溶出曲线的相似性f2因子法评价 时间点 n 输入 n=8 时间点Rt(参比制剂平均累积释放度)Tt(受试制剂平均累积释放度) 1129 21712 32615 43728 55038 66652 77361 88473 f2=49 相似性判断:不相似 方法说明: Rt与Tt分别代表参比和受试制剂第t时间点的平均累积释放度,n为测试点数。其中如果两条溶出曲线完全一致,则:f2=50×lg(100)=100;如果一批样品释药完全,而另一批尚未开始释药,则有:
因此,f2的值的范围在0~100,而且f2越大,两条曲线的相似性越高。 事实上即使是相同处方的产品,其批次不同,在溶出曲线上也会有一定的差异。如果受试与参比制剂溶出曲线的差异不大于参比制剂间溶出曲线的差异,那么就可以认为受试与参比制剂溶出曲线具有相似性。通常认为,同一处方不同批次的样品,在任一取样点释放度的平均差异不超过10%,是可以接受的。将10%代入式中计算: 因此,FDA与EMEA规定:若受试与参比制剂的溶出曲线间的f2值不小于50,则认为两者相似。 f2因子的应用条件及注意事项: 1.在进行参比与受试制剂的溶出曲线比较的过程中,时间点间隔无需相等,但两者所取各时间点必须一致,一般除0时外,选择3点以上,即n≥3。 2.f2计算公式只适用于受试与参比制剂的平均累积释放度差值<100时的溶出曲线比较(如果二者的差值>100,就会得到一个负值),普通口服制剂要保证药物溶出90%以上,缓释制剂、肠溶制剂药物释放需达到80%以上,或达到释放平台。 3.受试与参比制剂释放曲线上各时间点的平均累积释放度差异,在平台区达到最小(如果外推到释放100%,差值将为0),在该区域上取样点的增加会直接导致f2值偏大。因此,受试或参比制剂的药物累积释放度在85%以上的取样点应不多于一个,否则,将会给判定结果带来误差。 4.f2因子比较一般选择每个处方的12个剂量单位的测定均值来进行处理。因为不考虑参比和受试制剂批内样本间差异,所以若参比或受试制剂批内样本间差异较大时,用f2因子来评价两者溶出曲线的相似性时需要谨慎,从第2个时间点至最后1个时间点溶出结果的变异系数应小于10%。 5.f2值与平均偏差之间成非线性关系,它只适用于描述参比与受试制剂溶出曲线的相似性,而不能用于评价受试制剂样本间差异。
普通口服固体制剂溶出曲线测定与比较指导原 则 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 一、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临床疗效差异的风险。 二、溶出试验方法的建立溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa 常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。
(一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50—75转/分钟,篮法选择50—100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择 应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。 在确定药物主成分稳定性满足测定方法要求的前提下,推荐选择不少于3种pH值的溶出介质进行溶出曲线考察,如选择pH 值、和的溶出介质。对于溶解度受pH值影响大的药物,可能需 在更多种pH值的溶出介质中进行考察。推荐使用的各种pH值溶出介质的制备方法见附件。
附件2 普通口服固体制剂溶出曲线测定与比较 指导原则 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 一、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临床疗效差异的风险。 二、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常
数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50—75转/分钟,篮法选择50—100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择 应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。 在确定药物主成分稳定性满足测定方法要求的前提下,推荐选择不少于3种pH值的溶出介质进行溶出曲线考察,如选择pH值1.2、4.5和6.8的溶出介质。对于溶解度受pH值影响大
发布日 20070806 期 栏目化药药物评价 标题采用f2因子法评价溶出曲线的相似性需注意的问题 正文审评四部审评八室马玉楠 在制剂的开发研究中,通过对比不同处方之间的溶出曲线,可以较准确地反映药物处方、工艺、生产场地及规模等因素变化对药物体外释放行为的影响。 近年来,国外针对溶出曲线的相似性评价方法报道很多,其中f2因子方法因 为计算简单、判定结果可靠,作为评价体外溶出曲线相似性的方法,已经被美 国FDA的CDER和欧盟EMEA收载并推荐使用。 F2因子的计算公式为: Rt与Tt分别代表参比和受试制剂第t时间点的平均累积释放度,n为测 试点数。其中如果两条溶出曲线完全一致,则:f2=50×lg(100)=100;如果一 批样品释药完全,而另一批尚未开始释药,则有: 。因此,f2的值的范围在0~100,而且f2越大,两条曲线的相似性越高。 事实上即使是相同处方的产品,其批次不同,在溶出曲线上也会有一定的差异。如果受试与参比制剂溶出曲线的差异不大于参比制剂间溶出曲线的差 异,那么就可以认为受试与参比制剂溶出曲线具有相似性。通常认为,同一处 方不同批次的样品,在任一取样点释放度的平均差异不超过10%,是可以接受 的。将10%代入式中计算: 。因此,FDA与EMEA规定:若受试与参比制剂的溶出曲线间的f2值不小于50,则认为两者相似。 在某些情况下,如果对于任一取样点释放度的平均差异的限定不是10%,则可通过计算得出相应的f2值(临界值)。表1提供了一些释放度平均差异与 相应的f2临界值。 表1 释放度平均差异与f2临界值表 Table 1 Average difference of drug release percent and f2 Limit 平均偏差(Average difference) 2% 5% 10% 15% 20% F2临界值(f2 Limit) 83 65 50 41 36 f2因子的应用条件及注意事项: 1.在进行参比与受试制剂的溶出曲线比较的过程中,时间点间隔无需相等,但
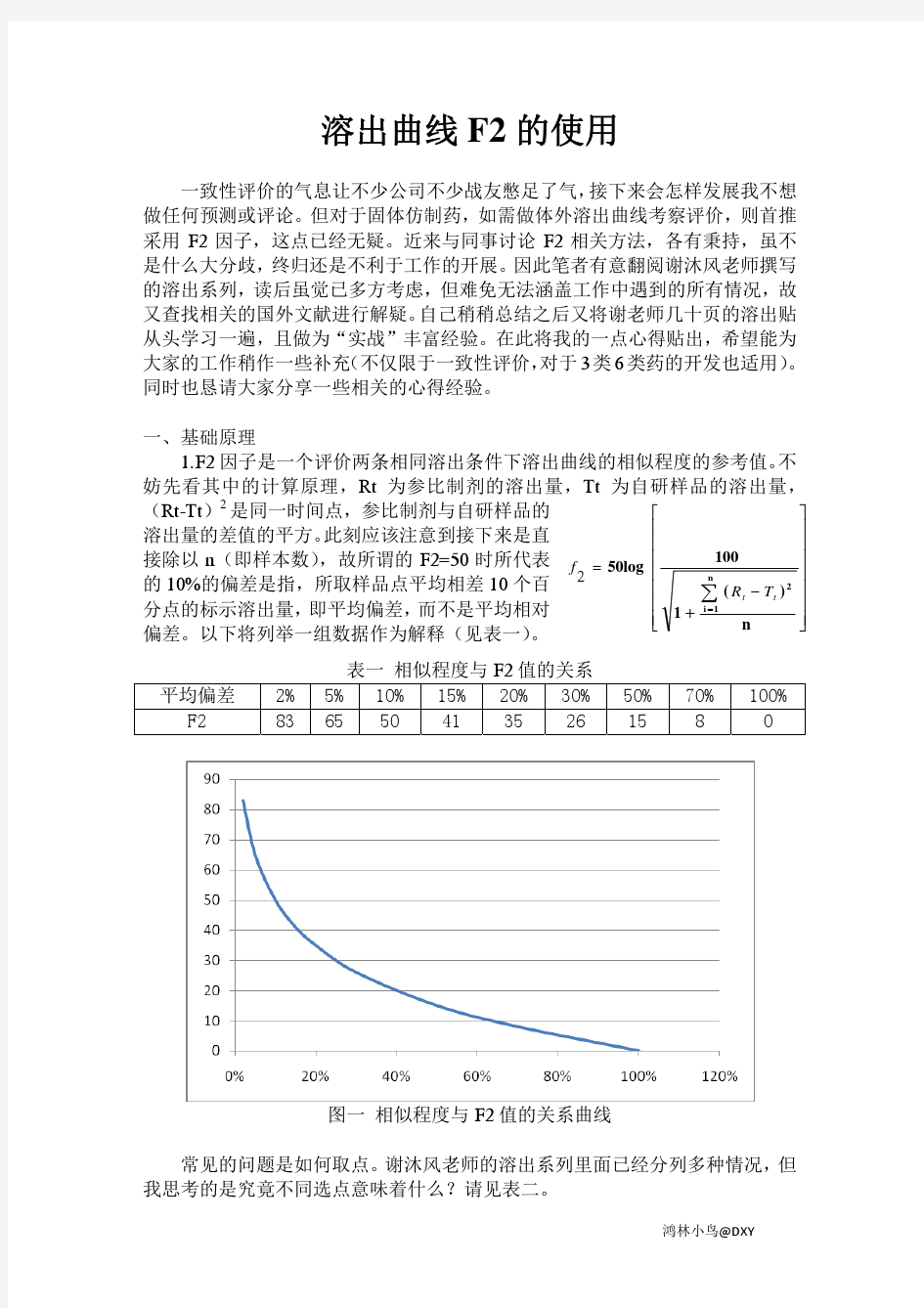
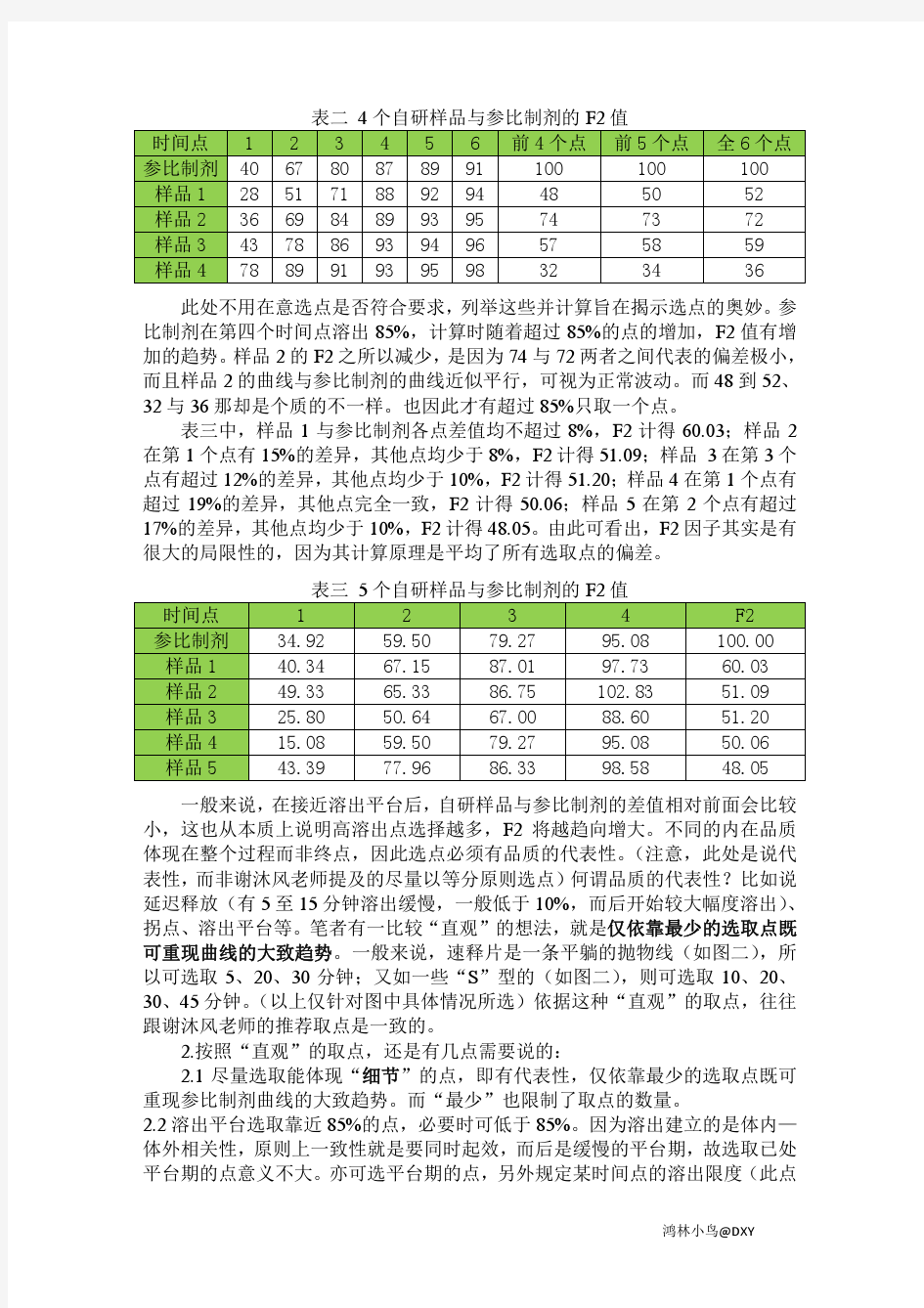
药物溶出曲线 一致性评价的气息让不少公司不少战友憋足了气,接下来会怎样发展我不想做任何预测或评论。但对于固体仿制药,如需做体外溶出曲线考察评价,则首推采用F2因子,这点已经无疑。近来与同事讨论F2相关方法,各有秉持,虽不是什么大分歧,终归还是不利于工作的开展。因此笔者有意翻阅谢沐风老师撰写的溶出系列,读后虽觉已多方考虑,但难免无法涵盖工作中遇到的所有情况,故又查找相关的国外文献进行解疑。自己稍稍总结之后又将谢老师几十页的溶出贴从头学习一遍,且做为“实战”丰富经验。在此将我的一点心得贴出,希望能为大家的工作稍作一些补充(不仅限于一致性评价,对于3类6类药的开发也适用)。同时也恳请大家分享一些相关的心得经验。 一、基础原理 1、F2因子是一个评价两条相同溶出条件下溶出曲线的相似程度的参考值。不妨先看其中的计算原理,Rt为参比制剂的溶出量,Tt为自研样品的溶出量,(Rt-Tt)2是同一时间点,参比制剂与自研样品的溶出量的差值的平方。此刻应该注意到接下来是直接除以n(即样本数),故所谓的F2=50时所代表的10%的偏差是指,所取样品点平均相差10个百分点的标示溶出量,即平均偏差,而不是平均相对偏差。以下将列举一组数据作为解释(见表一)。
常见的问题是如何取点。谢沐风老师的溶出系列里面已经分列多种情况,但我思考的是究竟不同选点意味着什么?请见表二。 表二4个自研样品与参比制剂的F2值 此处不用在意选点是否符合要求,列举这些并计算旨在揭示选点的奥妙。参比制剂在第四个时间点溶出85%,计算时随着超过85%的点的增加,F2值有增加的趋势。样品2的F2之所以减少,是因为74与72两者之间代表的偏差极小,而且样品2的曲线与参比制剂的曲线近似平行,可视为正常波动。而48到52、32与36那却是个质的不一样。也因此才有超过85%只取一个点。 表三中,样品1与参比制剂各点差值均不超过8%,F2计得60.03;样品2在第1个点有15%的差异,其他点均少于8%,F2计得51.09;样品3在第3个点有超过12%的差异,其他点均少于10%,F2计得51.20;样品4在第1个点有超过19%的差异,其他点完全一致,F2计得50.06;样品5在第2个点有超过17%的差异,其他点均少于10%,F2计得48.05。由此可看出,F2因子其实是有很大的局限性的,因为其计算原理是平均了所有选取点的偏差。 表三5个自研样品与参比制剂的F2值 一般来说,在接近溶出平台后,自研样品与参比制剂的差值相对前面会比较小,这也从本质上说明高溶出点选择越多,F2将越趋向增大。不同的内在品质体现在整个过程而非终点,因此选点必须有品质的代表性。(注意,此处是说代表性,而非谢沐风老师提及的尽量以等分原则选点)何谓品质的代表性?比如说延迟释放(有5至15分钟溶出缓慢,一般低于10%,而后开始较大幅度溶出)、拐点、溶出平台等。笔者有一比较“直观”的想法,就是仅依靠最
上海市药品检验所
谢沐风
撰写
【No.5 —— 溶出曲线的测定】
—— 上海市药品检验所 谢沐风 撰写
1. 关于测定时间点和结束时间点的设定 对于测定时间点,普通制剂与肠溶制剂可为 5、10、15、20、30、45、60、90、120 分钟,此后每隔 1 小时直至 6 小时止;缓控释制剂可为 15、30、45、60、90、120 分钟,3、 4、5、6、8、10、12、24 小时。当连续两点溶出率均达 90%(调释制剂为 85%)以上、且 差值在 5%以内时,试验则可提前结束。 对于结束时间点,在酸性介质中(如 pH 值 1.0)最长测定时间为 2 小时,在其他各 pH 值介质中普通制剂为 6 小时,缓控释制剂为 24 小时。
2. 其他事项 (1) 试验样品 用于比较的两种制剂含量差值应在 5%以内;每个品种各取 12 个单位。 取三个批号样品,在最终溶出率均可达 90%以上的溶出介质
(2) 参比制剂标准批号的选择
中,取溶出率在约 70%处、位于中间批号的样品进行试验。 在进行仿制药研发时,考虑到原研品批间差异与耐受性,建议从市场流通渠道获得有效 期内不同时间段的 3~5 批样品,分别测定后,取结果均值用于比较;并同时确定参比制剂在 各 pH 值溶出曲线的波动范围,以更为有效地评估原研制剂内在质量和自身仿制制剂的研发 深入程度。 如果主成分是在溶解状态下进行溶出度试验的(如一些散剂、颗粒剂) ,则适当选择某一 批号,即可。 (3) 试验样品的生产规模 由于固体制剂生物利用度与生产规模密切相关, 故一般情况下应不
少于今后工业化最大生产规模的 1/10 或不少于 10 万个单位。
3. 累积释放度校正计算公式 在多次取样时、可采取及时补充相同体积同温度溶出介质亦可采取不补液两种方式,但 必须保证每次抽取体积的固定性。累积校正计算公式如下: (1)补液时:
(C n?1 + ? ? ? ? ? ? +C 2 + C1 ) × V1 Cn L / V2 各时间点校正后的累积溶出量(%) = [ + ] × 100% L / V2 V2
其中 Cn为各时间点取出后的样品浓度(即稀释前的) ;
上海市药品检验所 谢沐风
L为制剂标示量(单位需与Cn一致)
撰写
1
手把手教你做出仿制药四条溶出曲线 原创2016-03-09书立 读完本文大约需要8分钟 2016 年 3 月 5 日,国务院办公厅印发了《关于开展仿制药质量和疗效一 致性评价的意见》,仿制药一致性评价工作正式展开。 仿制药一致性评价工作中,首先需要评价的是仿制制剂与参比制剂在体外溶 出曲线要一致。然而,将仿制制剂与参比制剂做到体外四条溶出曲线一致, 并不是一件容易的工作。 作者将平日的工作经验总结出来,欲与大家交流分享。 开始前的准备 将 BCS 再次分类 生物药剂学分类系统(BCS,biopharmaceutics classification system)是 1995 年由 Amidon 提出的基于药物溶解性质和渗透性差异的分类系统,分为四类。 对于体外四条溶出曲线而言,溶解性性质比渗透性更实用,因此根据溶解性质的差异将BCS 再次分类,分为 A 类(Ⅰ和Ⅲ)和 B 类(Ⅱ和Ⅳ)。之所以这样二次分类,是因为Ⅰ和Ⅲ、Ⅱ和Ⅳ分别在体外呈现出相同的溶解度性质。 将化合物根据pH-溶解度差异来分类
《仿制药质量一致性评价·口服固体制剂溶出曲线测定与比较指导原则》中提出,在进行溶出度实验之前,建议绘制化合物 pH-溶解度图。 那么根据 pH-溶解度的差异性,也可以将化合物分为两类: 一类是溶解度不存在 pH 依赖性或差异性。暂且将饱和溶解度无 pH 依赖性的原料药分为 a 类。 另一类是溶解度存在 pH 依赖性或差异性,其饱和溶解度随 pH 值增加而增加,或随 pH 值增加而降低。将这类化合物分为 b 类,比如 NAISD 类的布洛芬、双氯芬酸钠等。 这样分类如何应用呢?举个例子。 如表 1 所示,双氯芬酸钠在不同介质中的饱和溶解度差异性较大,再结合根据上述 BCS 的二次分类,那么可将双氯芬酸钠可定义为 Bb 类化合物。 之所以这样区分,是为了建立自我工作模型,以后在工作遇到相同的化合物,直接进行套用,从而降低工作量。 如何快速有效地做出四条溶出曲线? 根据化合物性质不同,其溶出曲线难易程度也是各有差别。 - Aa 类- 首先,最简单的化合物模型属于 Aa 类,即高溶解性无 pH 依赖性药物。 如果Aa 类药物的参比制剂(RLD)呈现出四条溶出曲线如图 1,那么在处方筛选工作中可选择任意一种介质作为区分介质。
【重磅推送】USP<1090>体内生物等效性试验指南第二部分 本文翻译自USP39-NF34 <1090>Assessment of drug product performance-Bioavailability, Bioequivalence, and Dissolution. 溶出度和体外产品性能 作为法定物质,USP专论提供了公开的质量标准,包括一系列检查方法,分析用对照以及限度标准。大多数口服固体制剂,包括口服悬浊液,需要进行溶出度或者药物释放度检查。药物溶出度和药物释放度检查分别在USP 通则溶出度<711>与释放度<724>章节中有描述。这些公开的质量标准用来进行质量控制检查以及上市获准。只有获得管理机构允许时,USP专论中的溶出度检查才与BA及BE相关联。如果没有这个关联,其将仅仅作为批放行的质量控制检查的方法。FDA的指导原则包括1.《行业指导原则-速释口服固体制剂溶出度检查Guidance for Industry—DissolutionTesting of Immediate Release Solid Oral Dosage Forms(1977)》(https://www.doczj.com/doc/5714292442.html,/; 请以文件名检索),2.《行业指导原则-延迟释放制口服制剂:开发、评估及体内外相关性的应用Guidance forIndustry—Extended Release Oral Dosage Forms: Development, Evaluation, andApplication of In Vitro/In Vivo Correlation(1977)》(https://www.doczj.com/doc/5714292442.html,/; 请以文件名检索)。 溶出度和体外生物利用度 药物溶出度和释放度检查在药物制剂开发过程中非常有用,可鉴别关键生产属性如辅料性质、生产工艺等对药物制剂特性的影响。在药物开发过程中,需要确定最优溶出度条件以辨别药物制剂处方及生产工艺变更。最终制剂成品获得批准上市后,药物溶出度和释放度检查在预测由于放大或上市后变更(SUPAC)造成的可能发生的特性变化方面非常有用。参考以下FDA指南: 《行业指导原则-速释口服固体制剂,放大及上市后变更:化学,生产及控制,体外溶出度试验及体内生物等效性证明Guidancefor Industry—Immediate Release Solid Oral Dosage Forms, Scale-Up andPostapproval Changes: Chemistry, Manufacturing, and Controls, In VitroDissolution Testing, and In Vivo Bioequivalence Documentation(1995)》(https://www.doczj.com/doc/5714292442.html,/; 请以文件名检索) 《行业指导原则-SUPAC-MR:调释固体口服制剂:放大及上市后变更:化学,生产及控制;体外溶出度试验及体内生物等效性证明Guidancefor Industry—SUPAC-MR: Modified-Release Solid Oral Dosage Forms: Scale-Up andPostapproval Changes: Chemistry, Manufacturing, and Controls; In VitroDissolution Testing and In Vivo Bioequivalence Documentation(1995)》(https://www.doczj.com/doc/5714292442.html,/; 请以文件名检索) 对于一些口服药物制剂,体外溶出度可能与体内表现相关,比如生物利用度和/或全身暴露量。USP通则章节制剂的体外和体内评价<1088>描述了不同的获得体外-体内相关性(IVIVC)的方法。 溶出度和体外等效性 溶出度测定方法是一种非常强有力的体外物理化学检查手段,可检测不同制剂产品的药物制剂质量和特性,例如口服固体制剂,透皮制剂,混悬液,特定半固体制剂。对于成品的USP检查可以分为两种类型:(1)药物制剂质量检查,(2)药物制剂特性检查。药物制剂质量检查是用于属性评估,例如含量测定,含量均匀度等;制剂特
上海市药品检验所 谢沐风撰写 xiemufeng@https://www.doczj.com/doc/5714292442.html, 本文版权归作者所有,任何个人或团体使用本文内容,请与作者联系。
【No.5 —— 溶出曲线的测定】
1. 关于测定时间点和结束时间点的设定 对于测定时间点,普通制剂与肠溶制剂可为 5、10、15、20、30、45、60、90、120 分钟,此后每隔 1 小时直至 6 小时止;缓控释制剂可为 15、30、45、60、90、120 分钟,3、 4、5、6、8、10、12、24 小时。当连续两点溶出率均达 90%(调释制剂为 85%)以上、且 差值在 5%以内时,试验则可提前结束。 对于结束时间点,在酸性介质中(如 pH 值 1.0)最长测定时间为 2 小时,在其他各 pH 值介质中普通制剂为 6 小时,缓控释制剂为 24 小时。
2. 其他事项 (1) 试验样品 用于比较的两种制剂含量差值应在 5%以内;每个品种各取 12 个单位。 取三个批号样品,在最终溶出率均可达 90%以上的溶出介质
(2) 参比制剂标准批号的选择
中,取溶出率在约 70%处、位于中间批号的样品进行试验。 在进行仿制药研发时,考虑到原研品批间差异与耐受性,建议从市场流通渠道获得有效 期内不同时间段的 3~5 批样品,分别测定后,取结果均值用于比较;并同时确定参比制剂在 各 pH 值溶出曲线的波动范围,以更为有效地评估原研制剂内在质量和自身仿制制剂的研发 深入程度。 如果主成分是在溶解状态下进行溶出度试验的(如一些散剂、颗粒剂) ,则适当选择某一 批号,即可。 (3) 试验样品的生产规模 由于固体制剂生物利用度与生产规模密切相关, 故一般情况下应不
少于今后工业化最大生产规模的 1/10 或不少于 10 万个单位。
1
附件2 普通口服固体制剂溶出曲线 测定与比较指导原则 一、概述 为进一步推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临
床疗效差异的风险。 三、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装臵、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50~75转/分钟,篮法选择50~100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择 应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。
普通口服固体制剂溶出曲线测定与比较指导原则 (2015-11-09 16:15:30) 分类: 普通口服固体制剂溶出曲线测定与比较指导原则 一、概述 为进一步推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出
曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临 床疗效差异的风险。 三、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50~75转/分钟,篮法选择50~100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择
美国和日本溶出曲线相似性判定方法介绍 来源:中国食品药品检定研究院 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出以及在胃肠道的渗透。由于药物的溶出对吸收具有重要影响,因此药物体外溶出度试验可能会与体内行为具有一定关联。 对于仿制药而言,与原研制剂体外溶出曲线具有相似性,虽然不能完全证明与原研制剂具有相同的生物等效性,但却可以大大提高生物等效性试验( BE 试验) 的成功率,而体外溶出曲线不相似,BE 试验的失败率将大大提高。 目前国外已有相关指导原则用于溶出曲线试验的指导。本文主要对美、日有关仿制药指导原则中溶出曲线相似性方法内容进行介绍,希望通过对两者的解读,能为我国仿制药质量一致性评价固体口服制剂体外评价方法提供借鉴。 1、美国溶出曲线相似性判定方法 FDA 在1997 年发布的普通口服固体制剂溶出度试验技术指导原则中,采用非模型依赖法和模型依赖法进行溶出曲线的比较。 1.1非模型依赖法( Model Independent Approaches) 差异因子( f1) 和相似因子( f2) 是一种简单的模型非依赖方法用于溶出曲线的比较{ A simple model independent approach uses a difference factor ( f1) and asimilarity factor( f2) to compare dissolution profiles}。差异因子( f1) 法是计算两条溶出曲线在每一时间点差异,是衡量两条曲线相对偏差的参数,计算公式如下: 其中n 为取样时间点个数,Rt为参比制剂( 或变更前产品) 在t 时刻的溶出度值,Tt为试验批次( 变更后样品) 在t 时刻的溶出度值。 相似因子( f2) 是衡量两条溶出曲线相似度的参数,计算公式如下: 其中n 为取样时间点个数,Rt为参比制剂( 或变更前产品,后面统称为参比制剂) 在t 时刻的溶出度值,Tt为试验批次( 变更后样品) 在t 时刻的溶出度值。 1.1.1差异因子和相似因子应用条件 ①分别取受试制剂和参比制剂各12 片( 粒) ,测定其溶出曲线。取12 片( 粒) 进行测定是其统计学计算所必须的最小单位。
普通口服固体制剂溶出曲线 测定与比较指导原则 一、概述 为进一步推进仿制药与原研药品质量和疗效一致性评价工作的开展,根据《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)要求,制定本指导原则。 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 二、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临床疗效差异的风险。
三、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪 溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50~75转/分钟,篮法选择50~100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择 应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。
采用f2因子法评价溶出曲线的相似性需注意的问题 发布日期20070806 栏目化药药物评价>>化药质量控制 审评四部审评八室马玉楠 在制剂的开发研究中,通过对比不同处方之间的溶出曲线,可以较准确地反映药物处方、工艺、生产场地及规模等因素变化对药物体外释放行为的影响。近年来,国外针对溶出曲线的相似性评价方法报道很多,其中f2因子方法因为计算简单、判定结果可靠,作为评价体外溶出曲线相似性的方法,已经被美国FDA的CDER和欧盟EMEA收载并推荐使用。 F2因子的计算公式为: Rt与Tt分别代表参比和受试制剂第t时间点的平均累积释放度,n为测试点数。其中如果两条溶出曲线完全一致,则:f2=50×lg(100)=100;如果一批样品释药完全,而另一批尚未开始释药,则有:。 因此,f2的值的范围在0~100,而且f2越大,两条曲线的相似性越高。 事实上即使是相同处方的产品,其批次不同,在溶出曲线上也会有一定的差异。如果受试与参比制剂溶出曲线的差异不大于参比制剂间溶出曲线的差异,那么就可以认为受试与参比制剂溶出曲线具有相似性。通常认为,同一处方不同批次的样品,在任一取样点释放度的平均差异不超过10%,是可以接受的。将10%代入式中计算: 。因此,FDA与EMEA规定:若受试与参比制剂的溶出曲线间的f2值不小于50,则认为两者相似。 在某些情况下,如果对于任一取样点释放度的平均差异的限定不是10%,则可通过计算得出相应的f2值(临界值)。表1提供了一些释放度平均差异与相应的f2临界值。
表1 释放度平均差异与f2临界值表 Table 1 Average difference of drug release percent and f2 Limit 平均偏差(Average difference)2% 5% 10% 15% 20% F2临界值(f2 Limit)83 65 50 41 36 f2因子的应用条件及注意事项: 1.在进行参比与受试制剂的溶出曲线比较的过程中,时间点间隔无需相等,但两者所取各时间点必须一致,一般除0时外,选择3点以上,即n≥3。 2.f2计算公式只适用于受试与参比制剂的平均累积释放度差值<100时的溶出曲线比较(如果二者的差值>100,就会得到一个负值),普通口服制剂要保证药物溶出90%以上,缓释制剂、肠溶制剂药物释放需达到80%以上,或达到释放平台。 3.受试与参比制剂释放曲线上各时间点的平均累积释放度差异,在平台区达到最小(如果外推到释放100%,差值将为0),在该区域上取样点的增加会直接导致f2值偏大。因此,受试或参比制剂的药物累积释放度在85%以上的取样点应不多于一个,否则,将会给判定结果带来误差。 4.f2因子比较一般选择每个处方的12个剂量单位的测定均值来进行处理。因为不考虑参比和受试制剂批内样本间差异,所以若参比或受试制剂批内样本间差异较大时,用f2因子来评价两者溶出曲线的相似性时需要谨慎,从第2个时间点至最后1个时间点溶出结果的变异系数应小于10%。 5.f2值与平均偏差之间成非线性关系,它只适用于描述参比与受试制剂溶出曲线的相似性,而不能用于评价受试制剂样本间差异。 总之,f2因子法作为定量描述制剂体外溶出曲线相似性的非模型依赖方法,简单易行、结果可靠。当药品处方、生产工艺、生产地点和生产规模等发生变更后,溶出度检查是比较变更前后制剂产品相似性或差异程度的重要工具和研究工作的重要内容,同时该方法也为口服固体制剂的处方筛选,产品质量控制、生物等效性评价等提供了有力的判定依据。在进行f2因子比较试验时要特别注意样本量、样本批间差异、溶出取样点等是否满足条件,以保证数据的可靠性。
普通口服固体制剂溶出曲 线测定与比较指导原则 The latest revision on November 22, 2020
附件2 普通口服固体制剂溶出曲线测定与比较 指导原则 本指导原则适用于仿制药质量一致性评价中普通口服固体制剂溶出曲线测定方法的建立和溶出曲线相似性的比较。 一、背景 固体制剂口服给药后,药物的吸收取决于药物从制剂中的溶出或释放、药物在生理条件下的溶解以及在胃肠道的渗透等,因此,药物的体内溶出和溶解对吸收具有重要影响。 体外溶出试验常用于指导药物制剂的研发、评价制剂批内批间质量的一致性、评价药品处方工艺变更前后质量和疗效的一致性等。 普通口服固体制剂,可采用比较仿制制剂与参比制剂体外多条溶出曲线相似性的方法,评价仿制制剂的质量。溶出曲线的相似并不意味着两者一定具有生物等效,但该法可降低两者出现临 床疗效差异的风险。 二、溶出试验方法的建立 溶出试验方法应能客观反映制剂特点、具有适当的灵敏度和区分力。可参考有关文献,了解药物的溶解性、渗透性、pKa常数等理化性质,考察溶出装置、介质、搅拌速率和取样间隔期等试验条件,确定适宜的试验方法。 (一)溶出仪
溶出仪需满足相关的技术要求,应能够通过机械验证及性能验证试验。必要时,可对溶出仪进行适当改装,但需充分评价其必要性和可行性。 溶出试验推荐使用桨法、篮法,一般桨法选择50—75转/分钟,篮法选择50—100转/分钟。在溶出试验方法建立的过程中,转速的选择推荐由低到高。若转速超出上述规定应提供充分说明。 (二)溶出介质 溶出介质的研究应根据药物的性质,充分考虑药物在体内的环境,选择多种溶出介质进行,必要时可考虑加入适量表面活性剂、酶等添加物。 1.介质的选择 应考察药物在不同pH值溶出介质中的溶解度,推荐绘制药物的pH-溶解度曲线。 在确定药物主成分稳定性满足测定方法要求的前提下,推荐选择不少于3种pH值的溶出介质进行溶出曲线考察,如选择pH值1.2、4.5和6.8的溶出介质。对于溶解度受pH值影响大的药物,可能需在更多种pH 值的溶出介质中进行考察。推荐使用的各种pH值溶出介质的制备方法见附件。 当采用pH7.5以上溶出介质进行试验时,应提供充分的依据。水可作为溶出介质,但使用时应考察其pH值和表面张力等因素对药物及辅料的影响。 2.介质体积 推荐选择500ml、900ml或1000ml。 (三)溶出曲线的测定