重磅药物:年销售额10亿美元
非小细胞肺癌:校正年龄后发病率为39.05/100000,其中男性41.43/100000,女性37.13/100000
埃克替尼用于晚期非小细胞肺癌二线治疗。
厄洛替尼单药适用于既往接受过至少一个化疗方案失败后的局部晚期或转移的非小细胞肺癌(NSCLC)。
克唑替尼胶囊可用于经CFDA 批准的检测方法确定的间变性淋巴瘤激酶(ALK)阳性的局部晚期或转移性非小细胞肺癌(NSCLC)患者的治疗。
易瑞沙:本品适用于治疗既往接受过化学治疗的局部晚期或转移性非小细胞肺癌(NSCLC)。
ZYKADIA适用为有间变性淋巴瘤激酶(ALK)-阳性转移对克唑替尼进展或不能耐受的非小细胞肺癌(NSCLC)患者的治疗。
区别:
1.目标靶点不同
2.临床试验结果不同。易瑞沙美国试验结果不佳而特罗凯良好,导
致易瑞沙美国销量很差。易瑞沙主要销售区域为中国。
3.
4.(学习的目的是增长知识,提高能力,相信一分耕耘一分
收获,努力就一定可以获得应有的回报)
5.
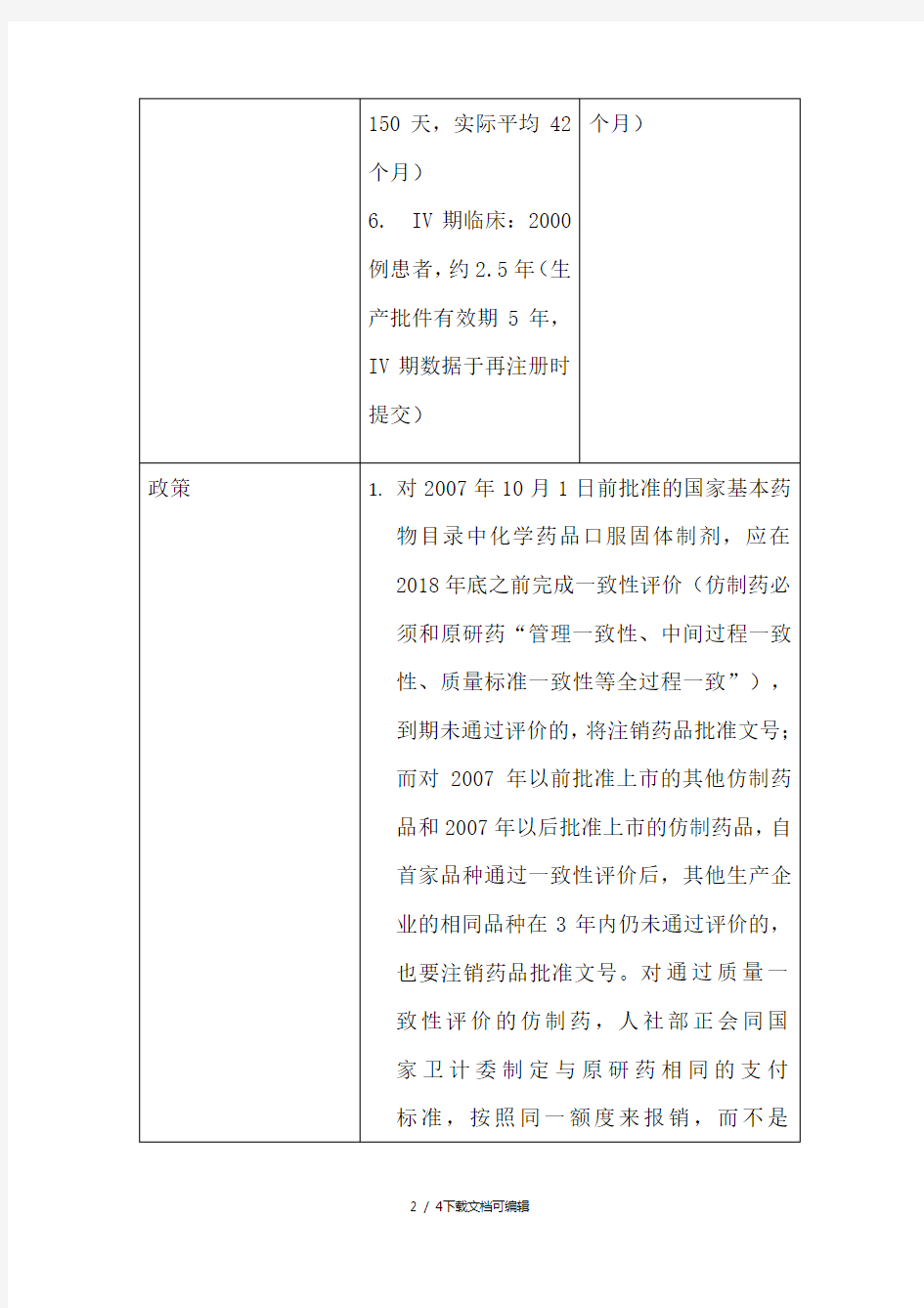
新药及仿制药制剂开发研究流程
28
28
28 / 3.
一、新注册管理办法对化药6 类的要求 根据药品注册管理办法附件二规定药物注册六类药物即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 第十二条仿制药申请,是指生产国家食品药品监督管理局已批准上市的已有国家标准的药品的注册申请;但是生物制品按照新药申请的程序申报。 第一百三十六条规定国家药品标准,是指国家食品药品监督管理局颁布的《中华人民共和国药典》、药品注册标准和其他药品标准,其内容包括质量指标、检验方法以及生产工艺等技术要求。药品注册标准,是指国家食品药品监督管理局批准给申请人特定药品的标准,生产该药品的药品生产企业必须执行该注册标准。药品注册标准不得低于中国药典的规定。 第七十四条仿制药应当与被仿制药具有同样的活性成份、给药途径、剂型、规格和相同的治疗作用。已有多家企业生产的品种,应当参照有关技术指导原则选择被仿制药进行对照研究。 第七十三条仿制药申请人应当是药品生产企业,其申请的药品应当与《药品生产许可证》载明的生产范围一致。 按管理办法理解,仿制药的概念明确突出了“制”,即制备、生产,是一种模仿的制备,意指该类药品是“模仿制备”出来的,体现了药品质量的监管上升到强化制备工艺的过程控制,实施药品质量全程控制的发展和提升。新版《药品注册管理办法》中工艺验证、生产现场检查、“三合一”(现场核查、审评结论、生产检查及药检报告)等要求的提出正是体现仿制药强调生产制备的理念。 同时,仿制药特点突出“同”字,即与被仿制品具有相同的活性成分,给药途径,剂型,规格和相同的治疗作用,要想达到这个目标必须保证仿制药与被仿制品物质基础一致和治疗作用一致,研究方向必须从这两方面下功夫。仿制药的申请单位必须是药品生产企业,并且与其生产许可证载明的范围一致,如果没有相应的剂型或车间,应该先在许可证增项后,购买相应的车间设备,在拟建车间生产样品,样品生产过程应符合《药品生产质量管理规范》的要求。 新法规对仿制药提出了更高的要求,主要体现在以下几点: 1. 对被仿制药品选择提出要求 注册管理办法第七十四条规定--仿制药应当与被仿制药具有同样的活性成份、给药途径、剂型、规格和相同的治疗作用。已有多家企业生产的品种,应当参照有关技术指导原则选择被仿制药进行对照研究。一般应首先选择以进口原研药,因为原发厂产品经过系统的非临床与临床研究,安全有效性得到确认,进口时对人种差异进行了研究。其次可考虑选用研究基础较好、临床应用较为广泛的非原研产品;没有进口原研产品的,有必要对市售品进行质量对比考查,择优选用,以确保仿制基础的可靠性。人体生物等效性试验与质量对比研究的参 比品应是同一厂家最好相同批次产品,以全面说明其物质基础及体内过程的一致性和等效性,为桥接其安全有效性奠定更为坚实的基础 2. 增加生产现场检查项目 注册管理办法第七十七条规定省、自治区、直辖市药品监督管理部门应当自受理
中国创新药咨询与服务先锋CRO 新药审批流程及审批时间
一. 原型药 原型药物:随着对生理生化机制的了解,得到了一些疾病治疗的突破性药物,这些药物不仅在医疗效果方面,而且在医药市场上也取得了较大的成功,这些药物通常被称为原型药物(prototype drug)。随之出现了大量的"me-too"药物。 二. 原研药 原研药:即指原创性的新药,经过对成千上万种化合物层层筛选和严格的临床试验才得以获准上市。需要花费15年左右的研发时间和数亿美元,目前只有大型跨国制药企业才有能力研制。在我国,“原研药”主要是指过了专利保护期的进口药。 三. Me - too”药物 "me-too"药物:特指具有自己知识产权的药物,其药效和同类的突破性的药物相当。这种旨在避开“专利”药物的产权保护的新药研究,大都以现有的药物为先导物进行研究。研究的要点是找到不受专利保护的相似的化学结构,这种研究有时可能得到比原“突破性”药物活性更好或有药代动力学特色的药物。 【例】最早治疗胃溃疡,是用胃舒平中和胃酸,此疗法治标不治本。后来科学家发现:人体中的胃组胺H2受体一旦激动,就会分泌胃酸。科学家因此发明了西米替丁药,阻断胃组胺H2受体,减少胃酸分泌,使溃疡逐渐愈合。此药一上市,就受到广泛追捧,科学家稍稍改变了西米替丁的化学结构,开发出了雷尼替丁、法莫替丁等胃药。这些后来的派生药,就被人称为Me-too药。 四. 新药(NewDrugs) 新药(New Drugs):系指未曾在中国境内上市销售的药品。对已上市药品改变剂型、改变给药途径、增加新适应症的药品注册按照新药申请的程序申报,但改变剂型但不改变给药途径,以及增加新适应症的注册申请获得批准后不发给新
详解仿制药研发具体流程 目录 一、综述2 二、仿制药研发项目汇总3 三、仿制药的研发具体步骤:5 (一)产品信息调研5 (二)前期准备(约一个月完成):5 1、参比制剂的采购5 2、原料采购5 3、色谱柱及对照品采购5 4、辅料采购:6 5、包材的采购:6 (三)处方工艺研究6 1、原辅料及参比制剂的检验6 2、处方工艺摸索6 3、初步验证工艺8 4、中试生产及工艺验证8 (四)质量研究9 1、质量研究项目的选择及方法初步确定9 2、质量标准的方法学验证10 3、质量对比研究12 4、质量标准的制定14 (五)稳定性研究(中试产品)14
(六)药理毒理研究16 (七)申报资料的撰写、整理16 (八)申报临床及申报现场核查17 (九)临床研究17 (十)申报生产17 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。
谈原研药与仿制药的不同 黄浪伍巧吉(海南通用三洋药业有限公司海口570302) 摘要:本文主要从新药法规、药价、质量和疗效等方面介绍原研药与仿制药的区别,并使公众对两者有更深入的了解。 关键词:原研药仿制药 中图分类号:R252.8文献标识码:B文章编号:1672-8351(2011)08-0083-02 A review of difference between Reference listed drug and Generic drug Abstract:This article mainly introduces the differences between Reference listed drug and Generic drug with the aspects of drug reg-ulations,price,quality and efficacy,and help the public have a further understandings of them. Key words:Reference listed drug Generic drug 原研药RLD(Reference listed drug),是指原创性的新药,亦可指全球首家上市的新药(ND,New Drug)。首家上市的新药几乎必定申请专利保护,亦即成为了众所周知的专利药(Patented drug)。所以,原研药和专利药基本上是同一意思。并且,一般专利药可享受20年的专利保护期。 而仿制药(Generic drug)是指以原研药为参考对比,在剂量、安全性、效力、质量、作用以及适应症上相同的一种仿制药品。 它们的区别主要在以下三个方面:1在药品的定义上 FDA只有2种,即原研药(RLD)和仿制药(ANDA)。其中,FDA对于仿制药的定义很严格,要求必须和被仿制产品的剂型、规格、给药途径一致。如不一致,则按新药申报(NDA)。而中国的药品分类定义则比较广泛,划分为6大类(此文指化学药品)。其中,对已上市药品改变剂型、改变给药途径、增加新适应症的药品,亦属于新药范畴,而且在审批、临床研究等方面却不同程度的简化。这样虽然能给国内的仿制药厂家节约一笔数额不菲的费用,也促进了仿制药的发展,但是同时也相 参考文献 [1]周月芬.补阳还五汤加味治疗乳腺癌术后上肢肿胀疗效观察[J].河北中医,2000,22(2):129. [2]吴玉华,万冬桂,张晓春.当归芍药散加味治疗乳腺癌术后同侧上肢肿胀38例[J].中医药学报,2003,31(1):45. [3]付烊.黄芪桂枝五物汤加减治疗乳腺癌术后同侧上肢水肿疗效观察[J].辽宁中医药大学学报,2010,12(5):180. [4]严善福,王树霞,周亚兵.桃红四物汤加味治疗乳腺癌术后上肢肿胀91例[J].上海中医药杂志,2009,43(6):39-40. [5]陈鹊汀,胡庚坤,王振东,等.桃红四物汤治疗乳腺癌术后皮瓣血运障碍40例[J].河北职工医学院学报,2003,20(1):29. [6]姬广伟,丁伟英,王颂歌.逐瘀利湿煎治疗乳腺癌术后上肢肿胀43例疗效观察[J].中国煤炭工业医学杂志,2010,13(2):277. [7]郑武,邹荣生.血府逐瘀汤加减结合功能锻炼治疗乳腺癌术后上肢水肿30例[J].福建中医药,2004,35(3):30. [8]陈海滨,应声闻.血府逐瘀汤预防乳腺癌术后皮瓣坏死体会[J].现代中西医结合杂志,2008,17(5):731. [9]许正国,刘加升,孟昭旭.血府逐瘀汤治疗乳腺癌根治术后并发症[J].辽宁中医杂志,2005,32(9):947. [10]余江利,吴军.补中益气汤协同新辅助化疗治疗乳腺癌48例[J].湖南中医杂志,2005,21(1):33-34. [11]张正习.加减逍遥散治疗中晚期乳腺癌32例临床观察[J].南中医药导报,2002,8(6):347-349. [12]文玲波,杨兰平,黄汉生.加味逍遥散改善乳腺癌术后化疗毒副反应的临床观察[J].湖南中医学院学报,2006,26(3):38-40. [13]罗雪冰.益气养阴化浊汤改善乳腺癌患者化疗恶心呕吐证的临床观察[J].第四军医大学学报,2007,28(18): 1725. [14]周斌,单泽松.旋覆代赭汤在乳腺癌化疗中的应用[J].江西中医药,2008,39(309):25-26. [15]陈彦.加味四君子汤对乳腺癌化疗毒副作用的影响[J].中医药导报,2009,15(1):38-40. [16]李茂林,孙显峰.益气升白汤治疗乳腺癌化疗后白细胞减少症109例[J].陕西中医,2010,31(2):182. [17]徐咏梅,杨国旺,王笑民,等.丹栀逍遥散加减治疗乳腺癌内分泌综合征65例临床观察[J].河北中医,2005,27(9):676. [18]刘展华,陆嵩,赵燕.柴胡疏肝散干预乳腺癌术后伴发抑郁症临床观察[J].新中医,2010,42(7):63-64. [19]王祥.六味地黄合甘麦大枣汤加味治疗乳腺癌内分泌治疗不良反应[J].黑龙江中医药,2008,1:29-30. [20]高绍荣,夏海平,张华,等.六味地黄汤加味治疗乳腺癌TAM疗后综合征68例[J].国医论坛,2005,20(2):24-25. [21]朱迪盈,李真喜,罗海英,等.小柴胡汤合六味地黄汤加减治疗乳腺癌术后更年期综合征[J].广东医学杂志,2000,21(8): 706. [22]张瑶,沈伟生.加味二仙汤治疗乳腺癌综合治疗后类更年期综合征32例[J].山东中医药大学学报,2007,31(2):137-138. [23]王兴春,李敏婕,李青兰,等.小柴胡汤配合化疗治疗晚期乳腺癌临床研究[J].山东中医杂志,2004,23(5):270-272. [24]苑艳娟,张芬梅,路标,等.柔肝健脾饮结合化疗治疗乳腺癌130例临床疗效观察[J].辽宁中医杂志,2008,35(3):395-396. [25]张莉,张仲海,杨赶梅,等.扶正消瘤汤对乳腺癌患者生活质量的影响[J].现代中西医结合杂志,2007,16(18):2494-2495. [26]陈彦.补气活血汤对乳腺癌新辅助化疗患者免疫功能的影响[J].中国实用医药,2009,4(10):159-160. [27]陈鹊汀,朱惠学,刘智勤,等.当归补血汤对乳腺癌术后化疗患者增效减毒作用的临床观察[J].河北职工医学院学报,2008,25(6):50-51. *指导老师
仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料 药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安 全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产 出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要 求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关 物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要 将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制 指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用 非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如 鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不 尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳 定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月) 项目项目内容所需时间一产品信息调研质量标准、工艺处方等一周二前期准备1、参比制剂的采购:一个月
我国仿制药的发展策略
我国仿制药的发展策略 摘要:一直以来,由于新药研发技术含量高,投入大、周期长、风险高、产出少,而我国医药企业的自主创新能力不足,新药研发方面相对较为落后。目前我国95%以上的制药企业长期在仿制药领域发展,因此可以说我国是仿制药大国,但却并不是仿制药强国。近年来,世界范围内许多药品专利陆续到期,即将失去市场独占权,垄断地位受到仿制药的挑战,给世界仿制药带来巨大的空间,同样也将对我国的医药市场带来无限机遇。无论从政策环境、社会需求、技术环境的角度看,仿制药的发展潜力都十分诱人,但我国要想从仿制药大国向仿制药强国转变,这中间还有许多路要走。本文主要就我国目前仿制药的发展现状及策略进行探讨。 关键词:仿制药;专利;现状;发展 根据IMS的数据,到2015年,将有1600亿美元规模的药品专利过期,给仿制药带来巨大的发展市场[1]。而我国虽为仿制药大国,但绝不是仿制药强国,生产的仿制药主要面向国内市场,部分仿制药出口,主要对象也是亚洲、非洲和拉丁美洲,而在全球医药消费比重较大的欧洲和北美洲所占份额较小[2]。我国要想从仿制药大国向仿制药强国转变,这中间还有许多路要走。 1.仿制药概述 仿制药( generic drug) 又叫通用名药、国际非专有名称药或非专利药。在我国是指国家食品药品监督管理局批准上市的已有国家标准的药品。国家规定仿制药应在5个方面与被仿制药达到一致,即活性成分、给药途径、剂型、规格、治疗作用[3]。但在美国,考虑到首家仿制药品的企业在仿制初期需要投入一定的研发资金,又从中划分出首仿药这一独立体,即第一个仿制申请者将拥有180天的市场专卖权,该期间FDA不再批准相同的ANDA( Abbreviated New Drug Appli- cation)上市,而且该药能够以新药80%的价格销售,从而保护该企业的利益,避免“一药多名”的产生[4]。从长远角度考虑,我国合理制定首仿药的相关政策势在必行。
仿制药(又称Generic Drug)是指与原研药(或称商品名药)在剂量、安全性和效力(strength)、质量、作用(performance)以及适应症(intended use)上相同的一种仿制品,又称通用名药、非专利药等。仿制研发的目标是实现临床应用上仿制药与原研药的“可替代性”。按照美国FDA的观点,能够获得批准的仿制药必须满足以下条件:和被仿制产品含有相同的活性成分,其中非活性成分可以不同;和被仿制产品的适应症、剂型、规格、给药途径一致;生物等效;质量符合相同的要求;生产的GMP标准和被仿制产品同样严格。 仿制药的上市,可以提供更加充足的临床供应,较大幅度地降低药价,缓解患者的经济负担,具有降低医疗支出、提高药品可及性、提升医疗服务水平等重要经济和社会效益。国外统计数据显示,随着仿制药上市数量的增加,药品价格最低将下降到原研药最初价格的9%左右。 尽管如此,仿制药与原研药的差异也须引起应有的关注并着力进行有效地控制。仿制药只是复制了原研药主要成份的分子结构,而原研药生产中关键工艺步骤、关键试剂、生产工艺的“设计空间”或关键辅料的质量控制等属于企业核心机密内容,是仿制企业难以合法拷贝的,导致仿制药的杂质谱、释药行为等关键质量属性,在有些情况下难以与原研药完全一致;同时,相关法规也未规定仿制药中其他成份(辅料)的添加与原研药必须相同;在仿制药许可中,其生物利用度应具有原研药的±20%左右等。这些因素导致仿制药的安全性有效性与原研药间的差异难以完全消除。美国家庭医师学会曾在研究报告中用事实来表明原研药的疗效和安全性并不是仿制药可以完全可替代的,尤其是在治疗危急患者和危急疾病时更是需要高度关注。事实上,如何保持仿制药与原研药的“一致”,如何研究求证仿制药与原研药的“差异”,如何准确评估并有效控制这些“差异”带来的风险,正是仿制药研发中需要高度关注的重点。 1、仿制研发的基本思路与策略 1.1设计并确保与原研药的“一致性”是仿制药研发的基本思路 仿制药是对已上市原研药的“仿制”,自1983年FDA通过的Waxman法案后,各国对于仿制药,不要求重复进行原研药批准之前进行的动物研究和人体临床研究,而是通过证明和原研药的生物等效性即可获得批准,实现与原研药的临床可替代。因此,仿制研发需要围绕如下几个关键问题进行研究和求证:拟仿产品的质量概况,尤其关键质量属性(Critical Quality Attributes,CQAs)包括哪些?仿制药与原研药关键质量属性是否一致?决定产品关键质量属性的关键工艺要素包括哪些?如何从关键的工艺要素和质量标准的关键质控项目确保与原研药关键质量属性的一致性?如何在产品贮藏过程中保持这种一致?建立的质量保障体系能否有效保障研制产品与上市原研药的一致性? 但是,仿制药与原研药的“一致性”并不仅仅是指产品检验结果的一致性。药品的质量不是检验出来的,而是通过科学的设计得以保障和实现的。药品质量首先来源于早期产品及工艺的设计,形成于药品的生产过程,研发早期的产品和工艺的设计情况即决定了产品的“先天”质量特征,通过具体的生产过程将实际质量状况赋予具体产品中,质量标准用于进一步论证、揭示产品的质量,是质量保证体系的重要组成部分,但不是唯一的;药品质量的保证还要靠遵循GMP、生产工艺、原材料和生产过程的控制、稳定性研究等;药品的质量需要质量标准的终点控制和生产过程控制相结合。同时,通过研究揭示药品在各种环境因素(如温度、湿度和光等条件)影响下,其质量随时间的变化情况,并由此确立有效期以及贮藏条件,以确保其质量。 ICH在Q8、Q9中引入了质量源于设计、质量风险管理和药品质量体系的概念,指出药品的质量不是检验赋予,而是来源于设计,并利用药品研发过程中所获得的信息,在生产过程中进行质量风险管理所获得。在综合国际标准化组织(ISO)质量概念的基础上,结合生产质量管理规范(GMP),在ICH Q10提出了药品质量体系概念,认为药品质量控制应
仿制药研发流程——即在药品研发中项目管理制定的依据标准和项目管理任务完成后的关键节点评估与质量放行标准一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。
研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月) 三、仿制药的研发具体步骤(即项目管理的任务要求与关键节点评估与质量放行的参考标准) (一) 产品信息调研(约一周完成) 是否有合法原料提供;临床资料、不良反应资料及产品说明书等相关资料;国内及进口制剂剂型及规格;产品质量标准(原研标准、国内首仿标准、药典标准);原研处方组成及工艺研究资料;药品的稳定性资料;专利情况;生产注册情况(产品原研厂家、国内生产申报厂家数情况);参比制剂来源等。
一、新药的研发过程 发明和研究安全有效的新药是一个长期、艰难和昂贵的进程。一种药物从最初的实验室研究到最终摆放到药柜销售平均需要花费12年的时间。进行临床前试验的5000种化合物中只有5种能进入到后续的临床试验,而仅其中的1种化合物可以得到最终的上市批准。新药研发阶段如下: (一)新药物实体的发现和确立。根据化学或生物学药物设计、天然药物、生物药物既有的经验理论、偶然的发现或现有临床的经验启发等确立研发靶标及新药物实体的来源方案。 (二)临床前试验。由制药公司进行的实验室和动物研究,以观察化合物针对目标疾病的生物活性,同时对化合物进行安全性评估。 (三)研发中新药申请(Investigational New Application, IND)。在临床前试验完成后,公司要向FDA提请一份IND,之后才能开始进行药物的人体试验。如果30天内FDA没有发出不予批准的申明,此IND即为有效。提出的IND需包括以下内容:先期的试验结果,后续研究的方式、地点以及研究对象;化合物的化学结构;在体内的作用机制;动物研究中发现的任何毒副作用以及化合物的生产工艺。另外,IND 必须得到制度审核部门(the Institutional Review Board)的审核和批准。同时,后续的临床研究需至少每年向FDA提交一份进展报告并得到准许。 (四)临床试验。
(1)Ⅰ期:此阶段大概需要1年时间,这些试验研究了药物的安全性方面,包括安全剂量范围。此阶段的研究同时确定了药物在体内的吸收、分布、代谢和排泄、以及药物的作用持续时间等项目。 (2)Ⅱ期:此阶段需要约100到300名志愿患者参与进行一些控制研究,以评价药物的疗效。这个阶段大约需要2年时间。 (3) Ⅲ期:此阶段持续约3年时间,医师通过对病患的监测以确定疗效和不良反应。 (五)新药申请(New Drug Application, NDA)。通过三个阶段的临床试验,将分析所有的试验数据。如数据能够成功证明药物的安全性和有效性,公司将向FDA提出新药申请。新药申请必须包括公司所掌握的一切相关科学信息。典型的新药申请有10万页甚至更多。根据法律,FDA审核一份NDA 的时限应该为6个月。但是几乎所有案例中的新药申请从首次提交到最终获得FDA批准的过程都超过这个时限。 (六)批准。FDA批准一份新药申请,此种新药就可以被医师用于处方。公司必须继续向FDA提交阶段性报告,包括所有的不良反应报告和一些质量控制记录。FDA还可能对一些药物要求做进一步的研究(Ⅳ期),以评价药物的长期疗效。 二、仿制药的研发过程 仿制药是已有国家药品标准的原料药或制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已得到充分证实,仿制药研发的目的是做到规模
制剂仿制药研发具体流程 一、综述 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”: 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将防腐剂含量测定定入质量标准。 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。动态上的对比为影响因素试验、加速试验的对比,即稳定
性对比研究。 2.有效性“同”: 对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。 3.晶型: 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度;在做成制剂的过程中,又不能保证晶型不产生变化。 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定性方面可通过影响因素试验和加速试验的对比来说明。 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
1 世界仿制药市场 最近在马尔塔召开了国际仿制药协会年会,从大会信息可以发现,由于社会老龄化,很多重磅炸弹式的药品专利到期,各国政府采取措施控制医疗费用上升,仿制药因此有很大的发展空间。但是,实际上,仿制药的销售并没有很大的增长,以出厂价计,根据IMS Health的数据,2004年全球仿制药市场仅为410亿美元,其中,包括美国、加拿大、英国、法国、德国、意大利、西班牙和日本在内的八国的仿制药销售总和为310亿美元,其余市场总计仅100亿美元。 数据进一步指出,美国的仿制药市场占全球的54%。美国、德国和英国仿制药的使用比较普遍,从数量而言,达到50%~53%,而德国仿制药的普遍使用是近年医疗体制改革的成果;在日本,病人不喜欢使用仿制药,仿制药的利用率才16%,这还是日本近年努力的结果。2003年日本仿制药销售仅为3800亿日元,占日本整个药品销售72500亿日元的%。日本使用仿制药少的原因,主要在于政府没有激励机制,没有参考价格制度,医生没有用通用名开处方的习惯,日本的用药文化就是相信品牌。在2004年,仿制药销售占全球药品销售的8%,而数量为23%。在过去5年中,仿制药的增长速度达到17%,远远超过商品名药物的销售增长速度。IMS估计,到2009年,尽管可能有降价等因素影响,但是仿制药销售仍将增加10~15%,达到660~820亿美元。另外,对于仿制药市场的增长因素,不确定因素还包括生物仿制药问题,美国表示反对,而欧洲的态度相对比较积极。 表1:2004年4月到2005年3月全球药品市场的情况 2 我国化学药品医院市场状况 2005年上半年我国化学药医院市场基本情况分析 根据统计所得,2005年1~6月化学药在医院购进金额较2004年同期增长%。在各大类药物中,抗感染药依然占据医院化学药市场的首位,但在国家降价措施以及对抗生素滥用的管理措施的出台后,该类药物的市场份额已由2004年上半年的%下降至2005年上半年的%,而市场份额处于上升的药物类别主要有血液及造血系统药物、心血管系统药物、抗肿瘤和免疫调节剂这三类,其他类别的药物市场份额基本处于窄幅波动。
仿制药研发具体流程 1 2 一、综述 3 4 根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全5 6 性、有效性已经得到较充分证实。 7 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 8 9 2、增加批准前生产现场的检查。 10 3、按照CTD格式要求提供申报资料,使申报规范,统一。 11 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 12 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出13 质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 14 15 分析上述新要求和参考指导原则,从而得出结论: 16 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求17 是做到“同”。方法为对比研究。 18 1.安全性“同”: 19 对于安全性,口服固体制剂控制的主要为有关物质,而液体制剂除控制有关物质外,还需对防腐剂、氧化剂等对人体有影响的物质进行控制。因此,必须要将 20 21 防腐剂含量测定定入质量标准。 22 研究的内容:静态上应包括杂质谱的对比,单个杂质的对比,杂质总量的对比。 动态上的对比为影响因素试验、加速试验的对比,即稳定性对比研究。 23 24 2.有效性“同”:
对于口服固体制剂,口服混悬剂(包括干混悬剂),溶出曲线是主要的控制指 25 26 标[1];对于口服溶液剂,防腐剂、矫味剂、氧化剂、增溶剂及稳定剂的选用非27 常重要,控制点为口感、渗透压、PH及有无絮凝现象;对于局部用制剂(如鼻28 喷雾剂),粒度分布、渗透压及黏度是主要控制指标。 29 研究的内容:分别进行溶出曲线对比;粒度分布对比;渗透压及黏度对比。30 3.晶型: 31 晶型的不同,药物的溶解度及稳定性有可能不相同,从而导致生物利用度不尽32 相同。而某个药物的晶型,文献资料很少;制剂中原料的晶型测定有一定的难度; 33 在做成制剂的过程中,又不能保证晶型不产生变化。 34 但是,鉴于仿制药研究的特点,溶解度方面可通过溶出曲线对比来说明;稳定35 性方面可通过影响因素试验和加速试验的对比来说明。 36 37 38 二、仿制药研发项目汇总(从立项到申报,时间为10—12个月)
详解仿制药研发具体流程
详解仿制药研发具体流程目录 一、综述2 二、仿制药研发项目汇总3 三、仿制药的研发具体步骤:5 (一)产品信息调研5 (二)前期准备(约一个月完成):5 1、参比制剂的采购5 2、原料采购5 3、色谱柱及对照品采购5 4、辅料采购:6 5、包材的采购:6 (三)处方工艺研究6 1、原辅料及参比制剂的检验6 2、处方工艺摸索6
3、初步验证工艺8 4、中试生产及工艺验证8 (四)质量研究9 1、质量研究项目的选择及方法初步确定9 2、质量标准的方法学验证10 3、质量对比研究12 4、质量标准的制定14 (五)稳定性研究(中试产品)14 (六)药理毒理研究16 (七)申报资料的撰写、整理16 (八)申报临床及申报现场核查17 (九)临床研究17 (十)申报生产17 一、综述
根据药品注册管理办法附件二的规定,仿制药即是已有国家药品标准的原料药或者制剂,该类药物国内已批准生产或上市销售,经过国内外广泛使用,其安全性、有效性已经得到较充分证实。 如今的新法规对仿制药提出了新的要求,主要是以下几点: 1、规范对被仿制药品的选择原则,即参比制剂的选择问题。 2、增加批准前生产现场的检查。 3、按照CTD格式要求提供申报资料,使申报规范,统一。 4、强调了对比研究,是判断两者质量是否一致的重要方法之一。 5、强化了工艺验证,目的是确保大生产时能始终如一地按照申报工艺生产出质量恒定的产品。 6、提出了晶型的要求,晶型的不同,溶解度和稳定性不同。 分析上述新要求和参考指导原则,从而得出结论: 仿制药研发的目的是做到规模化生产,强调本地化,以实现“替代性”。要求是做到“同”。方法为对比研究。 1.安全性“同”:
各药企对工艺研究都大同小异,我认为目前较为关键但是有普遍被多数企业所忽视的立项工作: 搞新药开发立项前查询是很重要的一步,在一个项目立项前对相关信息调研清楚了可以起到事半功倍的效果,如果调研工作做不好,以后的研发过程中将可能很被动。所以新药研发立项前查询工作是不容忽视的一个环节。但这项工作中涉及的面非常广,其中有许多问题需要注意,现在就请大家谈谈对立项前查询的看法: 1、立项前查询在新药研发过程中起到什么作用,是否有必要? 2、立项前查询中应该注意哪些问题? 本人不是搞立项的,本人认为立项前查询在新药研发过程中起到关键性的作用,相当必要,立项前查询中注意下面问题: 一、了解对国内外医药市场发展。 二、复方制剂的组方考虑。1.处方中的组分是否有配伍禁忌现象。2.处方中是否存在毒性较大,不良反应明显的药物。3.对作用机制相关性是否进行周密的考虑。4.对“个体化给药”是否给予充分的重视。 三、剂型的选择依据是否全面。 四、药物作用机制的设计是否严格。 五、是否处考察安全。 六、基础研究工作中是否科学,严肃。 七、新药类别的判断是否准确: 1、首研(或全球市场最大供应者)单位的查询,看该公司该品种的销售数据的增长趋势,由此一般可以判断该品种是处于发展中的那一个阶段,如成长期,成熟期,还是衰退期。 2、适应症,整个适应症的用药,如该品种不是该适应症的主要用药,那它的作用机制是否独特? 3、发病人群,是否有区域和经济文化背景的差异? 4、剂型,针对适应症的那一个人群市场??剂型设计是否新颖有吸引力? 5、不良反应,针对不同适应症,不良反应的要求是不一样的。显然,治疗感冒的药物和一个肿瘤用药的不良反应要求是不一样的。
新药/仿制药注册申报资料形式审核要求 申请注册新药: 按照《申报资料项目表》的要求报送资料项目1~30(资料项目6除外)。临床试验完成后报送的资料项目包括重新整理的综述资料1~6、资料项目12和14、临床试验资料28~32以及重新整理的与变更相关的资料和补充的资料,并按申报资料项目顺序排列。 对于注册分类1的品种,临床试验完成后应根据临床期间进行的各项研究的结果,重新整理报送资料项目1~30的全部资料 同时申请注册属于注册分类3的原料药和属于注册分类6的制剂的,其原料药的注册申请应当符合申报生产的要求。 申请注册仿制药品: 按照《申报资料项目表》的要求报送资料项目1~16和28~30。需进行临床试验的,在临床试验完成后报送资料项目28~32以及其他变更和补充的资料,并按申报资料项目顺序排列。 注册申报分三类情况: A:申报临床 B:申报生产 C:仿制药的申报 A、(申报临床)申报资料项目: (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书、起草说明及相关参考文献。 6、包装、标签设计样稿。(可不附) (二)药学研究资料 7、药学研究资料综述。 8、原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。 11、药品标准及起草说明,并提供标准品或者对照品。
12、样品的检验报告书。 13、原料药、辅料的来源及质量标准、检验报告书。 14、药物稳定性研究的试验资料及文献资料。 15、直接接触药品的包装材料和容器的选择依据及质量标准。(三)药理毒理研究资料 16、药理毒理研究资料综述。 17、主要药效学试验资料及文献资料。 18、一般药理学的试验资料及文献资料。 19、急性毒性试验资料及文献资料。 20、长期毒性试验资料及文献资料。 21、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等特殊安全性试验资料和文献资料。 22、复方制剂中多种成份药效、毒性、药代动力学相互影响的试验资料及文献资料。 23、致突变试验资料及文献资料。 24、生殖毒性试验资料及文献资料。 25、致癌试验资料及文献资料。 26、依赖性试验资料及文献资料。 27、非临床药代动力学试验资料及文献资料。 (四)临床试验资料 28、国内外相关的临床试验资料综述。 29、临床试验计划及研究方案。 30、临床研究者手册。 31、知情同意书样稿、伦理委员会批准件。(无) 32、临床试验报告。(无) B、(临床结束后报生产)申报资料项目: (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书、起草说明及相关参考文献。 6、包装、标签设计样稿。 (二)药学研究资料
仿制药-选题立项--汇总
————————————————————————————————作者:————————————————————————————————日期:
各药企对工艺研究都大同小异,我认为目前较为关键但是有普遍被多数企业所忽视的立项工作: 搞新药开发立项前查询是很重要的一步,在一个项目立项前对相关信息 调研清楚了可以起到事半功倍的效果,如果调研工作做不好,以后的研发过程 中将可能很被动。所以新药研发立项前查询工作是不容忽视的一个环节。但这 项工作中涉及的面非常广,其中有许多问题需要注意,现在就请大家谈谈对立项 前查询的看法: 1、立项前查询在新药研发过程中起到什么作用,是否有必要? 2、立项前查询中应该注意哪些问题? 本人不是搞立项的,本人认为立项前查询在新药研发过程中起到关键性的作 用,相当必要,立项前查询中注意下面问题: 一、了解对国内外医药市场发展。 二、复方制剂的组方考虑。1.处方中的组分是否有配伍禁忌现象。2.处方 中是否存在毒性较大,不良反应明显的药物。3.对作用机制相关性是否进行周密 的考虑。4.对“个体化给药”是否给予充分的重视。 三、剂型的选择依据是否全面。 四、药物作用机制的设计是否严格。 五、是否处考察安全。 六、基础研究工作中是否科学,严肃。 七、新药类别的判断是否准确: 1、首研(或全球市场最大供应者)单位的查询,看该公司该品种的销售数据 的增长趋势,由此一般可以判断该品种是处于发展中的那一个阶段,如成长期,成 熟期,还是衰退期。 2、适应症,整个适应症的用药,如该品种不是该适应症的主要用药,那它的作 用机制是否独特? 3、发病人群,是否有区域和经济文化背景的差异? 4、剂型,针对适应症的那一个人群市场??剂型设计是否新颖有吸引力? 5、不良反应,针对不同适应症,不良反应的要求是不一样的。显然,治疗感 冒的药物和一个肿瘤用药的不良反应要求是不一样的。
新药仿制药注册申报资料形式审核要求篇一:仿制药注册申报流程及资料 新法规对仿制药的要求 新法规对仿制药提出了更高的要求,主要体现在以下几点: 1. 对被仿制药品选择提出要求 注册管理办法第七十四条规定--仿制药应当与被仿制药具有同样的活性成份、给药途径、剂型、规格和相同的治疗作用。已有多家企业生产的品种,应当参照有关技术指导原则选择被仿制药进行对照研究。一般应首先选择以进口原研药,因为原发厂产品经过系统的非临床与临床研究,安全有效性得到确认,进口时对人种差异进行了研究。其次可考虑选用研究基础较好、临床应用较为广泛的非原研产品;没有进口原研产品的,有必要对市售品进行质量对比考查,择优选用,以确保仿制基础的可靠性。人体生物等效性试验与质量对比研究的参比品应是同一厂家最好相同批次产品,以全面说明其物质基础及体内过程的一致性和等效性,为桥接其安全有效性奠定更为坚实的基础。 2. 增加生产现场检查项目 注册管理办法第七十七条规定省、自治区、直辖市药品监督管理部门应当自受理申请之日起5 日内组织对研制情况和原始资料进行现场核查,并应当根据申请人提供的生
产工艺和质量标准组织进行生产现场检查,现场抽取连续生产的3 批样品,送药品检验所检验。样品的生产应当符合本办法第六十三条的规定。 原法规对于药物通过小试后达到中试的研究水平后批准生产,由于工艺不成熟,在验证的时候修订处方工艺后真正进入大生产;现法规规定药物由小试研究后通过中试规模完善处方工艺,用完善的处方 工艺进行验证试验,保证始终如一按既定处方工艺能生产出均一稳定 的样品后省局现场核查,抽取样品检验合格后,国家局对申报材料进 行审评,综合结合研制现场检查和生产现场核查结果才能批准生产。 这样的审批程序可解决既往申报工艺与大生产工艺不一致、大生 产不可行的弊端,同时保证申报工艺的大生产可行性,质量标准针对 大生产样品的适用性。只有现场核查、药审中心审评结论、生产检查 及药检报告三项都过关,才能拿到药品批准文号,加强了监管力度, 体现了仿制药的过程控制理念。