
开环易位聚合的研究进展
摘要:本文综述了开环易位聚合(Ring-opening metathesis polymerization,ROMP)的研究进展,详述了研究者们合成新的开环易位聚合催化剂的研究工作和利用开环易位聚合制得具有优异性能的聚合物的研究工作。
关键词:开环易位聚合;催化剂;降冰片烯及其衍生物
引言
开环易位聚合( Ring-opening metathesis polymerization, 简写为ROMP) 反应由于具有活性聚合的特点, 已经得到越来越多的关注[1]。ROMP的起源可以追溯到20世纪50年代中期。近年来,研究者们证明了很多烯烃易位反应的中间体,使得ROMP技术得到了广泛的普及和应用。新型的活性ROMP催化剂的研究及发展,使得这种活性聚合反应可以在常温、常压等温和条件下进行,这给该方法增添了新的活力。目前, 该领域的研究主要集中在合成高效的ROMP反应催化剂[2]和基于ROMP反应制备多功能的新材料[3]等方面。
1.烯烃易位反应简介
2005年,法国石油学院的伊夫·肖万(Y. Chauvin)、美国麻省理工学院的罗伯特·格拉布(Robert H. Grubbs)和加利福尼亚州加州理工学院的理查德·施罗克(Richard R. Schrock)三位科学家获得了诺贝尔化学奖。现在,越来越多的结构明确、稳定高效的催化剂被合成,使得烯烃易位反应能够和传统的碳-碳键的形成的合成方法相媲美。因此,与烯烃易位反应相关的研究已成为化学界极为重要的课题。
1.1烯烃易位反应基本概念
易位反应是指两种物质互相交换成分生成两种新的物质的反应。例如:AB+CD →AC+BD。同样的,两种烯烃互相交换双键两端的基团,从而生成两种新的烯烃的反应便是烯烃易位反应。更直观的表示如图1.1:
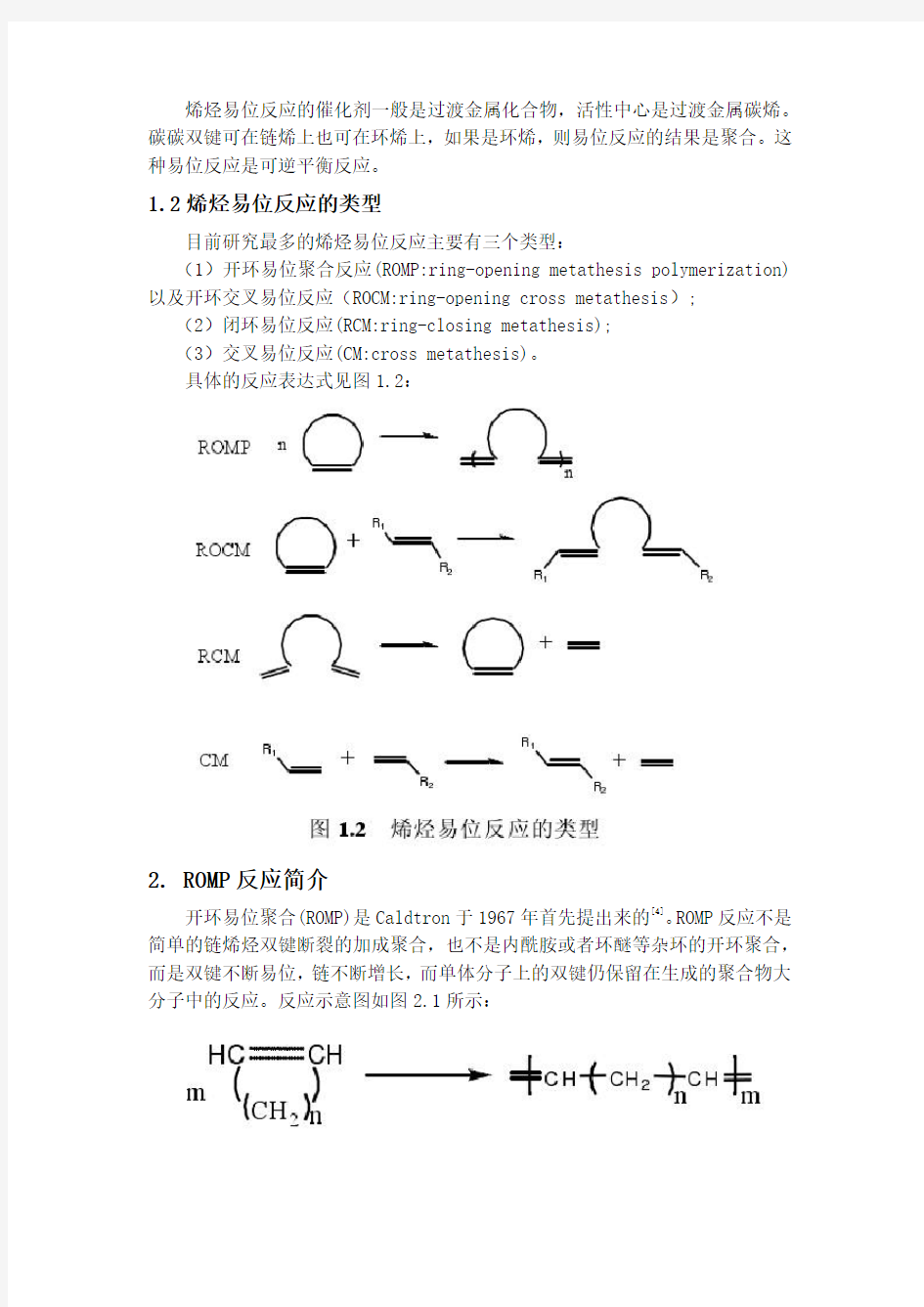
烯烃易位反应的催化剂一般是过渡金属化合物,活性中心是过渡金属碳烯。碳碳双键可在链烯上也可在环烯上,如果是环烯,则易位反应的结果是聚合。这种易位反应是可逆平衡反应。
1.2烯烃易位反应的类型
目前研究最多的烯烃易位反应主要有三个类型:
(1)开环易位聚合反应(ROMP:ring-opening metathesis polymerization)以及开环交叉易位反应(ROCM:ring-opening cross metathesis);
(2)闭环易位反应(RCM:ring-closing metathesis);
(3)交叉易位反应(CM:cross metathesis)。
具体的反应表达式见图1.2:
2. ROMP反应简介
开环易位聚合(ROMP)是Caldtron于1967年首先提出来的[4]。ROMP反应不是简单的链烯烃双键断裂的加成聚合,也不是内酰胺或者环醚等杂环的开环聚合,而是双键不断易位,链不断增长,而单体分子上的双键仍保留在生成的聚合物大分子中的反应。反应示意图如图2.1所示:
图2.1 ROMP反应示意图
ROMP反应条件比较温和,反应的速率很快。大多数情况下,ROMP反应中几乎没有链转移反应和链终止反应,因此它是一种活性聚合。利用ROMP反应可制得许多有特殊结构的新型官能化的聚合物。近年来,研究者们利用ROMP反应已研究出了一大批具有优异性能的新型的高分子材料,比如聚双环戊二烯、聚降冰片烯和聚环辛烯等,并且上述三种产品已经进行工业化生产。因此,ROMP已经成为制备高分子材料的重要的聚合方法之一。
2.1 ROMP反应的单体
环烯烃是否可以进行ROMP反应可以通过热力学来判断。在ROMP中,聚合热焓主要来自环张力能的释出,所以环的张力能是决定能否进行开环易位聚合的主要因素。环的张力越大,单体越活泼。单体可以是单环烯,如环丁烯、环辛烯、环戊烯和环庚烯等。单体也可以是双环烯,如降冰片烯及其衍生物,这也是目前研究最多的单体[5-6]。单体还可以是三环烯。单环烯与双环烯共聚可以制得高度交联的体形聚合物。
2.2 ROMP反应的催化剂
按照发展的进程,一般将催化剂分为如下三类:
(1)传统催化剂这是目前研究的最透彻的一类催化剂[7]。从20世纪50年代中期直到80年代早期,烯烃易位反应都是通过有限控制结构的多组分均相与异相催化体系的催化完成的。这些体系都是由过渡金属络合物吸附于惰性金属氧化物上而组成。一些经典的催化剂如WCl6/Bu4Sn,WOCl4/EtAlCl2,MoO3/SiO2和Re2O7/Al2O3等。因为这些催化体系的成本比较低而且容易制备,所以它们在烯烃易位聚合反应的商业应用中有着非常重要的地位。
(2)水溶性催化剂主要代表是K2RuCl3·H2O,其主要应用于2,3-双官能度取代的降冰片烯和7-氧化降冰片烯的聚合。
(3)卡宾型催化剂也称为碳烯型或亚烷基型催化剂,其催化活性高,是目前研究和发展的最重要的ROMP催化剂。类型有六种,如图2.2所示:
图2.2 卡宾型催化剂
在这类催化剂中,最重要的且应用最多的是Schrock催化剂[8]和Grubbs催化剂[9]。
目前的很多研究工作都致力于合成新型的效率更高的催化剂。
谢美然等[10]合成了离子液体负载的钌催化剂, 考察了该催化剂在离子液体中对极性环烯烃单体的ROMP反应规律。首先合成含离子液体的吡啶配体1, 2-二甲基-3-己氧基吡啶六氟磷酸盐咪唑。然后利用其与Grubbs第二代催化剂配位反应,制备离子液体负载的钌催化剂。最后,通过1H,13C NMR等方法对合成的化合物和催化剂进行表征。催化剂中与钌连接的苯亚甲基上(Ru=CH—Ph) 氢的振动峰由原来Grubbs第二代催化剂中的δ19.2移至δ18.6, 这表明得到了新的催化剂,ICP测定催化剂混合物中纯催化剂的质量分数为36.2%。该催化剂易溶于丙酮、甲醇及咪唑类离子液体等极性溶剂,解决了Grubbs催化剂不溶于离子液体的问题,实现了在纯离子液体中的均相ROMP反应。
Kotohiro Nomura等[11]研究了在[Ph3C][B(C6F5)4]存在的情况下,以V(NR)(CH2SiMe3)(OAr)2为催化剂的四氢呋喃的开环易位聚合,得到了低PDI值的高分子量的聚合物。PDI值低是因为起始效率受到催化剂配体丙烯酰胺和芳氧基的影响。25℃时,在PMe3存在的条件下,由V(NAd)(CH2SiMe3)2(OC6F5)通过正己烷α氢消除反应得到一个新的钒亚烷基V (CHSiMe3)(NAd)(OC6F5)(PMe3)2,展示了对降冰片烯的开环易位聚合反应的非凡的催化效率。即25℃时的催化活性比那些已经报道的钒亚烷基和传统的催化剂Mo(CHCMe2Ph)(N-2,6-i Pr2-C6H3)(O t Bu)2要高。
Danielle F. Sedbrook等[12]描述了一个二苯并环辛炔的开环易位聚合的非常好的引发剂。这种反应生成低分散性和可预测分子量的活性聚合物。通过酚醛
配体的σ电子取代激活已经广为人知的前催化剂[(N(tBu)Ar)3Mo≡CCH2CH3],证明了这种配体的螯合能力和传播中心钼的本质一样对聚合反应的结果产生了明显的影响。
2.3 ROMP的反应机理
由于催化剂组分之间的反应比较复杂,因此目前尚不能明确说明开环易位聚合的反应机理到底是什么样的[13-14]。研究者们对于开环易位聚合的反应机理的认识,主要有以下几种:
(1)置换反应的成对机理(由Calderson提出)如图2.3所示:
图2.3
(2)过渡金属环戊环中间物的机理(由Biefield提出)
(3)金属-碳烯配位化合物引发和增长的机理(由Herisson提出),这是目前普遍接受的一种机理,下述反应式可以清晰地反映出催化机理。
链引发和链增长反应活性中心为金属-碳烯,聚合反应机理如图2.4所示:
图2.4
绝大多数聚合情况如图2.5所示:
图2.5
3.应用
3.1 制备功能材料
随着研究的不断深入和发展,开环易位聚合已经获得了广泛的应用。如制备可控聚合物、合成光折变功能聚合物、制备纳米颗粒[15]等。
栗磊[16]利用酯化的方法,设计合成了叔丁基保护的降冰片烯衍生物单体。并且,使用活性ROMP方法使其聚合,得到了均聚物。研究了该聚合物的分子量及分子量分布,并且进行了脱保护研究,证实脱保护可以进行。在此基础上,使用该单体和亚胺单体进行共聚合,得到了嵌段共聚物。研究了两个单体聚合的先后顺序对得到的嵌段共聚物分子量及分子量分布的影响,阐明了该影响是保护基团的空间位阻原因造成的。对嵌段共聚物进行脱保护,得到了两亲性嵌段共聚物。利用选择性溶剂的方法对该两亲性嵌段共聚物进行自组装,得到了球形纳米材料。
王晋[17]将可聚合的降冰片烯官能团引入到含强极性次甲基生色团的咔唑类光折变功能化合物中,通过引入长的柔性侧基来改善聚合物玻璃化转变温度Tg,用Grubbs 2nd催化剂对所得到的单体进行开环易位聚合,得到降冰片烯类光折变聚合物。采用1H-NMR、IR、UV-vis、GPC等手段对所得聚合物进行了表征。实验结果显示,得到的聚合物的最大紫外吸收出现在410nm处,数均分子量为5.4×104。在此基础上通过改变生色团的种类,引入偶氮硝基苯生色团,合成了咔唑基含偶氮硝基苯生色团的降冰片烯类光折变功能单体。并分别使用Grubbs 1st,和Grubbs 2nd催化剂对所得单体进行聚合,得到两个聚合物P2和P2’。用1H-NMR、IR、UV-vis、DSC,TG及GPC等手段对单体及聚合物进行了表征。1H-NMR和UV-vis 显示所得各个化合物及聚合物为目标产物。DSC结果显示两种聚合物的玻璃化转变温度分别为75℃和43℃。热重分析结果表明聚合物具有良好的热稳定性。GPC 结果显示二者的数均分子量达到了104数量级。
Ki-Young Yoon等[18]报道了一个从由环辛四烯开环易位聚合反应合成得到的简单的聚乙炔二嵌段共聚物直接制备超分子一步路线法。这种成对聚合体的原地纳米粒子化比传统的自组装工序有优势,因为这种方法不需要任何沉闷的后期人工处理。而且,这种直接的方法提供了有趣的超分子的一种独一无二的跟毛虫类似的纳米结构。此外,这种新的超分子是非常稳定的生成物,因为聚乙炔核为自
组装提供了一个特别强的剥离力。尽管,原始的聚乙炔在空气中不稳定,但在纳米毛虫结构中是稳定的,因为被超分子壳保护着。
Zhen Yao等[19]采用第二代拉布催化剂通过开环易位聚合反应合成了二环戊二烯和环戊烯(CPE)的共聚物。研究了单体成分、催化剂浓度、反应温度、硬化时间以及单体转化率的影响。此外,反应采用注射成型工艺生产二环戊二烯和CPE的共聚物。研究发现二次硬化过程需要达到100%的单体转化率。讨论了CPE 的含量、催化剂的浓度以及二次硬化温度对热稳定性和力学性能的影响。共聚物中含有少量的CPE可以减少做TGA时的重量损失率并提高力学性能。然而过多的CPE会减少交联度导致强度的降低。
Wen-Yong Lai等[9]采用Grubbs第三代催化剂通过开环易位聚合合成含降冰片烯衍生物的有磷光的树枝状分子的聚合物。这个树枝状分子包含了含有两个2-苯基吡啶配体、一个苯基三唑基配体、联苯基树状分子和2-乙基己氧基表面基团的第三代铱复配物。苯三唑基配体提供了聚合物骨架的附着点,两种树枝状分子以不同数量的树状分子附着到2-苯基吡啶配体上。每个配体上含有一个或者两个树状分子的聚合物被称为单树枝化或者双树枝化。发现这样的树状分子含有相对狭窄的分散性,大约为1.4,粘度接近喷墨打印的要求,并且可以经过处理形成薄膜。这种树状分子在控制材料的光物理性能方面有重要的作用。母体聚合物中不含能附着配体的树状分子的荧光损失量为48%。增加单树枝化和双树枝化材料的树状分子的含量,发现荧光损失量分别增加到65%和71%。这种增加是因为链内的生色团相互作用降低了。观察到固态时的母体、单树枝化和双树枝化的聚合物的荧光损失分别为2%、44%和58%,证明了树枝分子对链内和链间的相互作用都能控制。
3.2 降冰片烯及其衍生物的研究
降冰片烯及其衍生物是研究开环易位聚合的最广泛的单体[20]。
杜创[21]用Grubbs 1st引发降冰片烯单体进行ROMP反应,研究了催化剂搅拌溶解时间、聚合反应中使用溶剂的极性和三苯基磷配体的加入对降冰片烯单体ROMP反应分子量及分子量分布的影响,得到降冰片烯单体ROMP反应的最佳条件。然后设计合成了N-propyl-norbornene-2,3-dicarboximide单体,得到了分子量可控的疏水聚合物。并且利用物理吸附方法将脂肪酶PSL成功地固定到该疏水聚合物上,研究了固定化PSL催化拆分手性2-辛醇反应的催化性质。第一次研究疏水聚合物分子量的不同对固定化PSL催化性质的影响。此外,当疏水聚合物的分子量大约是40kDa时,得到了固定化PSL催化拆分手性2-辛醇的最优化条件。此外还设计合成了含有聚乙二醇侧链的大分子单体,制备了分子量可控的梳形接枝聚合物,研究了它的水溶性。使用该大分子单体和降冰片烯进行共聚,得到了两亲性梳形嵌段共聚物。研究了单体聚合的先后顺序等因素对得到的嵌段共聚物分子量及分子量分布的影响。使用该两亲性梳形嵌段共聚物进行自组装,研究了自组装纳米粒子的形貌及尺寸。并且,初步的将该两亲性梳形嵌段共聚物应用在药物的承载和缓释等领域。
张丹枫等[22]以一系列含氧、含烯丙基等的取代二氯二茂钛络合物 CpCp′TiCl2(Cp′为取代的环戊二烯基)与甲基锂CH3Li 组成的催化体系对降冰片烯的ROMP进行了研究, 结果发现茂环上取代基的极性和大小对催化体系的活性有很大影响, 但对聚合物结构主链中双键顺反含量的影响不是很大。
陈凤香等[23]设计合成了含氧化乙烯重复单元和咪唑盐侧基的降冰片烯类单体,用Grubbs催化剂(PCy3)2Ru(CHPh)Cl2(1)和(SIMes) (PCy3)Ru(CHPh)Cl2(2)对单体进行了开环易位聚合(ROMP)。考察了单体在不同的条件下(溶剂、温度等)的聚合反应,特别是在离子液体中的ROMP的聚合特征。结果表明,2的催化活性比1的高;2催化单体在有机溶剂中聚合所得聚合物的分子量不可控,而在离子液体[BMIm]PF6中能够顺利进行均相聚合反应,且对聚合物的分子量可控性较好。用核磁共振谱(NMR)对合成的单体及聚合物的结构进行了表征。用差示扫描量热法(DSC)测定聚合物的玻璃化转变温度为-17.34℃,采用循环伏安法测得聚合物的电化学稳定窗口为3.0 V。
董薇等[24]先用酯化的方法合成了含有聚乙二醇(PEG)的降冰片烯大分子单体,再用开环易位聚合方法使大分子单体聚合,得到了PEG取代的聚降冰片烯接枝共聚物。并通过凝胶渗透色谱法(GPC)研究合成的接枝共聚物分子量及分子量分布情况。结果表明: 聚合物的数均分子量为1.0万-4.4万;分子量分布为1.11-1.22,并且聚合物的分子量分布随[M]/[I]的增加而变窄。
4.结语
开环易位聚合反应条件温和且反应速率快,新的催化剂的开发使得一些复杂分子的合成变得容易,并且符合“绿色化学”的要求,所以ROMP反应已经成为高分子合成领域中强有力的武器。
参考文献
[1] 陈凤香. 离子液体负载钌络合物的合成及其催化环烯烃开环易位聚合[D].华东师范大
学硕士学位论文,2007,6.
[2] Mohammed Al-Hashimi,Chayanant Hongfa,Beena George,et al. A Phase-Separable Seco nd-Generation Hoveyda-Grubbs Catalyst for Ring-Opening Metathesis Polymerization[J]. JOU RNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2012, 50:3954–3959.
[3] Amit A. Nagarkar, Aurelien Crochet, Katharina M. Fromm,et al. Efficient Amine End-F unctionalization of Living Ring-Opening Metathesis Polymers[J]. Macromolecules, 2012, 45: 4447? 4453.
[4] 张留成,闫卫东,王家喜.高分子材料进展[M].化学工业出版社,2005,6.
[5] Kyung-Hwan Yoon, Kyung Oh Kim, Mark Schaefer,et al. Synthesis and characterization of hydrogenated poly(norbornene endo-dicarboximide)s prepared by ring opening metathe-sis polymerization[J]. Polymer 2012, 53:2290-2297.
[6] Anne-Claire Le Meur,Cyril Aymonier,Valerie Heroguez. Crosslinked Polynorbornene Parti cles Synthesis by Ring-Opening Metathesis Polymerization in Dispersion[J]. JOURNAL OF P OLYMER SCIENCE PART A: POLYMER CHEMISTRY 2012, 50:1746–1754.
[7] Margaret M. Flook, Janna Bo ?r ner, Stefan M. Kilyanek,et al. Five-Coordinate Rearrangem ents of Metallacyclobutane Intermediates during Ring-Opening Metathesis Polymerization o f 2,3-Dicarboalkoxynorbornenes by Molybdenum and Tungsten Monoalkoxide Pyrrolide Initi ators[J]. Organometallics 2012, 31:6231? 6243.
[8] Rajendar Bandari, Michael R. Buchmeiser. Functional Monolithic Materials for Boronat eAffi nity Chromatography via Schrock Catalyst-Triggered Ring-Opening Metathesis Polymeri zation[J]. Macromol. Rapid Commun. 2012, 33:1399?1403.
[9] Wen-Yong Lai,Michael N. Balfour,Jack W. Levell,et al. Poly(dendrimers) with Phosphore scent Iridium(III) Complex-Based Side Chains Prepared via Ring-Opening Metathesis Polyme rization[J]. Macromolecules 2012, 45:2963? 2971.
[10] 谢美然,马卓,韩会景等. 含吡啶配体的钌催化剂合成及在离子液体中开环易位聚合反应[J].高等学校化学学报,2009,30(2):396-402.
[11] Kotohiro Nomura, Ken Suzuki,Shohei Katao,et al.Ring-Opening Polymerization of THF by Aryloxo-Modified(Imido)vanadium(V)-alkyl Complexes and Ring-Opening Metathesis Poly merization by Highly Active V(CHSiMe3)(NAd)(OC6F5)(PMe3)2[J]. Organometallics,2012,31:511 4?5120
[12] Danielle F. Sedbrook,Daniel W. Paley,Michael L. Steigerwald,et al. Bidentate Phenoxid es as Ideal Activating Ligands for Living Ring-Opening Alkyne Metathesis Polymerization[J]. Macromolecules 2012, 45:5040? 5044.
[13] 焦书科. 环烯烃开环聚合[J]. 石油化工,1981,10(9):625-633.
[14] 胡炳镛,陈德铨. 环烯烃开环聚合易位反应机理[J]. 合成橡胶工业,1998,7(5):3 93-397.
[15] Lixia Ren, Jiuyang Zhang, Christopher G. Hardy,et al.Cobaltocenium-Contai ning Block Copolymers: Ring-Opening Metathesis Polymerization, Self-Assembly an d Precursors for Template Synthesis of Inorganic Nanoparticles[J].Macromol. Ra pid Commun. 2012, 33:510?516.
[16] 栗磊. 开环易位聚合方法制备两亲性嵌段共聚物[D].吉林大学硕士学位论文,2009,5.
[17] 王晋. 开环易位聚合反应合成光折变功能聚合物[D].郑州大学硕士学位论文,2011,6.
[18] Ki-Young Yoon,In-Hwan Lee,Kyung Oh Kim,et al. One-Pot in Situ Fabrication of Stabl
e Nanocaterpillars Directly from Polyacetylene Diblock Copolymers Synthesized by Mild Ri ng-Opening Metathesis Polymerization[J].J. Am. Chem. Soc. 2012, 134: 14291? 14294. [19] Zhen Yao,Li-wu Z hou,Bin-bin Dai,et al. Ring-Opening Metathesis Copolymerization o
f Dicyclopentadiene and Cyclopentene Through Reaction Injection Moldin
g Process[J]. Wiley
Periodicals, Inc. J Appl Polym Sci 125, 2012:2489–2493.
[20] Kyung-Hwan Yoon,Kyung Oh Kim,Chengqing Wang,et al. Synthesis and Structure–Prop erty Comparisons of Hydrogenated Poly(oxanorbornene-imide)s and Poly(norbornene-imide) s Prepared by Ring-Opening Metathesis Polymerization[J]. JOURNAL OF POLYMER SCIENCE PART A: POLYMER CHEMISTRY 2012, 50:3914–3921.
[21] 杜创.开环易位聚合方法制备可控聚合物及其应用[D].吉林大学博士学位论文,2008,5.
[22] 张丹枫.降冰片烯及其衍生物开环易位聚合的研究进展[J].高分子材料科学与工程,2000,16(1):13-15.
[23] 陈凤香,史佳鑫,韩会景等.7-氧杂降冰片烯的官能化及其开环易位聚合[J].高分子学报,2007,4:383-388.
[24] 董薇,张秋平,杜创等.降冰片烯大分子单体开环易位聚合制备PEG取代的接枝共聚物[J].吉林大学学报,2012,50(3):567-570.
原子转移自由基聚合及其应用新进展 原子转移自由基聚合(ATRP),是近几年迅速发展并有着重要应用价值的一种活性聚合技术。自从1956 年Szwarc[1]等报道了一种没有链转移和链终止的负离子聚合技术以来,活性聚合的研究性得到了巨大的发展,并一直是高分子学术界高度重视的领域。1983年Webster等[2]成功地实现了适用于丙烯酸酯类单体的基团转移聚合。随后又成功的实现了开环聚合[3]、活性正离子聚合[4,5]、络合负离子聚合[6] 以及无金属离子的活性负离子聚合[7]。1993年Xerox公司在苯乙烯的普通自由基聚合体系中加入有机自由基捕捉剂(Tempo体系)[8],使反应体系在聚合过程中自由基保持较低的浓度,从而抑制了自由基的副反应。第一次实现了" 活性"自由基聚合。与此同时,1995年《美国化学会志》报道了CarnegieMellon大学Matyjaszewski教授和王锦山博士共同开发的原子转移自由基聚合(ATRP)[9],成功地实现了真正意义上的"活性"/可控自由基聚合,取得了活性自由基聚合领域的历史性突破。 1. ATRP基本原理 ATRP的基本原理如Figure 1.1所示: Figure 1.1 Mechanism of atom transfer radical polymerization
式中,R-X是引发剂卤代烃(X-般为Cl或Br),M t n为过渡金属络合物,它由过渡金属离子和配位剂构成。在引发阶段,处于低氧化态的过渡金属络合物(盐)M t n从一有机卤化物-X中夺取卤原子X,生成引发自由基R·及处于高氧化态的金属络合物(盐) M t n + 1 -X。R·引发可给出卤原子X,即M t n + 1-X 与R·/R-M·发生减活反应生成R-X/R-M-X。如果R-Mn-X (n = 1, 2, ...)与R-X-样可与M t n发生促活反应生成相应的R-Mn及M t n + 1-X,同时若R-Mn·与M t n + 1-X又可反过来发生减活反应生成R-Mn-X及M t n,在自由基聚合反应进行的同时,就会始终伴随着一个自由基活性种Mn·与有机大分子卤化物休眠种Mn-X的可逆转换平衡反应。卤原子的可逆转移控制着[Mn·],而一个快速的卤原子转换速率将控制着分子量及分子量分布。图示表明:ATRP的基本原理其实是通过一个交替的“活化—去活”可逆反应使得体系中游离基浓度处于极低,迫使不可逆终止反应被降低到最低程度,而链增长反应仍可进行,从而实现“活性”聚合[10]。由于在这种聚合反应中,只是将自由基活性种的浓度加以控制,链终止和链转移被极大地抑制了,所以这种聚合反应只能是可控聚合或“活性”聚合,而不是真正的活性聚合。同时,在这种可控聚合反应中包含着卤原子从卤化物到金属络合物(盐)、再从金属卤化物转移到自由基这样一个反复循环的原子转移过程,加之反应活性种为自由基,所以称为原子转移自由基聚合。由于已有实验证明某些基团也可发生类似的转移自由基反应,故王锦山等把这样一种反应称为“原子(基团)转移自由基聚合”[11]。 ATRP研究大致可以分成两个体系:一个是美国Carnegie-Mellon
基团转移聚合 摘要:介绍了基团转移聚合的机理和近几年来基团转移聚合在聚合单体、引发剂、催化剂、 聚合实施方法等方面的新进展。基团转移聚合技术在各种特定结构高分子合成中起到重要作用。 关键词:基团转移聚合,活性,单体,引发剂,催化剂 自1956年提出活性聚合的概念以来,活性聚合技术得到迅速发展。活性聚合技术的发展为合成结构和组成可控的聚合物材料提供了可能,使聚合物材料的应用范围进一步扩大,成为21世纪材料科学发展的基础。目前,活性聚合方法主要有活性阴离子聚合、阳离子聚合、自由基聚合、配位聚合和基团转移聚合等[1-2]。 1983年美国杜邦公司的O.w.webster在美国化学会186次会上宣读了“基团转移聚合反应”(Group Transfer Polymerization简称GTP)“,立即在世界上引起强烈反应[3]。GTP它是以α、β—不饱和酯、酮、酰胺和腈为单体,以带有硅、锗、锡烷基基团的化合物为引发剂,用阴离子型或路易斯酸型化合物作催化剂,选用适当的有机物作溶剂,通过催化剂与引发剂端基的硅、锗、锡原子配位,激发硅、锗、锡原子,使之与单体的羰基氧或氮结合成共价键单体中的双键完成加成反应,硅、锗、锡烷基基团移至末端形成“活性”化合物。以上过程反复进行,得到相应的聚合物。此项反应技术被认为是继本世纪50年代Ziegler 发现用配位催化剂使烯烃定向聚合之后的又一重要的新聚合技术。 GTP在这短短的几年里发展迅速,在控制聚合物分子量、分子量分布、端基官能化和反应条件等方面比通常的聚合方法具有更多的优越性,从而为“高分子的分子设计”又增添了新内容;同时已引用此种技术生产汽车面漆、合成液晶聚合物和一些特殊的聚合物,如嵌段、遥爪型高分子材料等。可以预料,在不久的将来,GTP技术会取得更大的进展[4-5]。 一、GTP反应机理 目前基团转移聚合中研究得最多的单体是甲基丙烯酸甲酯(MMA)和丙烯酸乙酯(EA)。由于MMA的活性最大,因此研究得更为深入。下面就以MMA为单体,乙烯酮的硅烷缩醛类化合物为引发剂来介绍反应机理。 1.链引发 亲核性阴离子催化剂Nu-先与引发剂或活性聚合物中活性端基上的硅原子配位,使硅原子活化,然后活化的硅原子与单体中羧基氧原子相连形成六配位硅的中间过渡态,最后形成中间体的端基上连着活性基团。引发剂与单体进行如下Michael加成反应,在端基上重新生成一个三甲基硅氧基和一个双键[6]。
第七章开环聚合 一、课程主要内容 开环聚合概述;开环聚合的特征;环状单体的种类及其聚合能力;环醚的开环聚合。 对第七章内容作一般了解。 二、习题与答案 本章试题有基本概念题、填空题和简答题。 ㈠基本概念题 ⒈开环聚合:开环聚合是指具有环状结构的单体经引发聚合后将环打开形成高分子化合物的一类聚合反应。 ⒉环醚:环中含有醚键-O-的环状化合物称为环醚。 ⒊环氧化合物:三节环醚又称为环氧化合物或氧化烯,如环氧乙烷又称为氧化乙烯,环氧丙烷又称为氧化丙烯。 ⒋环缩醛:在环中含有-CH2O-基团的环状化合物。 ⒌内酯:环中含有酯基-COO-的环状化合物,称为内酯或环酯。 ⒍内酰胺:环中含有酰胺基-CONH-的环状化合物,称为内酰胺或环酰胺。 ㈡填空题 ⒈环状类单体主要有环醚类、环酰胺类、环酯类和环烯烃等几类。 ⒉环醚单体主要有环氧乙烷、环氧丙烷、氧杂环丁烷、3,3’-二氯甲基氧杂环丁烷、和四氢呋喃等。 ⒊环氧化合物有环氧乙烷、环氧丙烷和环氧氯丙烷等。 ⒋环缩醛有三聚甲醛和四聚甲醛。 ⒌含硅的环状化合物有1,1’,3,3’,-四甲基-1.3-二硅环丁烷和2,2’,4,4’,6,6’,8,8’-八甲基 -2,4,6,8-四硅氧杂环辛烷(D4) 。 ㈢简答题 ⒈写出开环聚合反应简式、聚合机理,并写出环氧丙烷开环聚合的聚合反应简式? 答案: 开环聚合反应简式可表示如下 n R [ R Z ]n Z 在环状单体中,R为烷基,Z为杂原子:O,S,N,P,Si或-CONH-,-COO-,-CH=CH-基团等。 绝大多数环状单体的开环聚合是按离子型聚合机理进行的,有少数环状单体的开环聚合是按水解聚合机理进行的。 环氧丙烷进行开环聚合的聚合反应简式为
第六章离子聚合 一、名称解释 1. 阳离子聚合:增长活性中心为带正电荷的阳离子的连锁聚合。 2. 活性聚合:当单体转化率达到100%时,聚合仍不终止,形成具有反应活性聚合物(活性聚合物)的聚合叫活性聚合。 3. 化学计量聚合:阴离子的活性聚合由于其聚合度可由单体和引发剂的浓度定量计算确定,因此也称为化学计量聚合。 4. 开环聚合:环状单体在引发剂作用下开环,形成线形聚合物的聚合反应。 5. Ziegler-Natta引发剂:Zigler-Natta引发剂是一大类引发体系的统称,通常有两个组份构成:主 引发剂是Ⅳ~Ⅷ族过渡金属化合物。共引发剂是Ⅰ~Ⅲ族的金属有机化合物。 6. 配位聚合:单体与引发剂经过配位方式进行的聚合反应。具体的说,采用具有配位(或络合)能力的引发剂、链增长(有时包括引发)都是单体先在活性种的空位上配位(络合)并活化,然手插入烷基—金属键中。配位聚合又有络合引发聚合或插入聚合之称。 7. 定向聚合:任何聚合过程(包括自由基、阳离子、阴离子、配位聚合)或任何聚合方法(如本体、悬浮、乳液和溶液等),只要它是经形成有规立构聚合物为主,都是定向聚合。定向聚 合等同于立构规整聚合。 二、选择题 1. 下列单体中哪一种最容易进行阳离子聚合反应---------------------------------------------( B ) A.CH2=CH2B.CH2=CHOCH3C.CH2=CHCl D.CH2=CHNO2 2. 下列哪种物质不能作为阳离子聚合的引发剂------------------------------------------------(B ) A.正碳离子盐B.有机碱金属C.质子酸D.Lewis酸 3. 四氢呋喃可以进行下列哪种聚合---------------------------------------------------------( C ) A.自由基聚合B.阴离子聚合C.阳离子聚合D.配位聚合 4. 在无终止的阴离子聚合中,阴离子无终止的原因是(C ) A 阴离子本身比较稳定 B 阴离子无双基终止而是单基终止 C 从活性链上脱出负氢离子困难 D 活化能低,在低温下聚合 5. 合成聚合物的几种方法中,能获得最窄相对分子质量分布的是( A ) A 阴离子聚合 B 阳离子聚合 C 自由基聚合D自由基共聚合 6. 能引发苯乙烯阴离子活性聚合,并且聚合度等于两倍的动力学链长的是(D) A. BuLi B. AIBN C. AlCl3+H2O D. 萘+钠 7. 制备分子量分别较窄的聚苯乙烯,应该选择(B) A阳离子聚合B阴离子聚合反应C配位聚合反应D自由基聚合反应
开环易位聚合的研究进展 摘要:本文综述了开环易位聚合(Ring-opening metathesis polymerization,ROMP)的研究进展,详述了研究者们合成新的开环易位聚合催化剂的研究工作和利用开环易位聚合制得具有优异性能的聚合物的研究工作。 关键词:开环易位聚合;催化剂;降冰片烯及其衍生物 引言 开环易位聚合( Ring-opening metathesis polymerization, 简写为ROMP) 反应由于具有活性聚合的特点, 已经得到越来越多的关注[1]。ROMP的起源可以追溯到20世纪50年代中期。近年来,研究者们证明了很多烯烃易位反应的中间体,使得ROMP技术得到了广泛的普及和应用。新型的活性ROMP催化剂的研究及发展,使得这种活性聚合反应可以在常温、常压等温和条件下进行,这给该方法增添了新的活力。目前, 该领域的研究主要集中在合成高效的ROMP反应催化剂[2]和基于ROMP反应制备多功能的新材料[3]等方面。 1.烯烃易位反应简介 2005年,法国石油学院的伊夫·肖万(Y. Chauvin)、美国麻省理工学院的罗伯特·格拉布(Robert H. Grubbs)和加利福尼亚州加州理工学院的理查德·施罗克(Richard R. Schrock)三位科学家获得了诺贝尔化学奖。现在,越来越多的结构明确、稳定高效的催化剂被合成,使得烯烃易位反应能够和传统的碳-碳键的形成的合成方法相媲美。因此,与烯烃易位反应相关的研究已成为化学界极为重要的课题。 1.1烯烃易位反应基本概念 易位反应是指两种物质互相交换成分生成两种新的物质的反应。例如:AB+CD →AC+BD。同样的,两种烯烃互相交换双键两端的基团,从而生成两种新的烯烃的反应便是烯烃易位反应。更直观的表示如图1.1:
功能高分子材料 ▲1、什么是功能高分子?什么是特种高分子?两者的区别和关系如何? (1)功能高分子:是指当有外部刺激时,能通过化学或物理的方法做出相应输出的高分子材料。 功能高分子材料是指既有传统高分子材料的机械性能,又有某些特殊功能的高分子材料。 (2)特种高分子材料:是指带有特殊物理、力学、化学性质和功能的高分子材料,其性能和特征都大大超出了原有通用高分子材料的范畴。 (3)功能高分子属于特种高分子材料的范畴。特种高分子材料可细分为功能高分子和高性能高分子两类。 ▲2、功能和性能有什么区别?功能高分子和高性能高分子有什么不同? (1)性能:材料对外部作用的抵抗特性。(2)功能:指从外部向材料输入信号时,材料内部发生质和量的变化而产生输出的特性。 (3)功能高分子:是指当有外部刺激时,能通过化学或物理的方法做出相应输出的高分子材料。 (4)高性能高分子:是对外力有特别强的抵抗能力的高分子材料。 (从实用的角度看,对功能材料来说,人们着眼于它们所具有的独特的功能; 而对高性能材料,人们关心的是它与通用材料在性能上的差异。) 3B、功能高分子材料的类型 (1)力学功能材料:①强化功能材料,②弹性功能材料。 (2)化学功能材料:①分离功能材料,②反应功能材料,③生物功能材料。 (3)物理化学功能材料:①耐高温高分子,②电学功能材料,③光学功能材料,④能量转换功能材料。 (4)生物化学功能材料:①人工脏器用材料,②高分子药物,③生物分解材料。 这一分类,实际上包括了所有特种高分子材料。国内一般采用按其性质、功能或实际用途划分为8种类型。 (1)反应性高分子材料,(2)光敏型高分子,(3)电性能高分子材料,(4)高分子分 离材料,(5)高分子吸附材料,(6)高分子 智能材料,(7)医药用高分子材料,(8)高 性能工程材料。 ▲1、什么是活性聚合?阴离子活性聚合的 特征是什么? (1)活性聚合:是指引发速度远远大于增 长速度,并且在特定条件下不存在链终止反 应和链转移反应,亦即活性中心不会自己消 失的反应。二氯乙基氯/乙酸乙酯引发 (2)阴离子活性聚合的基本特点:①聚合 反应速度极快;②单体对引发剂有强烈的选 择性;③无链终止反应;④多种活性种共存; ⑤相对分子质量分布很窄。 ▲2、通过哪些途径可实现阳离子活性聚 合?哪些单体适合进行阳离子活性聚合? (1)途径①设计匹配性亲核反离子,如 采用HI/I2引发体系引发烷基乙烯基醚进行 阴离子活性聚合②适当的lewis酸碱配对 引发,如采用二氯乙基铝/乙酸乙酯引发 (2)目前,烷基乙烯基醚、异丁烯、苯乙 烯及其衍生物、1, 3 —戊二烯、茚和α-蒎烯 等都已经实现了阳离子活性聚合。 ▲3、为什么基团转移聚合也属于活性聚合 范畴? 基团转移聚合与阴离子型聚合一样,属“活 性聚合”范畴。基团转移聚合是以不饱和酯、 酮、酰胺和腈类等化合物为单体,以带有硅、 锗、锡烷基等基团的化合物为引发剂,用阴 离子型或路易士酸型化合物作催化剂,选用 适当的有机物为溶剂,通过催化剂与引发剂 之间的配位,激发硅、锗、锡等原子与单体 羰基上的氧原子结合成共价键,单体中的双 键与引发剂中的双键完成加成反应,硅、锗、 锡烷基团移至末端形成“活性”化合物的过 程。 包括①链引发反应,②链增长反应,③链终 止反应。 ▲4、自由基活性可控聚合有哪几类? 阴离子活性聚合、阳离子可控聚合、基团转 移聚合、原子转移自由基聚合、活性开环聚 合、活性开环歧化聚合等 ▲5、什么是高分子的化学反应?他们与小 分子的化学反应有什么异同点?影响高分 子化学反应的因素有哪些? (1)高分子的化学反应:可以将天然和合 成的通用高分子转变为具有新型结构与功 能的聚合物的化学反应。 (2)与小分子的化学反应的相同点: 高分子可以进行与低分子同系物相同的化 学反应。例如含羟基高分子的乙酰化反应和 乙醇的乙酰化反应相同;聚乙烯的氯化反应 和己烷的氯化反应类似。 (3)与小分子的化学反应的不同点: ①在低分子化学中,副反应仅使主产物产率 降低。而在高分子反应中,副反应却在同一 分子上发生,主产物和副产物无法分离,因 此形成的产物实际上具有类似于共聚物的 结构。 (4)高分子的反应活性的影响因素: ①聚集态结构因素:结晶和无定形聚集态结 构、交联结构与线性结构、均相溶液与非均 向溶液等结构因素均会对高分子的化学反 应造成影响。 ②化学结构因素:a)几率效应:当高分子 的化学反应涉及分子中相邻基团作无规成 对反映时,某些基团由于反应几率的关系而 不能参与反应,结果在高分子的分子链上留 下孤立的单个基团,使转化程度受到限制。 b)邻近结构效应:分子链上邻近结构的某 些作用,如静电作用和位阻效应,均可使基 团的反应能力降低或增加。 6、有哪些制备特种与功能高分子的制备方 法?各有什么优缺点? (1)功能高分子的制备方法主要有以下四 种类型: ①功能性小分子的高分子化;②已有高分子 材料的功能化;③多功能材料的复合;④已 有功能高分子的功能扩展。 (2)制备方法各自的优缺点: ①功能性小分子的高分子化:对功能性小分 子进行高分子化反应,赋予其高分子的功能 特点。 包括:a)带有功能性基团的单体的聚合,b) 带有功能性基团的小分子与高分子骨架的 结合,c)功能性小分子通过聚合包埋与高 分子材料结合。 主要优点是可以使生成的功能高分子功能 基分布均匀,聚合物结构可以通过聚合机理 预先设计,产物的稳定性较好。 精品文档
基金项目:浙江省科技支撑计划(2007C21G 2150010)和浙江省高等学校特聘教授基金(2005Z J008); 作者简介:邱化玉(1963-),理学博士,钱江学者特聘教授,博士研究生导师,研究方向为有机硅化学; 3通讯联系人,E 2mail :hyqiu @https://www.doczj.com/doc/53642793.html,. 环硅氧烷开环聚合反应的机理及动力学研究 吕素芳,李美江,邬继荣,蒋剑雄,来国桥,邱化玉 3(杭州师范大学有机硅化学及材料技术教育部重点实验室和浙江省有机硅 材料技术重点实验室,杭州 310012) 摘要:环硅氧烷在亲核或亲电催化剂、温度或辐射作用下,可开环聚合生成线型聚硅氧烷,聚合方法主要有 本体聚合和乳液聚合。本体聚合可分为阴离子聚合和阳离子聚合,阴离子聚合就是在碱性催化剂(亲核试剂) 作用下,使环硅氧烷开环聚合成线型聚硅氧烷的过程;阳离子聚合就是环硅氧烷在酸性催化剂(亲电试剂)作用下的开环聚合反应。乳液聚合则是单体和水(或其它分散介质)并用乳化剂配成乳液状态进行聚合,按所采用的乳化剂种类不同,主要有阴离子型和阳离子型两种类型。本文总结了近几年国内外环硅氧烷本体聚合和乳液聚合的开环聚合机理及动力学研究情况,并对今后此方面的研究进行了展望。 关键词:环硅氧烷;阴离子聚合;阳离子聚合;反应机理;动力学引言 环硅氧烷是有机硅工业中最主要的原料,大多数有机硅高分子都是由环硅氧烷开环聚合得到的,强酸或强碱引发环硅氧烷开环聚合是合成线型聚硅氧烷常用的方法。常用的环硅氧烷为六甲基环三硅氧烷(D 3)和八甲基环四硅氧烷(D 4)。环硅氧烷开环聚合的方法主要有本体聚合和乳液聚合,二者均可分为阴离子聚合和阳离子聚合两种。本体聚合是单体本身加入少量其它介质聚合;乳液聚合则是单体和水(或其它分散介质)并用乳化剂配成乳液状态进行聚合。 环硅氧烷在亲核或亲电催化剂、温度或辐射作用下,可开环聚合生成线型聚硅氧烷。聚合过程由4 个阶段组成[1]:(1)聚合引发阶段,形成反应中心;(2)链增长阶段;(3)链终止阶段(活性中心消失);(4)链 转移形成新的活性点。 线形聚硅氧烷是有机硅聚合物最基本的形式,研究最多,应用也最广。聚硅氧烷的聚合方法从反应类型上来区分,可分为以两端有活性官能团封端的线形聚硅氧烷为基本原料的缩合聚合和以环硅氧烷为单体的开环聚合两大类。由于开环聚合所用的单体价廉易得、聚合过程简易可行,因此对开环聚合的研究、应用最多,而对环硅氧烷开环聚合的机理及动力学进行深入的研究,可为聚硅氧烷的工程设计及生产控制提供理论依据。本文旨在近年来国内外对环硅氧烷的本体聚合和乳液聚合的开环聚合机理及动力学研究进行综述,分别对本体聚合的阴离子聚合和阳离子聚合的机理及动力学进行综述,对环硅氧烷的乳液聚合的开环聚合机理及动力学研究进行综述。 1 本体聚合 111 阴离子催化开环聚合反应 11111 机理研究 阴离子催化开环聚合反应,就是在碱性催化剂(亲核试剂)作用下,使环硅氧烷开环聚合成线型聚硅氧烷的过程。以K OH 催化D 4开环聚合为例,反应开始时,K OH 中OH - 阴离子与D 4硅原子上的3d 轨道配位,导致D 4内电子云密度重新分布,在加热下引起Si —O —Si 键断裂(开环),生成链端含
第八章 开环聚合 8.1 概述 高分子化学中,以环状单体通过开环聚合来合成聚合物,同样具有重要的地位。在这种聚合过程中,增长链通过不断地打开环状结构,形成高聚物: 以环醚为例,环氧乙烷经开环聚合反应,得到一种聚醚,即聚氧化乙烯。这在工业上已得到应用。 能够进行开环聚合的单体很多,如环状烯烃,以及内酯、内酰胺、环醚、环硅氧烷等环内含有一个或多个杂原子的杂环化合物。开环聚合既具有某些加成聚合的特征,也具有缩合聚合的特征。由开环聚合得到的聚合物,重复单元与环状单体开裂时的结构相同,这与加成聚合相似;而聚合物主链中往往含有醚键、酯键、酰胺键等,与缩聚反应得到的聚合物常具有相同的结构,只是无小分子放出。开环聚合与缩聚反应相比,还具有聚合条件温和、能够自动保持官能团等物质的量等特点,因此开环聚合所得聚合物的平均分子质量,通常要比缩聚物高得多。有些单体如乳酸,采用缩聚反应无法得到高分子质量的聚合物;而采用乳交酯的开环聚合,就能够获得高分子质量的聚乳酸。但是,与缩聚反应相比,开环聚合可供选择的单体较少,例如二元酸与二元醇能够通过缩聚获得聚酯;而开环聚合,只有相当于α,ω-羟基酸的环内酯可供选择。聚酰胺的情况也是如此。另外,有些环状单体合成困难,因此由开环聚合所得到的聚合物品种受到限制。开环聚合就机理而言,有些属于逐步聚合,有些属于连锁聚合。 8.1.1 聚合范围及单体可聚性 如前所述,环醚、环酯、环酰胺、环硅氧烷等能够进行开环聚合。此外,环胺、环硫化物、环烯烃、以及N-羧基-α-氨基酸酐等同样也能进行开环聚合。 环状单体能否转变为聚合物,取决于聚合过程中自由能的变化情况,与环状单体和线形聚合物的相对稳定性有关。Dainton 以环烷烃作为环状单体的母体,研究了环大小与聚合能力的关系。表6-1列出了环烷烃在假想开环聚合时的自由能变化ΔG lc 0、焓变ΔH lc 0、及熵变ΔS lc 0。 R X [ R X ]n n [ CH 2 CH 2 O ]n n H 2C CH 2O
低表面能聚合物的聚合进展 罗正鸿*1,何腾云1,蔺存国2,戴李宗1 (1.厦门大学化学化工学院,厦门 361005; 2.海洋腐蚀与防护国防科技重点实验室,中国船舶重工集团第七二五研究所,青岛 266071) 摘要:低表面能聚合物材料是低表面能材料中重要的一类。这类材料性能独特,用途广泛,尤其体现在涂料领域,用作防污涂层。目前,低表面能聚合物主要有氟碳树脂、有机硅树脂及氟硅树脂三种。有关这三种材料的合成研究十分活跃,出现了众多的合成研究报道。按合成过程对应的聚合机理划分,主要有基团转移聚合、阳离子聚合、阴离子聚合、自由基聚合、活性官能团间的反应。本文按聚合机理划分方法对低表面能聚合物近期的合成研究进展进行综述。 关键词:低表面能聚合物;接触角;聚合机理 引言 通常所说的低表面能是指表面能低于100mN m的材料[1]。材料表面能越低,附着力越小,材料表面与液体的接触角也就越大[2]。目前出现了众多的低表面能材料,其中最重要一类是低表面能聚合物。这类材料用途广泛,在航空航天、印刷、生物化学、传感器、环境防污、金属冶炼、海洋防污等领域均有应用[3,4]。其中,重要应用领域在涂料领域,用作防污涂层。一般认为,涂料的表面能只有在低于25mN m 时(也就是涂料表面与液体的接触角大于98 )才具有防污效果[5]。 低表面能聚合物主要有氟碳树脂、有机硅树脂及氟硅树脂三大类:(1)氟碳树脂:将F原子引入到聚合物链中可降低聚合物表面能。主要原因有:C F键键能比C H键键能大,且F原子电子云对C C 键的屏蔽较H原子强;此外,C H键的电子云分布使得含C H键的物质能与油污发生作用,而C F键中电子被紧紧束缚在原子核周围,综合作用使得氟化合物具有低表面能。例如氟含量很高的聚四氟乙烯表面能约为20m N m[6];(2)有机硅树脂:有机硅树脂中最主要的是聚硅氧烷,该类聚合物有Si O骨架而具有独特的性能,比如低表面能,热稳定性低玻璃化转变温度等[7]。用碳酸钙填充的聚二甲基硅氧烷涂料其表面能低至22m N m[8];(3)氟硅树脂:利用氟碳树脂和有机硅树脂各自优点可得到性能更优良的低表面能材料[9]。 由于这三类低表面能聚合物用途广泛,已出现了众多的合成研究报道。近期有关低表面能聚合物的合成方法,按合成过程对应的聚合机理划分,主要有:基团转移聚合、阳离子聚合、阴离子聚合、自由基聚合、活性官能团间的反应。本文按聚合机理划分方法对低表面能聚合物近期合成进展进行综述。 1 低表面能聚合物的合成 1 1 基团转移聚合 某些(甲基)丙烯酸酯类的单体在类似于三甲代甲硅烷基二甲基乙烯酮乙缩醛(MTDA)引发剂和四丁基铵酯类催化剂组成的催化体系作用下能发生基团转移聚合,可合成嵌段共聚物,但由于受反应机理的约束,可发生该类反应的单体不多,这样大大限制了其应用范围;用基团转移聚合可得到预期的聚合物结 基金项目:海洋腐蚀与防护国防科技重点实验室基金项目(No.51449020205QT8703)、福建省科技计划重点项目(No. 2005H040); 作者简介:罗正鸿,男,博士、副教授,研究方向为高分子材料及聚合工程; *通信联系人。E mail:luozh@https://www.doczj.com/doc/53642793.html,.
开环易位聚合(ROMP)论文:开环易位聚合物纳米粒子的制备及表征 【中文摘要】聚合物纳米粒子因其独特的结构而呈现出诸多新奇的物理、化学特性,在光学、食品工业、高性能涂料、高分子催化剂、生物医用材料等方面有广泛的应用前景,因此引起了普遍的关注。本论文采用简便易行的方法—聚合诱导自组装,得到了粒径均一的“核-壳”结构的聚合物纳米粒子。在选择性溶剂甲苯中,采用开环易位聚合(ROMP)的方法,用第一代钉卡宾催化剂(Ru-I)引发,首先加入单体—2,3-二异丁酰溴甲氧基-5-降冰片烯(BNBE)反应一段时间得到均聚物(PBNBE),然后加入单体—7-氧代降冰片烯二甲酯(ONBDM)再反应一段时间。由于第一个单体的均聚物在甲苯中溶解性很好,而第二个单体的均聚物在甲苯中溶解性很差,利用两个嵌段的溶解性差异,直接 得到分散性好,粒径均一的以PBNBE为壳,以PONBDM为核的壳官能化的胶束。考察了不同投料比,浓度对胶束结构形态的影响。用核磁(NMR),元素分析(EA),凝胶渗透色谱法(GPC),动态光散射(DLS),原子力显微镜(AFM),透射电镜(TEM)对其组成与形态进行表征。结果表明:聚合物胶束的直径随着PBNBE嵌段的增长而增大,随着PONBDM嵌段的增长反而减小;通过此方法可简便的得到了分散性... 【英文摘要】Polymeric core-shell nanoparticles with special structure and some novel physical and chemical properties have attracted considerable research interest
2009年第38卷第1l期石油化工 PETROCHEMICALTECHNOLoGY ?1141?以可逆基团转移配位聚合法进一步 扩展聚烯烃品种 吕立新 (中国石油化工股份有限公司北京化工研究院,北京100叭3) [摘要]介绍了在用单活性中心催化剂进行乙烯、丙烯和1一烯烃配位聚合时,双分子间的可逆基团转移配位聚合新工艺,包括配位链转移聚合、链穿梭聚合和配位再生基团转移聚合。在此类聚合反应中,大分子链、化学基团(如甲基)、元素(如氯)在过渡金属活性中心与主族金属烷基化合物之间进行快速、可逆的转移。其中,主族金属烷基化合物作为链穿梭剂或增长大分子链的暂时寄存场所(代存物)。这些新工艺的引入不仅促进了聚烯烃新品种(特别是嵌段共聚物)的发展,而且使原先只能通过活性聚合才能制备出的聚烯烃材料,现在有可能在工业生产中实现。 [关键词]可逆基团转移聚合;配位聚合;链转移聚合;嵌段共聚物;聚烯烃 [文章编号]1000—8144(2009)1l—1141—10[中图分类号]TQ325.1[文献标识码]A ExpandingtheSpectrumofPolyolel盖nsThroughReVersibleGroupTransfer CoordinationPolymerization L西L抚加 (Be幻ingResearchInstituteofchemicalIndus仃y,sINOPEc,Be巧inglo0013,china) lAbstractJTheintentofmisreViewistoproVideasynopsisofadVancesrelatedtot11ei删uction aIldcontrolofreverSiblebiHl01eculargroup—transferpolymerizationpmcessesforsin91e—sitecoordinationpolymerizationofemene,pmpene,a11d1一a11(ene.Outlookonmeirfuturepmspectsisalsowithilltlle anicle.弧eaboveⅡlentionedprocessesincludingcoordinatiVechain一廿ansferp01ym耐zation, chain—shuttlingpolymerizationanddegeneratiVegroup一仃ansferpolymerizationoperatedbyfastreVersiblegmups(polymerylgrouporchernjcalgroupsuchasmetllylgmup,oratomsuchaschl嘶ne)仃ansfer betweenactivetransition-metalcentersa11d majn—gmup—metalalkylcompounds.Thelattercanserveeitlleras“chainshuttling"agentsoras”surrogate”chain-growmsites.RecentintroductionofmeSepmcesses furthernotonlyprovidethebasisfornewparadigmsfornew poly01efins(especially blockcopolymers)discoveryanddevelopmentbutalsocanproVidememechanismsbywmchprecisionpoly01efinsfo肌erlypreparedonlyunderliVingconditionscannowpotentiallybe brou曲ttocommercialreahzation.[Keywords]reVersiblegroup—transferpolymedzation;coodination polymerization;chain-transferpolymerization;blockcopolymer;polyolefin 当前,聚烯烃已成为高分子材料中应用面最广、产量和消费量最大的一个品种。目前通过催化 剂选择和聚合工艺的变化,这类聚合物的微观结构 在许多方面已可以灵活调控和定制,包括相对分子 质量大小及其分布、立构规整性、共聚单体含量和 支链结构。而控制共聚单体在主链上的分布(无 规、嵌段、交替)以改变树脂的性能,仍是尚待解决的问题。过去的研究工作更多集中于活性配位聚合,取得了一定的进展,也合成出一些全新的共聚物…2I;但正如其他的活性聚合一样,一个催化剂的[收稿日期]2009一03—16;[修改稿日期]2009—07一06。[作者简介]吕立新(1929一),男,江苏省苏州市人,大学,教授级高级工程师,电邮Iulixin@brici.ac.cn。 飞00多 一评一~述一~约一一特一∥甚k万方数据
【最新整理,下载后即可编辑】 第六章 开环聚合 习题参考答案 1. 试讨论环状单体环的大小与开环聚合反应倾向的关系。 解答: 环状单体能否转变为聚合物,取决于聚合过程中自由能的变化情况,与环状单体和线形聚合物的相对稳定性有关。以环烷烃为例,由液态的环烷烃(I )转变为无定型的聚合物(c ): 聚合过程中的自由能变化: ΔG lc 0 =ΔH lc 0 — T ΔS lc 0≤ 0 由表6-1可以看出,除六元环外,其他环烷烃的ΔG lc 0均小于0,开环聚合在热力学上是有利的。除六元环烷烃外,其他环烷烃的聚合可行性为:三元环,四元环>八元环>五元环,七元环。对于三元环、四元环来讲,ΔH lc 0是决定ΔG lc 0的主要因素,是开环聚合的主要推动力;而对于五元环、六元环和七元环来说,ΔH lc 0和ΔS lc 0对ΔG lc 0的贡献都重要。随着环节数的增加,熵变对自由能变化的贡献增大,十二元环以上的环状单体,熵变是开环聚合的主要推动力。 以上仅是通过热力学分析的结果,事实上环烷烃的开环聚合通常难于进行,主要是因为环烷烃的结构中不存在容易被引发物种进攻的键,这是动力学原因。其他的环状单体如内酰胺、内酯、环醚等杂环单体与环烷烃不同,由于杂原子的存在提供了可接受引发物种亲核或亲电进攻的部位,从而能够进行开环聚合。 2. 氧化丙烯的负离子聚合通常仅能得到低分子量的聚合物,试讨论原因。 解答: 在氧化丙烯的负离子开环聚合过程中,由于存在副反应如交换反应、向单体的转移反应等,使得聚合物的相对分子质量降低,仅能得到低聚物。具体原因如下: (CH 2)n x x n (CH 2)[](l) (c)
第六章离子聚合 思考题 6.1试从单体结构来解释丙烯腈和异丁烯离子聚合行为的差异,选用何种引发剂?丙烯酸、烯丙醇、丙烯酰胺、氯乙烯能否进行离子聚合,为什么? 答丙烯腈中氰基为吸电子基团,同时与双键形成丌-丌共轭,能使双键上的电子云密度减弱,有利于阴离子的进攻,并使所形成的碳阴离子的电子云密度分散而稳定,因此丙烯腈能够进行阴离子聚合。进行阴离子聚合时,可选用碱金属、碱金属化合物、碱金属烷基化合物、碱金属烷氧化合物等作为引发剂。 异丁烯中两个甲基为推电子基团,能使双键上的电子云密度增加,有利于阳离子的进攻,并使所形成的碳阳离子的电子云密度分散而稳定,因此异丁烯能够进行阳离子聚合。进行阳离子聚合时,通常采用质子酸、lewis酸及其相应的共引发剂进行引发。 丙烯酸、烯丙醇、丙烯酰胺、氯乙烯不能进行离子聚合,因为没有强烈的推电子基团和吸电子基团。 思考题6.2下列单体选用哪一引发剂才能聚合?指出聚合机理类型。 答苯乙烯三种机理均可,可以选用表中5种引发剂的任一种。 偏二腈乙烯,阴离子聚合,选用Na+萘或n-C4H9Li引发。 异丁烯,阳离子聚合,选用SnCl4+ H2O或BF3+H2O。 丁基乙烯基醚,阳离子聚合,选用SnCl4+ H2O或BF3+H2O。 CH2=C(CH3)CO2CH3,阴离子聚合和自由基聚合。阴离子聚合,选用Na+萘或n-C4H9Li 引发;自由基聚合选用(C6H5CO)2O2作引发剂。 思考题6.3下列引发剂可以引发哪些单体聚合?选择一种单体作代表,写出引发反应式。 (1)KNH2(2)A1C13+HCl (3)SnCl4+C2H5Cl (4)CH3ONa 答(1) KNH2是一类高活性的阴离子引发剂,可以引发大多数阴离子聚合的单体进行聚合。如引发苯乙烯进行聚合 (2) A1C13活性高,用微量水作共引发剂即可。A1C13+HCl配合时,C1-亲核性过强,易与阳离子共价终止,因此很少采用。 (3) SnCl4+C2H5Cl以引发异丁烯、乙烯基烷基醚及共轭烯烃进行阳离子聚合 (4) CH3ONa可以引发高活性和较高活性的单体进行阴离子聚合。高活性单体如硝基乙烯、偏二氰乙烯。较高活性单体如丙烯腈、甲基丙烯腈等,以及环氧烷烃(如环氧乙烷、环氧丙烷等)的开环聚合。 思考题6.4在离子聚合中,活性种离子和反离子之间的结合可能有几种形式?其存在形式受哪些因素影响?不同形式对单体的聚合机理、活性和定向能力有何影响? 答离子聚合中,活性种离子近旁总伴有反离子。它们之间的结合,可以是共价键、离子对,乃至自由离子,彼此处于平衡之中。如下所示,结合形式和活性种的数量受溶剂性质、温度及反离子等因素的影响。 Bδ-Aδ+,?B-A+ ?B-║A+ ?B- + A+ 极化共价键紧密接触溶剂隔离自由离子
练习一 1、传统聚合反应包括自由基聚合反应、离子聚合反应、配位聚合反应、逐步聚合反应反应。 2、基团转移聚合的引发剂通常为带有硅、锗和锡烷基基团的化合物。 3、开环易位聚合的产物立体异构的主要原因之一碳碳双键C=C在聚合物中保持不变。 4、当前真正大规模工业化应用的活性聚合是活性阴离子聚合。 5、低温等离子体聚合大致可分等离子体聚合、等离子体引发聚合和等离子体表面处理三大类。 6、模板聚合的过程包括模板(T)与单体(M)形成复合物(单体模板化)、模板聚合和模板与“复制”高分子的分离三个步骤。 (将确定的模板化合物溶解或分散于单体和交联剂中进行共聚合,然后用特定溶剂将模板化合物溶解洗脱,最后在聚合物骨架上留下与模板分子大小和形状相匹配的孔穴。这种有模板参加、以获得模板聚合物为目的的交联共聚反应称为“模板聚合”。) 7、超临界二氧化碳在聚合物合成中应用最多的反应类型是自由基反应。 8、液晶的出现主要是因为刚性结构和强作用力(分子间作用力,如氢键)。 9、双光子聚合的核心部分是双光子聚合的光敏引发体系。 10、制备抗凝血材料的重要方法之一是对现有物理力学性能较好的聚合物材料进行表面改性并通过物理覆盖等方法实现。 1、活性/可控聚合不存在链终止反应。(×)【只有活性聚合不存在链终止】 2、开环易位聚合反应是活性聚合。(√) 3、高温等离子体不适应于高分子化学合成,因为电子与气体之间不存在热平衡。(×) 4、预辐射接枝法中聚合物自由基的利用率较高。(×)【较低】 5、超分子化学是以非共价键的形成是分子组装,从而制备新功能性分子结合体。(√) 6、液晶高分子在分子量上存在多分散性。(√)【分子分布宽】 7、等离子体引发单体进行聚合反应得到的聚合物产物组成纯净不含引发剂等杂质。(√) 8、根据共聚物大分子的序列结构。共聚物可分为交替、嵌段、接枝和无规四大类。(√) 9、导电高分子中高分子基料的作用是提供载流子。(×)【结构型导电高分子才是,复合导电高分子不是】 10、前线聚合所需的外供能量是短暂一次性的,聚合启动后即可停止供热。(√) 练习二 1、活性/可控自由基聚合可分为稳定自由基调控聚合法、引发链转移终止法、可逆加成-裂解链转移活性自由基聚合、原子转移自由基聚合四类。 2、GTP过程分为链引发、链增长和链终止三步。 3、液晶高分子往往是由小分子液晶基元键合而成,这些液晶基元可分为棒状、碟状和双亲分子。 4、按照单体物态分类,辐射聚合可分为溶液聚合、固相辐射聚合和气相辐射聚合。(如果四个空再加上乳液辐射聚合) 5、第一个实现活性/可控聚合的是(活性)阴离子聚合。 6、导电高分子根据其组成可分为结构型导电高分子和复合型导电高分子。 7、功能高分子化合物主要两种合成方法高分子功能化和功能单体高分子化。 8、高分子药物与低分子药物相比,具有低毒,高效,缓释,长效,可定点、定向释放等优点。
第六章离子聚合 思考题试从单体结构来解释丙烯腈和异丁烯离子聚合行为的差异,选用何种引发剂丙烯酸、烯丙醇、丙烯酰胺、氯乙烯能否进行离子聚合,为什么 答丙烯腈中氰基为吸电子基团,同时与双键形成丌-丌共轭,能使双键上的电子云密度减弱,有利于阴离子的进攻,并使所形成的碳阴离子的电子云密度分散而稳定,因此丙烯腈能够进行阴离子聚合。进行阴离子聚合时,可选用碱金属、碱金属化合物、碱金属烷基化合物、碱金属烷氧化合物等作为引发剂。 异丁烯中两个甲基为推电子基团,能使双键上的电子云密度增加,有利于阳离子的进攻,并使所形成的碳阳离子的电子云密度分散而稳定,因此异丁烯能够进行阳离子聚合。进行阳离子聚合时,通常采用质子酸、lewis酸及其相应的共引发剂进行引发。 丙烯酸、烯丙醇、丙烯酰胺、氯乙烯不能进行离子聚合,因为没有强烈的推电子基团和吸电子基团。 思考题下列单体选用哪一引发剂才能聚合指出聚合机理类型。 答苯乙烯三种机理均可,可以选用表中5种引发剂的任一种。 偏二腈乙烯,阴离子聚合,选用Na+萘或n-C4H9Li引发。 异丁烯,阳离子聚合,选用SnCl4+ H2O或BF3+H2O。 丁基乙烯基醚,阳离子聚合,选用SnCl4+ H2O或BF3+H2O。 CH2=C(CH3)CO2CH3,阴离子聚合和自由基聚合。阴离子聚合,选用Na+萘或n-C4H9Li引发;自由基聚合选用(C6H5CO)2O2作引发剂。 思考题下列引发剂可以引发哪些单体聚合选择一种单体作代表,写出引发反应式。 (1)KNH2 (2)A1C13+HCl (3)SnCl4+C2H5Cl (4)CH3ONa 答 (1) KNH2是一类高活性的阴离子引发剂,可以引发大多数阴离子聚合的单体进行聚
烯烃易位反应 1.易位反应 烯烃易位反应基本概念:易位反应是指两种物质互相交换成分生成两种新的物质的反应。同样的,两种烯烃互相交换双键两端的基团,从而生成两种新的烯烃的反应便是烯烃易位反应。 烯烃易位反应的催化剂:一般是过渡金属化合物,活性中心是过渡金属碳烯。碳碳双键可在链烯上也可在环烯上,如果是环烯,则易位反应的结果是聚合。这种易位反应是可逆平衡反应。 目前研究最多的烯烃易位反应主要有三个类型: (1)开环易位聚合反应(ROMP:ring-opening metathesis polymerization) 以及开环交叉易位反应(ROCM:ring-opening cross metathesis); (2)闭环易位反应(RCM:ring-closing metathesis); (3)交叉易位反应(CM:cross metathesis)。 具体的反应表达式见下图: 2.开环易位聚合 开环易位聚合反应不是简单的链烯烃双键断裂的加成聚合,也不是内酰胺或者环醚等杂环的开环聚合,而是双键不断易位,链不断增长,而单体分子上的双键仍保留在生成的聚合物大分子中的反应。 环烯烃是否可以进行ROMP反应可以通过热力学来判断。在ROMP中,聚合热焓主要来自环张力能的释出,所以环的张力能是决定能否进行开环易位聚合的主要因素。环的张力越大,单体越活泼。单体可以是单环烯,如环丁烯、环辛烯、环戊烯和环庚烯等。单体也可以是双环烯,如降冰片烯及其衍生物,这也是目前研究最多的单体。单体还可以是三环烯。单环烯与双环烯共聚可以制得高度交联的体形聚合物。 一般将ROMP反应的催化剂分为如下三类:(1)传统催化剂如:WCl6/Bu4Sn,WOCl4/EtAlCl2,MoO3/SiO2和Re2O7/Al2O3等。因为这些催化体系的