力
图12 F + H2反应的HF(v'=3)--H(a)、F + HD 反应的HF(v'=3)--D(b)一维绝热势阱
红线为XXZ 势能面, 蓝线为FXZ势能面[53]学的研究无论从实验上还是理论上都达到了前所未有的光谱级的精度.
3.4 HF(v′=3)前向散射的机理
尽管邱明辉等人通过观测HF(v′=2)的前向散射峰及其随能量变化的共振峰而证实了F + H2反应中共振态的存在及动力学机理, 但李远哲等人在1980年代观测到的HF(v′=3)前向散射的形成机理还没有给出解答. 当时, 李远哲他们认为该反应中的前向散射是由F + H2反应中的共振态引起的, 可是后来的理论研究不能很好地支持这个说法. 2008王兴安等人利用交叉分子束-里德堡氢原子飞行时间谱技术研究了基态F原子与H2分子的反应, 获得了碰撞能在0.50~1.0 kcal/mol范围内的全量子态分辨的微分反应截面[55]. 实验发现, 产物HF(v′=3)在碰撞能恰好大于反应阈值0.52 kcal/mol时就开始产生, 然后HF(v′=3)的积分截面随碰撞能增加单调上升, 说明产物通道没有出口势垒. 图13(a)是产物HF(v′=3)在碰撞能为0.94 kcal/mol时的散射微分截面示意图, 显示出一个
图13 F(2P3/2) + H2(j = 0) → HF(v′ = 3, j′) + H反应在碰撞能为0.94 kcal/mol处产物微分截面实验 (a) 与理论 (b) 结果[55]
1096
中国科学B辑: 化学 2009年第39卷第10期
图14 FXZ势能面得到的HF(v′ = 3)-H一维振动绝热分波势能面[55]
很明显的前向散射峰, 这一实验结果与李远哲等人在1985年的实验观测相符合. 对于HF(v′=3)反应通道, 理论计算表面, 在碰撞能为0.94 kcal/mol时, 总角动量J = 10的分波对前向散射贡献最大, J = 9的分波的贡献次之, 其余分波对前向散射的贡献较小. 图13(b)是在FXZ势能面上量子动力学计算的结果, 理论和实验符合得很好. 图14是FXZ势能面得到的HF(v′=3) --H一维振动绝热分波势能面. 从图中可以看出, 随着碰撞参数的增大, HF(v′=3)反应通道出口处的离心势垒逐渐形成并增大. 反应体系在0.94 kcal/mol能量下恰好可以隧穿J = 10的离心势垒, 并且刚好高于J = 9的离心势垒. 这说明实验观测到的F + H2反应的产物HF(v′=3)前向散射来自于反应体系在过渡区域出口处放慢通过HF(v′=3)--H′振动绝热通道上因离心势场形成的势垒所导致的时间延迟. 这与H + HD反应中的前向散射机理相似, 但放慢通过的区域不同. 这一结果终于对这一困惑反应动力学领域20多年的科学问题给出了一个清晰的答案.
3.5转动激发对反应共振态的影响
现实世界中的化学过程, 反应物大多都具有一定的转动能, 所以研究反应物转动激发对反应动力学的影响具有重要的意义. 2008年任泽峰等人测量了F + H2(j = 1)反应在0.56和0.19 kcal/mol两个碰撞能下的转动态分辨的微分散射截面[56,57]. 与前述F + H2(j = 0)反应测量结果形成对比的是, F + H2(j = 1)反应产物HF(v′=2)在0.56 kcal/mol碰撞能下产物分布没有前向散射峰, 而在0.19 kcal/mol碰撞能下却观测到较大的前向散射峰(图15). 将F + H2(j = 1)的产物HF(v′ = 2)前向散射信号对碰撞能的依赖关系与F + H2(j = 0)的相对应关系相比较发现二者比较类似, 只是F + H2(j = 1)的峰值位置向低能方向平移了0.35 kcal/mol, 这个数值近似等于H2分子j = 1的能级. 全量子散射计算的结果也得出了与实验结果相符合的结论, 说明F + H2(j = 1)反应之所以在碰撞能为0.19 kcal/mol附件发生共振是因为该碰撞能与H2(j = 1)转动能0.34 kal/mol之和符合F + H2(j = 0)反应的共振能.
图15 实验测得的F + H2(j′ = 1)反应产物HF微分截面示意图
(a) 0.56 kcal/mol; (b) 0.19 kal/mol[56]
4总结和展望
从本文可以看出, 经过近几年的努力, 科学家们现在对H + H2和F + H2这两个化学反应动力学教科书式的样本体系的反应过程和过渡态结构及其动力学
1097
戴东旭等: 化学反应过渡态的结构和动力学
性质有了比较清楚的认识.
(1) H + H2及其同位素变型反应的过渡态区域是一个相对简单的势垒, 沿着反应坐标方向不存在准束缚结构的动力学共振态, 但是在垂直于反应坐标方向上存在分立结构的量子化瓶颈态. 当反应体系在演化过程中, 体系的能量恰好和所经历的量子化瓶颈态的能垒符合而减慢经过过渡态的时间, 就可能导致产物的前向散射分布. 而当体系经过不同的量子化瓶颈态时, 各个量子化瓶颈态通道之间会发生量子干涉. 通过反应物分子的转动取向, 可以选择反应体系所经历的量子化瓶颈态.
(2) F + H2及其同位素变型反应是一个典型的存在动力学共振态的反应体系, 但在这个反应体系中却存在不同的导致产物前向散射的机制. 在较低反应能下, 主要是HF(v′=3)-H势能面上的两个反应共振态在起作用并导致产物HF(v′=2)发生前向散射. 而在较高的反应能, 则是因为HF(v′=3)--H产物出口通道的离心势垒导致产物HF(v′=3)发生前向散射.
今后, 人们将会将研究重点转向N(2D) + H2等三原子体系以及例如H + CH4等较为复杂的基元反应的动力学和过渡态性质的研究, 这要求我们将态态化学反应动力学的研究方法和成果应用到更复杂的化学体系中, 达到认识化学过程的基本机制, 控制或部分控制化学过程的目的.
参考文献
1 Polanyi J C, Zewail A H. Direct observation of the transition state. Acc Chem Res, 1995, 28(3): 119—132[DOI]
2 Friedman R S, Truhlar D G. Chemical reaction thresholds are resonances. Chem Phys Lett, 1991, 183(6): 539—546[DOI]
3 Neumark D M, Wodtke A M, Robinson G N, Hayden C C, Lee Y T. Molecular beam studies of the F + H2 reaction. J Chem Phys,
1985, 82(7): 3045—3066[DOI]
4 Neumark D M, Wodtke A M, Robinson G N, Hayden C C, Shobatake K, Sparks R K, Schafer T P, Lee Y T. Molecular beam studies
of the F + D2 and F+HD reactions. J Chem Phys, 1985, 82(7): 3067—3077[DOI]
5 Liu K. Crossed-beam studies of neutral reactions: State-specific differential cross sections. Ann Rev Phys Chem, 2001, 52(1):
139—164[DOI]
6 Qiu M, Che L, Ren Z, Dai D, Wang X, Yang X. High resolution time-of-flight spectrometer for crossed molecular beam study of
elementary chemical reactions. Rev Sci Instr, 2005, 76(8): 083107—5[DOI]
7 Yang X. State-to-state dynamics of elementary chemical reactions using Rydberg H-atom translational spectroscopy. Inter Rev Phys
Chem, 2005, 24(1): 37—98[DOI]
8 Yang X. State-to-state dynamics of elementary bimolecular reactions. Ann Rev Phys Chem, 2007, 58(1): 433—459[DOI]
9 Yang X, Zhang D H. Dynamical Resonances in the fluorine atom reaction with the hydrogen molecule. Acc Chem Res, 2008, 41(8):
981—989[DOI]
10 Fernández-Alonso F, Zare R N. Scattering resonances in the simplest chemical reaction. Ann Rev Phys Chem, 2002, 53(1): 67—99[DOI]
11 Schnieder L, Seekamp-Rahn K, Wrede E, Welge K H. Experimental determination of quantum state resolved differential cross
sections for the hydrogen exchange reaction H + D2→ HD + D. J Chem Phys, 1997, 107(16): 6175—6195[DOI]
12 Kitsopoulos T N, Buntine M A, Baldwin D P, Zare R N, Chandler D W. Reaction product imaging: The H + D2 reaction. Science,
1993, 260: 1605—1610[DOI]
13 Schnieder L, Seekamprahn K, Borkowski J, Wrede E, Welge K H, Aoiz F J, Banares L, Dmello M J, Herrero V J, Rabanos V S, Wyatt
R E. Experimental studies and theoretical predictions for the H + D → HD + D reaction. Science, 1995, 269(5221): 207—210[DOI] 14 Buntin S A, Giese C F, Gentry W R. State-resolved differential cross sections for the reaction D + H2 → HD + H. J Chem Phys, 1987,
87(2): 1443—1445[DOI]
15 Kliner D a V, Adelman D E, Zare R N. Comparison of experimental and theoretical integral cross sections for D + H2(v=1, j=1) →
HD(v'=1, j′) + H. J Chem Phys, 1991, 95(3): 1648—1662[DOI]
16 Pack R T, Parker G A. Quantum reactive scattering in three dimensions using hyperspherical (APH) coordinates. Theory. J Chem
Phys, 1987, 87(7): 3888—3921[DOI]
17 Zhang J Z H, Miller W H. Quantum reactive scattering via the S-matrix version of the Kohn variational principle: Differential and 1098
中国科学B辑: 化学 2009年第39卷第10期integral cross sections for D + H2→ HD + H. J Chem Phys, 1989, 91(3): 1528—1547[DOI]
18 Zhao M, Mladenovic M, Truhlar D G, Schwenke D W, Sharafeddin O, Sun Y, Kouri D J. Spectroscopic analysis of transition state
energy levels: Bending-rotational spectrum and lifetime analysis of H3 quasibound states. J Chem Phys, 1989, 91(9): 5302—5309[DOI] 19 Zhao M, Truhlar D G, Schwenke D W, Kouri D J. Effect of rotational excitation on state-to-state differential cross sections: D + H2
→ HD + H. J Phys Chem, 1990, 94(18): 7074—7090[DOI]
20 D’mello M, Manolopoulos D E, Wyatt R E. Quantum dynamics of the H + D2→ D + HD reaction: Comparison with experiment. J
Chem Phys, 1991, 94(9): 5985—5993[DOI]
21 Levine R D, Wu S F. Resonances in reactive collisions: Computational study of the H + H2 collision. Chem Phys Lett, 1971, 11(5):
557—561[DOI]
22 Nieh J C, Valentini J J. Experimental observation of dynamical resonances in the H + H2reaction. Phys Rev Lett, 1988, 60(6):
519[DOI]
23 Kendrick B K, Jayasinghe L, Moser S, Auzinsh M, Shafer-Ray N. Observation of predicted resonance structure in the H+D2→
HD(v'=0, j'=7) + D reaction at a collision energy of 0.94 eV. Phys Rev Lett, 2000, 84(19): 4325[DOI]
24 Kendrick B K, Jayasinghe L, Moser S, Auzinsh M, Shafer-Ray N. Erratum: Observation of predicted resonance structure in the H +
D2→ HD(v'=0, j'=7) + D reaction at a collision energy of 0.94 eV [Phys. Rev. Lett. 84, 4325 (2000)]. Phys Rev Lett, 2001, 86(11): 2482[DOI]
25 Fernández-Alonso F, Bean B D, Ayers J D, Pomerantz A E, Zare R N, Ba?ares L, Aoiz F J. Evidence for scattering resonances in the
H + D2 Reaction. Angew Chem Inter Ed, 2000, 39(15): 2748—2752[DOI]
26 Althorpe S C, Fernández-Alonso F, Bean B D, Ayers J D, Pomerantz a E, Zare R N, Wrede E. Observation and interpretation of a
time-delayed mechanism in the hydrogen exchange reaction. Nature, 2002, 416(6876): 67—70[DOI]
27 Harich S A, Dai D, Wang C C, Yang X, Chao S D, Skodje R T. Forward scattering due to slow-down of the intermediate in the H +
HD → D + H2 reaction. Nature, 2002, 419: 281—284[DOI]
28 Lovejoy E R, Kim S K, Moore C B. Observation of transition-state vibrational thresholds in the rate of dissociation of ketene. Science,
1992, 256(5063): 1541—1544[DOI]
29 Kim S K, Lovejoy E R, Moore C B. Transition state vibrational level thresholds for the dissociation of triplet ketene. J Chem Phys,
1995, 102(8): 3202—3219[DOI]
30 Gezelter J D, Miller W H. Dynamics of the photodissociation of triplet ketene. J Chem Phys, 1996, 104(10): 3546—3554[DOI]
31 Kaledin a L, Seong J, Morokuma K. Predominance of nonequilibrium dynamics in the photodissociation of ketene in the triplet state.
J Phys Chem A, 2001, 105(12): 2731—2737[DOI]
32 King R A, Allen W D, Ma B, Iii H F S. Fragmentation surface of triplet ketene. Faraday Discuss 1998, 110: 23—50[DOI]
33 Dai D, Wang C C, Harich S A, Wang X, Yang X, Chao S D, Skodje R T. Interference of quantized transition-state pathways in the H
+ D2 -> D + HD chemical reaction. Science, 2003, 300: 1730—1734[DOI]
34 Zhang J, Dai D, Wang C C, Harich S A, Wang X, Yang X, Gustafsson M, Skodje R T. State to state to state dynamics of the D + H2
→ HD + H reaction: Control of transition-state pathways via reagent orientation. Phys Rev Lett, 2006, 96(9): 093201—4[DOI]
35 Schatz G C, Bowman J M, Kuppermann A. Large quantum effects in the collinear F + H2→ FH + H reaction. J Chem Phys, 1973,
58(9): 4023—4025[DOI]
36 Schatz G C, Bowman J M, Kuppermann A. Exact quantum, quasiclassical, and semiclassical reaction probabilities for the collinear F +
H2→ FH + H reaction. J Chem Phys, 1975, 63(2): 674—684[DOI]
37 Schatz G C, Bowman J M, Kuppermann A. Exact quantum, quasiclassical, and semiclassical reaction probabilities for the collinear F +
D2 → FD + D reaction. J Chem Phys, 1975, 63(2): 685—696[DOI]
38 Wu S F, Johnson B R, Levine R D. Quantum mechanical computational studies of chemical reactions : III. Collinear A BC reaction
with some model potential energy surfaces. Mol Phys, 1973, 25(4): 839—856[DOI]
39 Connor J N L, Jakubetz W, Manz J. The F + H2 (v = 0) → FH (v' ≤ 3) + H reaction: Quantum collinear reaction probabilities on
three different potential energy surfaces. Mol Phys, 1978, 35(5): 1301—1323[DOI]
40 Connor J N L, Jakubetz W, Manz J. Quantum collinear reaction probabilities for vibrationally excited reactants : F + H2(v≤2) →
FH(v'≤5) + H. Mol Phys, 1980, 39(4): 799—816[DOI]
1099
戴东旭等: 化学反应过渡态的结构和动力学
41 Neumark D M, Wodtke A M, Robinson G N, Hayden C C, Lee Y T. Experimental investigation of resonances in reactive scattering:
The F + H2 reaction. Phys Rev Lett, 1984, 53(3): 226[DOI]
42 Stark K, Werner H J. An accurate multireference configuration interaction calculation of the potential energy surface for the F + H2
→ HF + H reaction. J Chem Phys, 1996, 104(17): 6515—6530[DOI]
43 Aoiz F J, Banares L, Herrero V J, Saez Rabanos V, Stark K, Werner H J. Classical dynamics for the F + H2→ HF + H reaction on a
new ab initio potential energy surface. A direct comparison with experiment. Chem Phys Lett, 1994, 223(3): 215—226[DOI]
44 Castillo J F, Manolopoulos D E, Stark K, Werner H-J. Quantum mechanical angular distributions for the F+H2 reaction. J Chem Phys,
1996, 104(17): 6531—6546[DOI]
45 Skodje R T, Skouteris D, Manolopoulos D E, Lee S-H, Dong F, Liu K. Resonance-mediated chemical reaction: F+HD --> HF+D.
Phys Rev Lett, 2000, 85(6): 1206[DOI]
46 Skodje R T, Skouteris D, Manolopoulos D E, Lee S-H, Dong F, Liu K. Observation of a transition state resonance in the integral
cross section of the F + HD reaction. J Chem Phys, 2000, 112(10): 4536—4552[DOI]
47 Dong F, Lee S H, Liu K. Reactive excitation functions for F + p-H2/n-H2]/D2 and the vibrational branching for F + HD. J Chem
Phys, 2000, 113(9): 3633—3640[DOI]
48 Nizkorodov S A, Harper W W, Chapman W B, Blackmon B W, Nesbitt D J. Energy-dependent cross sections and nonadiabatic
reaction dynamics in F(2P3/2,2P1/2) + n--H2→ HF(v, J) + H. J Chem Phys, 1999, 111(18): 8404—8416[DOI]
49 Rusin L Y, Sevryuk M B, Toennies J P. Comparison of experimental time-of-flight spectra of the HF products from the F + H2
reaction with exact quantum mechanical calculations. J Chem Phys, 2005, 122(13): 134314—9[DOI]
50 Hayes M, Gustafsson M, Mebel A M, Skodje R T. An improved potential energy surface for the F+H2 reaction. Chem Phys, 2005,
308(3): 259—266[DOI]
51 Qiu M, Ren Z, Che L, Dai D, Harich S A, Wang X, Yang X, Xu C, Xie D, Gustafsson M, Skodje R T, Sun Z, Zhang D H.
Observation of Feshbach Resonances in the F + H2→ HF + H Reaction. Science, 2006, 311(5766): 1440—1443[DOI]
52 Xu C, Xie D, Zhang D H. A global ab initio potential energy surface for F + H2→ HF + F. Chin J Chem Phys, 2006, 19(2): 96—98
53 Ren Z, Che L, Qiu M, Wang X, Dong W, Dai D, Wang X, Yang X, Sun Z, Fu B, Lee S-Y, Xu X, Zhang D H. Probing the resonance
potential in the F atom reaction with hydrogen deuteride with spectroscopic accuracy. Proceedings of the National Academy of Sciences, 2008, 105(35): 12662—12666[DOI]
54 Fu B, Xu X, Zhang D H. A hierarchical construction scheme for accurate potential energy surface generation: An application to the F
+ H2 reaction. J Chem Phys, 2008, 129(1): 011103—4[DOI]
55 Wang X, Dong W, Qiu M, Ren Z, Che L, Dai D, Wang X, Yang X, Sun Z, Fu B, Lee S-Y, Xu X, Zhang D H. HF(v' = 3) forward
scattering in the F + H2reaction: Shape resonance and slow-down mechanism. Proceedings of the National Academy of Sciences, 2008, 105(17): 6227—6231[DOI]
56 Ren Z, Che L, Qiu M, Wang X, Dai D, Harich S A, Wang X, Yang X, Xu C, Xie D, Sun Z, Zhang D H. Probing Feshbach resonances
in F + H2(j = 1) → HF + H: Dynamical effect of single quantum H2-rotation. J Chem Phys, 2006, 125(15): 151102—4[DOI]
57 Yang X, Xie D, Zhang D H. Dynamical resonance in F+H2 chemical reaction and rotational excitation effect. Chin Sci Bull, 2007,
52(8): 1009—1012[DOI]
1100
中国科学B辑: 化学 2009年第39卷第10期
Transition state structure and dynamics of chemical reaction
DAI DongXu & YANG XueMing
State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
Abstract: The concept of transition state has played a crucial role in the field of chemical kinetics and reaction dy-namics. However, detecting and characterizing the transition states have been a major challenge in both experiment and theory. In recent years, two benchmark reaction systems, H + H2 and F + H2, have been studied on the full quan-tum state resolved level by using crossed molecular beams method with H-atom Rydberg tagging technique and high accuracy calculation of quantum dynamics. The strong interaction between theory and experiment has significantly enhanced our understanding of the dynamics of these reactions. Clear physical pictures for the reaction transition states and dynamics in these benchmark reactions have emerged, providing the textbook examples of dynamical resonances in elementary chemical reactions.
Keywords: transition state, chemical reaction dynamics, differential reaction cross section, dynamical resonance, crossed molecular beams
1101
第十章化学动力学基础(一) 返回上一页 1. 298 K时N2O5(g)分解反应半衰期t1/2为5.7 h,此值与N2O5的起始浓度无关,试求: (1) 该反应的速率常数. (2) 作用完成90%时所须的时间. 2. 某人工放射性元素放出α粒子,半衰期为15 min ,试问该试样有80%分解,需时若干? 3. 把一定量的PH3(g)迅速引入温度为950 K的已抽空的容器中,待反应物达到该温度时开始计时(此时已有部分分解),测得实验数据如下: t/s 0 58 108 ∞ P/kPa 35.00 36.34 36.68 36.85 已知反应 4pH3(g) P4(g) + 6H2(g) 为一级反应,求该反应的速率常数k值(设在t=∞时反应基本完成) 4. 在某化学反应中随时检测物质A的含量,1小时后,发现A已作用了75%,试问2小时后A还剩余多少没有作用?若该反应对A 来说是: (1) 一级反应. (2) 二级反应(设A与另一反应物B起始浓度相同) (3) 零级反应(求A作用完所用时间) 5. 在298 K时, NaOH与CH3COOCH3皂化作用的速率常数k2与NaOH与CH3COOC2H5皂化作用的速率常数k2' 的关系为k2=2.8k2' .试问在相同的实验条件下,当有90% CH3COOCH3被分解时, CH3COOC2H5的分解百分数为若干?
6. 对反应2NO(g) +2H2(g)---> N2(g) +2H2O(l) 进行了研究,起始时NO与H2的物质的量相等.采用不同的起始压力相应的有不同的半衰期,实验数据为: p0 /kPa 47.20 45.40 38.40 33.46 26.93 t1/2/min 81 102 140 180 224 求该反应级数为若干? 7. 反应A+B P的动力学实验数据如下, [A]0/(mol·dm-3) 1.0 2.0 3.0 1.0 1.0 [B]0/(mol·dm-3) 1.0 1.0 1.0 2.0 3.0 r0/(mol·dm-3·s-1) 0.15 0.30 0.45 0.15 0.15 若该反应的速率方程为 ,求x和y的值. 8. 碳的放射性同位素在自然界树木中的分布基本保持为总碳量的 1.10×%.某考古队在一山洞中发现一些古代木头燃烧的灰烬,经分析的含 量为总碳量的9.87×%,已知的半衰期为5700年,试计算这灰距今约有多少年? 9. 某抗菌素在人体血液中呈现简单级数的反应,如果给病人在上午8点注射一针抗菌素,然后在不同时刻t测定抗菌素在血液中的浓度c(以mg/100 cm3表示),得到以下数据 t/h 4 8 12 16 c /(mg/100 cm3) 0.480 0.326 0.222 0.151 (1) 确定反应的级数. (2) 求反应的速率常数k和半衰期t1/2.
第三章 水溶液中的离子平衡 一、电解质的有关定义 物质 单质 化合物 电解质 非电解质:大多数非金属氧化物和有机物。如SO 3、CO 2、C 6H 12O 6、CCl 4、CH 2=CH 2…… 强电解质:强酸、强碱、绝大多数金属氧化物和盐。如HCl 、NaOH 、NaCl 、BaSO 4 弱电解质:弱酸、弱碱和水。如HClO 、NH 3·H 2O 、Cu(OH)2、H 2O …… 混和物 纯净物 1、电解质与非电解质本质区别: 在一定条件下(溶于水或熔化)能否电离(以能否导电来证明是否电离) 电解质——离子化合物或共价化合物 非电解质——共价化合物 离子化合物与共价化合物鉴别方法:熔融状态下能否导电 2、强电解质与弱电质的本质区别:在水溶液中是否完全电离(或是否存在电离平衡) 注意:①电解质、非电解质都是化合物 ②SO 2、NH 3、CO 2等属于非电解质 ③强电解质不等于易溶于水的化合物(如BaSO 4不溶于水,但溶于水的BaSO 4全部电离,故BaSO 4为强电解质) 4、强酸(HA )与弱酸(HB )的区别:(1)溶液的物质的量浓度相同时,pH (HA)<pH (HB) (2)pH 值相同时,溶液的浓度C HA <C HB (3)pH 相同时,加水稀释同等倍数后,pH HA >pH HB 二、水的电离和溶液的酸碱性 1、水的电离平衡:H 2O H + + OH - 水的离子积:K W = [H + ]·[OH -] 25℃时, [H + ]=[OH -] =10-7 mol/L ; K W = [H + ]·[OH -] = 10-14 注意:K W 只与温度有关,温度一定,则K W 值一定; K W 不仅适用于纯水,适用于任何溶液(酸、碱、盐)。 2、水电离特点:(1)可逆 (2)吸热 (3)极弱 3、影响水电离平衡的外界因素: (1)酸、碱 :抑制水的电离(pH 之和为14的酸和碱的水溶液中水的电离被同等的抑制)(2)温度:促进水的电离(水的电离是吸热的)(3)易水解的盐:促进水的电离(pH 之和为14两种水解盐溶液中水的电离被同等的促进) 4、溶液的酸碱性和pH : (1)pH= -lg[H + ] 注意:①酸性溶液不一定是酸溶液(可能是 溶液) ;②pH <7 溶液不一定是酸性溶液(只有温度为常温才对); ③碱性溶液不一定是碱溶液(可能是 溶液)。 (2)pH 的测定方法:酸碱指示剂——甲基橙、石蕊、酚酞 pH 试纸 ——最简单的方法。 操作:将一小块pH 试纸放在洁净的玻璃片上,用玻璃棒蘸取未知液点试纸中部,然后与标准比色卡比较读数即可。 注意:①事先不能用水湿润PH 试纸;②只能读取整数值或范围
第二章 反应动力学基础 一、填空题 1. 生成主产物的反应称为 主反应 ,其它的均为 副反应 。 2. 化学反应的总级数为n ,如用浓度表示的速率常数为C K ,用逸度表示的速率常数f K ,则C K =n f K 。 3. 化学反应的总级数为n ,如用浓度表示的速率常数为C K ,用气体摩尔分率表示的速率常数y K , 则C K = n p RT ???? ?? y K 。 4. 化学反应速率式为βαB A C A C C K r =-,用浓度表示的速率常数为C K ,假定符合理想气体状态方程,如用压力表示的速率常数P K ,则C K =____)()(βα+RT ___P K 。 5. 反应A + B → C ,已知115.0-=s k ,则反应级数n= 1 。 6. 反应3A → P ,已知s l mol k ?=/15.0,则反应级数n=___0____。 7. 活化能的大小直接反映了 反应速率 对温度的敏感程度。 8. 对于一非恒容均相化学反应B A B A αα?,反应组分A 的化学反应速率=-A r Vdt dn r A A -=- 。( V d t dn r A A -=-、 Vdt dn r B A -=-、dt dC r A A -=-、dt dC r B A -=-) 9. 气相反应A + B → 3P + S 进料时无惰性气体,A 与B 以1∶1摩尔比进料,则膨胀因子A δ=____2___。 10. 气相反应3A + B → P + S 进料时无惰性气体,A 与B 以2∶1摩尔比进料,则膨胀因子A δ=___-2/3____ 11. 在一间歇恒容反应器中进行如下平行反应12k k A P A S ??→??→,P 为目的产物,已知0A c 的单位为[]/mol L ,1k 的单位为1s -????,2k 的单位为[]/L mol s ?,活化能12E E >。则R A = )(221A A C k C k +- 。目的产物P 的瞬时选择性P S = 1212A A A k c k c k c + ,为了提高P S ,A c 要控制得较 低 ,T 要控制得较 高 。
化学动力学基础(习题课) 1. 某金属的同位素进行β放射,经14d(1d=1天后,同位素的活性降低6.85%。求此同位素的蜕变常数和半衰期;要分解 90.0%,需经多长时间? 解:设反应开始时物质的质量为100%,14d后剩余未分解者为100%-6.85%,则 代入半衰期公式得 一、是非题 下列各题中的叙述是否正确?正确的选“√”,错误的选“×”。 √× 1.反应速率系数k A与反应物A的浓度有关。 √× 2.反应级数不可能为负值。 √× 3.对二级反应来说,反应物转化同一百分数时,若反应物的初始浓度愈低,则所需时间愈短。 √× 4.对同一反应,活化能一定,则反应的起始温度愈低,反应的速率系数对温度的变化愈 敏感。 √× 5. Arrhenius活化能的定义是。
√× 6.若反应A?Y,对A为零级,则A的半衰期。 二、选择题 选择正确答案的编号: 某反应,A → Y,其速率系数k A=6.93min-1,则该反应物A的浓度从1.0mol×dm-3变到0.5 mol×dm-3所需时间是: (A)0.2min;(B)0.1min;(C)1min;(D)以上答案均不正确。 某反应,A → Y,如果反应物A的浓度减少一半,它的半衰期也缩短一半,则该反应的级数 为: (A)零级;(B)一级;(C)二级;(D)以上答案均不正确。 三、填空题 在以下各小题的“ 1.某化学反应经证明是一级反应,它的速率系数在298K时是k=( 2.303/3600)s-1,c0=1mol×dm-3。 (A)该反应初始速率u0为 (B)该反应的半衰期t1/2 (C)设反应进行了1h,在这一时刻反应速率u1为 2.只有一种反应物的二级反应的半衰期与反应的初始浓度的关系为 3.反应A → B+D中,反应物A初始浓度c A,0=1mol×dm-3,初速度u A,0=0.01mol×dm-3×s-1,假定该反 应为二级,则其速度常数k A为t1/2为。 4.某反应的速率系数k=4.62′10-2min-1,则反应的半衰期为 5.反应活化能E a=250kJ×mol-1,反应温度从300K升高到310K时,速率系数k增加
高中化学全部知识点(化学口诀+总结) 一、化学计算 化学式子要配平,必须纯量代方程,单位上下要统一,左右倍数要相等。 质量单位若用克,标况气体对应升,遇到两个已知量,应照不足来进行。 含量损失与产量,乘除多少应分清。 二、气体制备 气体制备首至尾,操作步骤各有位,发生装置位于头,洗涤装置紧随后,除杂装置分干湿,干燥装置把水留; 集气要分气和水,性质实验分先后,有毒气体必除尽,吸气试剂选对头。 有时装置少几个,基本顺序不可丢,偶尔出现小变化,相对位置仔细求。 三、氢气还原氧化铜 试管被夹向下倾,实验开始先通氢,空气排尽再点灯,冷至室温再停氢 先点灯,会爆炸,先停氢,会氧化,由黑变红即变化,云长脸上笑哈哈。 一、化合价口诀 一价钾钠氟氢银,二价氧钙钡镁锌,三铝四硅五价磷;二三铁,二四碳。二四六硫都齐全,铜汞二价最常见。 二、溶解性口诀 钾钠铵盐溶水快①硫酸盐除去钡铅钙②氯化物不溶氯化银,硝酸盐溶液都透明。③口诀中未有皆下沉。④ 注:①钾钠铵盐都溶于水;②硫酸盐中只有硫酸钡、硫酸铅、硫酸钙不溶;③硝酸盐都溶于水; ④口诀中没有涉及的盐类都不溶于水; 三、1—20号元素顺序口诀 氢氦锂铍硼,碳氮氧氟氖;钠镁铝硅磷,硫氯氩钾钙。 青孩你别蹦,炭蛋养沸奶,那妹雨归淋,牛鹿鸭呷莱。 四、金属活动性口诀 钾钙钠镁铝。锌铁锡铅氢,铜汞银铂金。 制氧气口诀: 二氧化锰氯酸钾;混和均匀把热加。制氧装置有特点;底高口低略倾斜。 集气口诀: 与水作用用排气法;根据密度定上下。不溶微溶排水法;所得气体纯度大。 电解水口诀:
正氧体小能助燃;负氢体大能燃烧。 化合价口诀: 常见元素的主要化合价:氟氯溴碘负一价;正一氢银与钾钠。氧的负二先记清;正二镁钙钡和锌。正三是铝正四硅;下面再把变价归。全部金属是正价;一二铜来二三铁。锰正二四与六七;碳的二四要牢记。非金属负主正不齐;氯的负一正一五七。氮磷负三与正五;不同磷三氮二四。有负二正四六;边记边用就会熟。 常见根价口诀 一价铵根硝酸根;氢卤酸根氢氧根。高锰酸根氯酸根;高氯酸根醋酸根。二价硫酸碳酸根;氢硫酸根锰酸根。暂记铵根为正价;负三有个磷酸根。 金属活动性顺序表: (初中)钾钙钠镁铝、锌铁锡铅氢、铜汞银铂金。(高中)钾钙钠镁铝锰锌、铬铁镍、锡铅氢;铜汞银铂金。 化合价口诀二: 一价氢氯钾钠银;二价氧钙钡镁锌,三铝四硅五氮磷;二三铁二四碳,二四六硫都齐;全铜以二价最常见。 盐的溶解性: 钾钠铵硝皆可溶、盐酸盐不溶银亚汞;硫酸盐不溶钡和铅、碳磷酸盐多不溶。多数酸溶碱少溶、只有钾钠铵钡溶 初中化学知识记忆方法 学习初中化学,“记忆”是其中的一个重要环节。下面谈一下记忆的方法。 一、简化记忆 化学需要记忆的内容多而复杂,同学们在处理时易东扯西拉,记不全面。克服它的有效方法是:在理解的基础上,通过几个关键的字或词组成一句话,或分几个要点,或列表来简化记忆。如:用六个字组成:“一点、二通、三加热”。这一句话概括氢气还原氧化铜的关键步骤及注意事项。在研究氧气化学性质时,同学们可把所有现象综合起来分析、归纳得出如下记忆要点:一、燃烧是否有火或火焰。二、是燃烧的产物是如何确定的看到、嗅到或通过其它辅助实验。 三、所有燃烧实验均放热。抓住这几点就大大简化了记忆量。氧气、氢气的实验室制法,同学们第一次接触,新奇但很陌生,不易掌握,可分如下几个步骤简化记忆。一、原理用什么药品制取该气体;二、装置;三、收集方法;四、如何鉴别。如此记忆,既简单明了,又对以后学习其它气体制取有帮助。 二、编顺口溜记忆 初中化学有不少知识容量大,记忆难,很适合用编顺口溜的方法来记忆。如刚开始学元素符号时可这样记忆:碳、氢、氧、氮、氯、硫、磷;钾、钙、钠、镁、铝、铁、锌;溴、碘、锰、钡、铜、硅、银;氦、氖、氩、氟、铂和金。记忆化合价也是同学们比较伤脑筋的问题,也可编这样的顺口溜:钾、钠、银、氢+1价;钙、镁、钡、锌+2价;氧、硫-2价;铝+3价。这样主要元素的化合价就记清楚了。 三、关键字词记忆 这是记忆概念的有效方法之一,在理解基础上,找出概念中几个关键字或词来记忆整个概念。如:能改变其它物质化学反应速度一变而本身的质量和化学性质在化学反应前后都不变二不变这一催化剂内涵可用“一变、二不变”几个关键的字来记忆。 对新旧知识中具有相似性和对立性的有关知识进行比较,找出异同点。如:学习“离子”概念时,可用第二章中所学过的“原子”概念在结构方面、所带电荷方面、性质方面、表示方面以及它们在一定条件下可以相互转化方面进行比较,找出它们的区别及联系,从而防止混淆加深记忆。另外离子的表示方法和元素化合价的表示方法也易混淆,应注
化学动力学基础(一) 一、简答题 1.反应Pb(C 2H 5)4=Pb+4C 2H 5是否可能为基元反应?为什么? 2.某反应物消耗掉50%和75%时所需要的时间分别为t 1/2和 t 1/4,若反应对该反应物分别是一级、二级和三级,则t 1/2: t 1/4的比值分别是多少? 3.请总结零级反应、一级反应和二级反应各有哪些特征?平行反应、对峙反应和连续反应又有哪些特征? 4.从反应机理推导速率方程时通常有哪几种近似方法?各有什么适用条件? 5.某一反应进行完全所需时间时有限的,且等于k c 0(C 0为反应物起始浓度),则该反应是几级反应? 6. 质量作用定律对于总反应式为什么不一定正确? 7. 根据质量作用定律写出下列基元反应速率表达式: (1)A+B→2P (2)2A+B→2P (3)A+2B→P+2s (4)2Cl 2+M→Cl 2+M 8.典型复杂反应的动力学特征如何? 9.什么是链反应?有哪几种? 10.如何解释支链反应引起爆炸的高界限和低界限? 11.催化剂加速化学反应的原因是什么? 二、证明题 1、某环氧烷受热分解,反应机理如下: 稳定产物?→??+?+??→??++??→??? +??→?432134 33k k k k CH R CH R CH RH CO CH R H R RH
证明反应速率方程为()()RH kc dt CH dc =4 2、证明对理想气体系统的n 级简单反应,其速率常数()n c p RT k k -=1。 三、计算题 1、反应2222SO Cl SO +Cl →为一级气相反应,320℃时512.210s k --=?。问在320℃ 加热90min ,22SO Cl 的分解百分数为若干?[答案:11.20%] 2、某二级反应A+B C →初速度为133105---???s dm mol ,两反应物的初浓度皆为 32.0-?dm mol ,求k 。[答案:11325.1---??=s mol dm k ] 3、781K 时22H +I 2HI →,反应的速率常数3-1-1HI 80.2dm mol s k =??,求2H k 。[答 案:113min 1.41---??=mol dm k ] 4、双光气分解反应32ClCOOCCl (g)2COCl (g)→可以进行完全,将反应物置于密 闭恒容容器中,保持280℃,于不同时间测得总压p 如下: [答案: 1.1581a =≈;-14-12.112h 5.8710s k -==?] 5、有正逆反应均为一级反应的对峙反应: D-R 1R 2R 32L-R 1R 2R 3CBr 已知半衰期均为10min ,今从D-R 1R 2R 3CBr 的物质的量为1.0mol 开始,试计算10min 之后,可得L-R 1R 2R 3CBr 若干?[答案:0.375mol] 6、在某温度时,一级反应A →B ,反应速率为0.10mol ·dm -3·s -1时A 的转化率 为75%,已知A 的初始浓度为0.50mol ·dm -3,求(1)起始反应初速率;(2)速率常数。[答案:r 0=0.40s -1 ; k = 0.80 dm 3·mol -1·s -1 ] 7、在某温度时,对于反应A+B →P ,当反应物初始浓度为0.446和0.166mol ·dm -3 时,测 得反应的半衰期分别为4.80和12.90min ,求反应级数。[答案:2] 8、某二级反应,已知两种反应物初始浓度均为0.1mol ·dm -3,反应15min 后变
化学反应原理知识点归 纳 Company number:【0089WT-8898YT-W8CCB-BUUT-202108】
专题一:化学反应与能量变化 一、反应热、焓变 1.反应热:化学反应过程中放出或吸收的热量,叫反应热。包括燃烧热和中和热。 电 离 : 注意: 水解 : 吸热反应的发生不一定需要 常见的吸热反应: 铵盐与碱的反应:如NH 4Cl 与Ba(OH)28H 2O 加热才能进行。 大多数的分解反应:CaCO 3== CaO + CO 2 生产水煤气:C + H 2O == CO+H 2 碳和二氧化碳的反应:C+CO 2=2CO 燃烧反应 金属与酸(或水)的反应 常见的放热反应: 酸碱中和反应 自发的氧化还原反应 CaO(Na 2O 、Na 2O 2)与水的反应 浓酸与强碱溶于水 2、焓变:在恒温恒压的条件下,化学反应过程中吸收或放出的热量称为反 应的焓变。 符号:用ΔH 表示 单位:kJ/mol 放热反应:ΔH= —QkJ/mol ;或ΔH<0 吸热反应:ΔH= +QkJ/mol ;或ΔH>0 3、反应热产生的原因: 宏观:反应物和生成物所具有的能量不同,ΔH=_____________________________ 微观:化学反应过程中化学键断裂吸收的能量与新化学键生成所放出的能量不同,ΔH=____________ 二、热化学方程式 1.热化学方程式的概念:能表示反应热的化学方程式,叫做热化学方程式。热化学方程式不仅表示了化学反应中的物质变化,也表明了化学反应中的能量变化。 2.书写热化学方程式时的注意点 (1)需注明ΔH 的“+”与“—”,“+”表示 ,“—”表示 ;比较ΔH 的大小时,要考虑ΔH 的正负。 (3)要注明反应物和生成物的状态:g 、 l 、s 、aq 注意: 放热反应不一定常温下就自发进行,可能需要加热或点燃条件。
5202 反应 2O 3→ 3O 2的速率方程为 - d[O 3]/d t = k [O 3]2[O 2]-1 , 或者 d[O 2]/d t = k '[O 3]2[O 2]-1,则速率常数 k 和 k ' 的关系是: ( ) (A) 2k = 3k ' (B) k = k ' (C) 3k = 2k ' (D) -k /2 = k '/3 5203 气相反应 A + 2B ─→ 2C ,A 和 B 的初始压力分别为 p A 和 p B ,反应开始时 并无 C ,若 p 为体系的总压力,当时间为 t 时,A 的分压为: ( ) (A) p A - p B (B) p - 2p A (C) p - p B (D) 2(p - p A ) - p B 5204 对于反应 2NO 2= 2NO + O 2,当选用不同的反应物和产物来表示反应速率时,其相互关系为:( ) (A) -2d[NO 2]/d t = 2d[NO]/d t = d[O 2]/d t (B) - d[NO 2]/2d t = d[NO]/2d t = d[O 2]/d t = d ξ /d t (C) - d[NO 2]/d t = d[NO]/d t = d[O 2]/d t (D) - d[NO 2]/2d t = d[NO]/2d t = d[O 2]/d t = 1/V d ξ /d t 5207 气相基元反应 2A k 1 B 在一恒容的容器中进行,p 0为 A 的初始压力, p t 为时间 t 时反应 体系总压,此反应速率方程 d p t / d t = 。 - k (2p t - p 0)2 5208 有一反应 mA → nB 是一简单反应,其动力学方程为 -d c A / d t = kc A m , c A 的单位为 mol ·dm -3, 时间单位为 s ,则: (1) k 的单位为 ___________ mol 1- m ·dm 3( m -1)·s -1 (2) 以d c B /d t 表达的反应速率方程和题中给的速率方程关系为 B A A A 1d 1d 'd d m m c c k c k c n t m t m =-== 5209 反应 2N 2O 5─→ 4NO 2+ O 2 在328 K 时,O 2(g)的生成速率为0.75×10-4 mol ·dm -3·s -1。 如其间任一中间物浓度极低, 难以测出, 则该反应的总包反应速率为 _______________mol ·dm -3·s -1, N 2O 5之消耗速率为__________ mol ·dm -3·s -1,NO 2之生成速率为_______________mol ·dm -3·s -1 。0.75×10-4, 1.50×10-4, 3.00×10-4 5210 O 3分解反应为 2O 3─→3O 2 ,在一定温度下, 2.0 dm 3容器中反应。实验测出O 3每秒消耗1.50×10-2 mol, 则反应速率为_______________mol ·dm -3·s -1氧的生成速率为_______________mol ·dm -3·s -1, d ξ /d t 为_______________ 0.75×10-2, 2.25×10-2, 1.50×10-2.。 5211 2A +B =2C 已知反应某一瞬间, r A =12.72 mol ·dm -3·h -1, 则 r B = , r C =_____________r B =6.36 mol ·dm -3·h -1, r C =12.72mol ·dm -3·h -1 5212分别用反应物和生成物表示反应A +3B =2C 的反应速率, 并写出它们间关系为: 。r A = 13r B =1 2 r C 5222 有关基元反应的描述在下列诸说法中哪一个是不正确的: ( ) (A) 基元反应的级数一定是整数 (B) 基元反应是“态-态”反应的统计平均结果 (C) 基元反应进行时无中间产物,一步完成 (D) 基元反应不一定符合质量作用定律 5223 400 K 时,某气相反应的速率常数k p = 10-3(kPa)-1·s -1,如速率常数用 k C 表示,则 k C 应为: (A) 3.326 (mol ·dm -3)-1·s -1 k C = k p (RT ) (B) 3.0×10-4 (mol ·dm -3)-1·s -1 (C) 3326 (mol ·dm -3)-1·s -1 (D) 3.0×10-7 (mol ·dm -3)-1·s -1 5224 如果反应 2A + B = 2D 的速率可表示为:
【 一、焓变、反应热 要点一:反应热(焓变)的概念及表示方法 化学反应过程中所释放或吸收的能量,都可以用热量来描述,叫做反应热,又称焓变,符号为ΔH,单位为kJ/mol,规定放热反应的ΔH为“—”,吸热反应的ΔH为“+”。 特别提醒: (1)描述此概念时,无论是用“反应热”、“焓变”或“ ΔH”表示,其后所用的数值必须带“+”或“—”。 (2)单位是kJ/mol,而不是kJ,热量的单位是kJ。 (3)在比较大小时,所带“+”“—”符号均参入比较。 要点二:放热反应和吸热反应 1.放热反应的ΔH为“—”或ΔH<0 ;吸热反应的ΔH为“+”或ΔH >0 ?H=E(生成物的总能量)-E(反应物的总能量) ?H=E(反应物的键能)-E(生成物的键能) 2.常见的放热反应和吸热反应 ①放热反应:活泼金属与水或酸的反应、酸碱中和反应、燃烧反应、多数化合反应。 ②吸热反应:多数的分解反应、氯化铵固体与氢氧化钡晶体的反应、水煤气的生成反应、炭与二氧化碳生成一氧化碳的反应 3.需要加热的反应,不一定是吸热反应;不需要加热的反应,不一定是放热反应 4.通过反应是放热还是吸热,可用来比较反应物和生成物的相对稳定性。 如C(石墨,s)C(金刚石,s)△H3= +1.9kJ/mol,该反应为吸热反应,金刚石的能量高,石墨比金属石稳定。 二、热化学方程式的书写 书写热化学方程式时,除了遵循化学方程式的书写要求外,还要注意以下几点: 1.反应物和生成物的聚集状态不同,反应热的数值和符号可能不同,因此必须注明反应物和生成物的聚集状态,用s、l、g分别表示固体、液体和气体,而不标“↓、↑”。 2.△H只能写在热化学方程式的右边,用空格隔开,△H值“—” 表示放热反应,△H值“+”表示吸热反应;单位为“kJ/mol”。 3.热化学方程式中各物质化学式前面的化学计量数仅表示该物质的物质的量,并不表示物质的分子数或原子数,因此,化学计量数可以是整数,也可以是分数。 4.△H的值要与热化学方程式中化学式前面的化学计量数相对应,如果化学计量数加倍,△H也要加倍。 5.正反应若为放热反应,则其逆反应必为吸热反应,二者△H的数值相等而符号相反。 三、燃烧热、中和热、能源 要点一:燃烧热、中和热及其异同
5202 反应 2O 3→ 3O 2的速率方程为 - d[O 3]/d t = k [O 3]2[O 2]-1 , 或者 d[O 2]/d t = k '[O 3]2[O 2]-1,则速率常数 k 和 k ' 的关系是: ( ) (A) 2k = 3k ' (B) k = k ' (C) 3k = 2k ' (D) -k /2 = k '/3 5203 气相反应 A + 2B ─→ 2C ,A 和 B 的初始压力分别为 p A 和 p B ,反应开始时 并无 C ,若 p 为体系的总压力,当时间为 t 时,A 的分压为: ( ) (A) p A - p B (B) p - 2p A (C) p - p B (D) 2(p - p A ) - p B 5204 对于反应 2NO 2= 2NO + O 2,当选用不同的反应物和产物来表示反应速率时,其相互关系为:( ) (A) -2d[NO 2]/d t = 2d[NO]/d t = d[O 2]/d t (B) - d[NO 2]/2d t = d[NO]/2d t = d[O 2]/d t = d ξ /d t (C) - d[NO 2]/d t = d[NO]/d t = d[O 2]/d t (D) - d[NO 2]/2d t = d[NO]/2d t = d[O 2]/d t = 1/V d ξ /d t 5207 气相基元反应 2A k 1 B 在一恒容的容器中进行,p 0为 A 的初始压力, p t 为时间 t 时反应 体系总压,此反应速率方程 d p t / d t = 。 - k (2p t - p 0)2 5208 有一反应 mA → nB 是一简单反应,其动力学方程为 -d c A / d t = kc A m , c A 的单位为 mol ·dm -3, 时间单位为 s ,则: (1) k 的单位为 ___________ mol 1- m ·dm 3( m -1)·s -1 (2) 以d c B /d t 表达的反应速率方程和题中给的速率方程关系为 B A A A 1d 1d 'd d m m c c k c k c n t m t m =-== 5209 反应 2N 2O 5─→ 4NO 2+ O 2 在328 K 时,O 2(g)的生成速率为0.75×10-4 mol ·dm -3·s -1。 如 其间任一中间物浓度极低, 难以测出, 则该反应的总包反应速率为 _______________mol ·dm -3·s -1, N 2O 5 之消耗速率为__________ mol ·dm -3·s -1,NO 2之生成速率为_______________mol ·dm -3·s -1 。0.75×10-4, 1.50×10-4, 3.00×10-4 5210 O 3分解反应为 2O 3─→3O 2 ,在一定温度下, 2.0 dm 3容器中反应。实验测出O 3每秒消耗1.50× 10-2 mol, 则反应速率为_______________mol ·dm -3·s -1氧的生成速率为_______________mol ·dm -3·s -1, d ξ /d t 为_______________ 0.75×10-2, 2.25×10-2, 1.50×10-2.。 5211 2A +B =2C 已知反应某一瞬间, r A =12.72 mol ·dm -3·h -1, 则 r B = , r C =_____________r B =6.36 mol ·dm -3·h -1, r C =12.72mol ·dm -3·h -1 5212分别用反应物和生成物表示反应A +3B =2C 的反应速率, 并写出它们间关系为: 。 r A =13r B =12 r C 5222 有关基元反应的描述在下列诸说法中哪一个是不正确的: ( ) (A) 基元反应的级数一定是整数 (B) 基元反应是“态-态”反应的统计平均结果 (C) 基元反应进行时无中间产物,一步完成 (D) 基元反应不一定符合质量作用定律 5223 400 K 时,某气相反应的速率常数k p = 10-3(kPa)-1·s -1,如速率常数用 k C 表示,则 k C 应为: (A) 3.326 (mol ·dm -3)-1·s -1 k C = k p (RT ) (B) 3.0×10-4 (mol ·dm -3)-1·s -1 (C) 3326 (mol ·dm -3)-1·s -1 (D) 3.0×10-7 (mol ·dm -3)-1·s -1 5224 如果反应 2A + B = 2D 的速率可表示为:
化学选修化学反应原理 知识点总结 集团档案编码:[YTTR-YTPT28-YTNTL98-UYTYNN08]
《化学反应原理》知识点总结 第一章:化学反应与能量变化 1、反应热与焓变:△H=H(产物)-H(反应物) 2、反应热与物质能量的关系 3 4 ①多数的分解反应 ② 2NH 4Cl(s)+Ba(OH)2·8H 2O(s)=BaCl 2+2NH 3+10H 2O ③ C(s)+ H 2O(g) 高温 CO+H 2 ④CO 2+ C 高温 2 CO 5、反应条件与吸热、放热的关系: 反应是吸热还是放热与反应的条件没有必然的联系,而取决与 反应物和产物具有的总能量(或焓)的相对大小。 6、书写热化学方程式除了遵循书写化学方程式的要求外,还应注意以下几点: ①放热反应△H 为“-”,吸热反应△H 为“+”,△H 的单位为kJ/mol ②反应热△H 与测定条件(温度、压强等)有关,因此应注意△H 的测定条件;绝大多数化学反应的△H 是在298K 、101Pa 下测定的,可不注明温度和压强。 ③热化学方程式中各物质化学式前面的系数仅表示该物质的物质的量,并不表示物质的分子或原子数,因此化学计量数可以是分数或小数。必须注明物质的聚集状态,热化学方程式是表示反 应已完成的数量,所以方程式中化学式前面的计量数必须与△H 相对应;当反应逆向进行时,反应热数值相等,符号相反。 7、利用盖斯定律进行简单的计算 8、电极反应的书写: 活性电极:电极本身失电子 ⑴电解:阳极:(与电源的正极相连)发生氧化反应 惰性电极:溶液中阴离子失电子 (放电顺序:I ->Br ->Cl ->OH - ) 阴极:(与电源的负极相连)发生还原反应,溶液中的阳离子得电子 (放电顺序:Ag +>Cu 2+>H +) 能量 反应物的总能量 生成物的总能量 反应过程 总能量 总能量
一、焓变、反应热 要点一:反应热(焓变)的概念及表示方法 化学反应过程中所释放或吸收的能量,都可以用热量来描述,叫做反应热,又称焓变,符号为ΔH,单位为kJ/mol,规定放热反应的ΔH为“—”,吸热反应的ΔH为“+”。 特别提醒:(1)描述此概念时,无论是用“反应热”、“焓变”或“ ΔH”表示,其后所用的数值必须带“+”或“—”。 (2)单位是kJ/mol,而不是kJ,热量的单位是kJ。 (3)在比较大小时,所带“+”“—”符号均参入比较。 要点二:放热反应和吸热反应 1.放热反应的ΔH为“—”或ΔH<0 ;吸热反应的ΔH为“+”或ΔH >0 ?H=E(生成物的总能量)-E(反应物的总能量) ?H=E(反应物的键能)-E(生成物的键能) 2.常见的放热反应和吸热反应 ①放热反应:活泼金属与水或酸的反应、酸碱中和反应、燃烧反应、多数化合反应。 ②吸热反应:多数的分解反应、氯化铵固体与氢氧化钡晶体的反应、水煤气的生成反应、炭与二氧化碳生成一氧化碳的反应 3.需要加热的反应,不一定是吸热反应;不需要加热的反应,不一定是放热反应 4.通过反应是放热还是吸热,可用来比较反应物和生成物的相对稳定性。 如C(石墨,s)C(金刚石,s)△H3= +1.9kJ/mol,该反应为吸热反应,金刚石的能量高,石墨比金属石稳定。 二、热化学方程式的书写 书写热化学方程式时,除了遵循化学方程式的书写要求外,还要注意以下几点: 1.反应物和生成物的聚集状态不同,反应热的数值和符号可能不同,因此必须注明反应物和生成物的聚集状态,用s、l、g分别表示固体、液体和气体,而不标“↓、↑”。 2.△H只能写在热化学方程式的右边,用空格隔开,△H值“—” 表示放热反应,△H值“+”表示吸热反应;单位为“kJ/mol”。 3.热化学方程式中各物质化学式前面的化学计量数仅表示该物质的物质的量,并不表示物质的分子数或原子数,因此,化学计量数可以是整数,也可以是分数。 4.△H的值要与热化学方程式中化学式前面的化学计量数相对应,如果化学计量数加倍,△H 也要加倍。 5.正反应若为放热反应,则其逆反应必为吸热反应,二者△H的数值相等而符号相反。 三、燃烧热、中和热、能源 要点一:燃烧热、中和热及其异同
《化学反应原理》知识点总结 第一章:化学反应与能量变化 1、反应热与焓变:△H=H(产物)-H(反应物) 2、反应热与物质能量的关系 3、反应热与键能的关系 △H=反应物的键能总和-生成物的键能总和 4、常见的吸热、放热反应 ⑴常见的放热反应: ①活泼金属与水或酸的反应 ②酸碱中和反应 ③燃烧反应 ④多数的化合反应 ⑤铝热反应 ⑵常见的吸热反应 ①多数的分解反应 ② 2NH 4Cl(s)+Ba(OH)2·8H 2O(s)=BaCl 2+2NH 3+10H 2O ③ C(s)+ H 2O(g) 高温 CO+H 2 ④CO 2+ C 高温 2 CO 5、反应条件与吸热、放热的关系: 反应是吸热还是放热与反应的条件没有必然的联系,而取决与反应物和产物具有的 总能量(或焓)的相对大小。 6、书写热化学方程式除了遵循书写化学方程式的要求外,还应注意以下几点: ①放热反应△H 为“-”,吸热反应△H 为“+”,△H 的单位为kJ/mol ②反应热△H 与测定条件(温度、压强等)有关,因此应注意△H 的测定条件;绝大多数化学反应的△H 是在298K 、101Pa 下测定的,可不注明温度和压强。 ③热化学方程式中各物质化学式前面的系数仅表示该物质的物质的量,并不表示物质的分子或原子数,因此化学计量数可以是分数或小数。必须注明物质的聚集状态,热化学方程式是表示反应已完成的数量,所以方程式中化学式前面的计量数必须与△H 相对应;当反应逆向进行时,反应热数值相等,符号相反。 7、利用盖斯定律进行简单的计算 8、电极反应的书写: 活性电极:电极本身失电子 能量 反应物的总能量 生成物的总能量 反应过程 总能量 总能量
化学反应动力学 第二章习题 1、The first-order gas reaction SO 2Cl 2 → SO 2 + Cl 2 has k = 2.20 ? 10-5 s -1 at 593K, (1) What percent of a sample of SO 2Cl 2 would be decomposed by heating at 593K for 1 hour? (2) How long will it take for half the SO 2Cl 2 to decompose? 解:一级反应动力学方程为: t k e Cl SO Cl SO ?-?=ο][][2222 ? t k e Cl SO Cl SO ?-=ο ][] [2222 (1) 反应达1小时时:60 601020.222225][][???--=e Cl SO Cl SO ο =0.924=92.4% 已分解的百分数为:100%-92.4%=7.6% (2) 当 21][][2222=οCl SO Cl SO 时,7.315062 1 ln 1=-=k t s 5 21102.2693 .0-?= t = 31500 s = 8.75 hour 2、T-butyl bromide is converted into t-butyl alcohol in a solvent containing 90 percent acetone and 10 percent water. The reaction is given by (CH 3)3CBr + H 2O → (CH 3)3COH + HBr The following table gives the data for the concentration of t-utyl bromide versus time: T(min) 0 9 18 24 40 54 72 105 (CH 3)CBr (mol/L) 0.1056 0.0961 0.0856 0.0767 0.0645 0.0536 0.0432 0.0270 (1) What is the order of the reaction? (2) What is the rate constant of the reaction? (3) What is the half-life of the reaction? 解: (1) 设反应级数为 n ,则 n A k dt A d ][] [=- ? kt A A n n =---1 1][1][1ο 若 n=1,则 ] [][ln 1A A t k ο = t = 9 01047.00961.01056.0ln 91==k , t = 18 01167.00856.01056 .0ln 181==k t = 24 01332.00767.01056.0ln 241== k , t = 40 01232.00645 .01056.0ln 401==k t = 54 01256.0=k , t = 72 01241.0=k , t = 105 01299.0=k
专题一:化学反应与能量变化 一、反应热、焓变 1.反应热:化学反应过程中放出或吸收的热量,叫反应热。包括燃烧热和中和热。 电离 : 注意: 水解 : 吸热反应的发生不一定需要 常见的吸热反应: 铵盐与碱的反应:如NH 4Cl 与Ba(OH)2?8H 2O 加热才能进行。 大多数的分解反应:CaCO 3== CaO + CO 2 生产水煤气:C + H 2O == CO+H 2 碳和二氧化碳的反应:C+CO 2=2CO 燃烧反应 金属与酸(或水)的反应 常见的放热反应: 酸碱中和反应 自发的氧化还原反应 CaO(Na 2O 、Na 2O 2)与水的反应 浓酸与强碱溶于水 2、焓变:在恒温恒压的条件下,化学反应过程中吸收或放出的热量称为反应的焓变。 符号:用ΔH 表示 单位:kJ/mol 放热反应:ΔH= —QkJ/mol ;或ΔH<0 吸热反应:ΔH= +QkJ/mol ;或ΔH>0 3、反应热产生的原因: 宏观:反应物和生成物所具有的能量不同,ΔH=_____________________________ 微观:化学反应过程中化学键断裂吸收的能量与新化学键生成所放出的能量不同,ΔH=____________ 二、热化学方程式 1.热化学方程式的概念:能表示反应热的化学方程式,叫做热化学方程式。热化学方程式不仅表示了化学反应中的物质变化,也表明了化学反应中的能量变化。 2.书写热化学方程式时的注意点 (1)需注明ΔH 的“+”与“—”,“+”表示 ,“—”表示 ;比较ΔH 的大小时,要考虑ΔH 的正负。 (3)要注明反应物和生成物的状态:g 、 l 、s 、aq (3)各物质前的化学计量数表示物质的量,不表示分子个数,因此,可以是整数也可以是分数,但系数与ΔH 的值一定要相对应。 (4)要注明反应温度和压强,但中学化学中所用ΔH 的数据一般都是在101kPa 和25℃时的数据,因此可不特别注明; (5)对于可逆反应,其ΔH 同样要与系数相对应,但若按系数投料反应,则由于可逆反应不能进行完全,其反应热的数值会比ΔH 的数值要小。 三、燃烧热、热值与中和热: 1.燃烧热:在1atm 下,1mol 物质完全燃烧的反应热叫做该物质的标准燃烧热。(物质完全燃烧是指含有 注意: 放热反应不一定常温下就自发进行,可能需要加热或点燃条件。