p53 Signaling
RT2 Profiler? PCR Array
p53 Signaling Pathway PCR Array
Cellular Senescence PCR Array
DNA Damage Signaling Pathway PCR Array
Cell Cycle PCR Array
SureSilencing RNAi
p53 Signaling Pathway Gene RNAi
Cellular Senescence Gene RNAi
DNA Damage Signaling Pathway Gene RNAi
Cell Cycle Gene RNAi
Cignal? Reporter Assays
p53 Pathway Reporter Assay Kit
E2F Reporter Assay Kit
EGR1 Reporter Kit
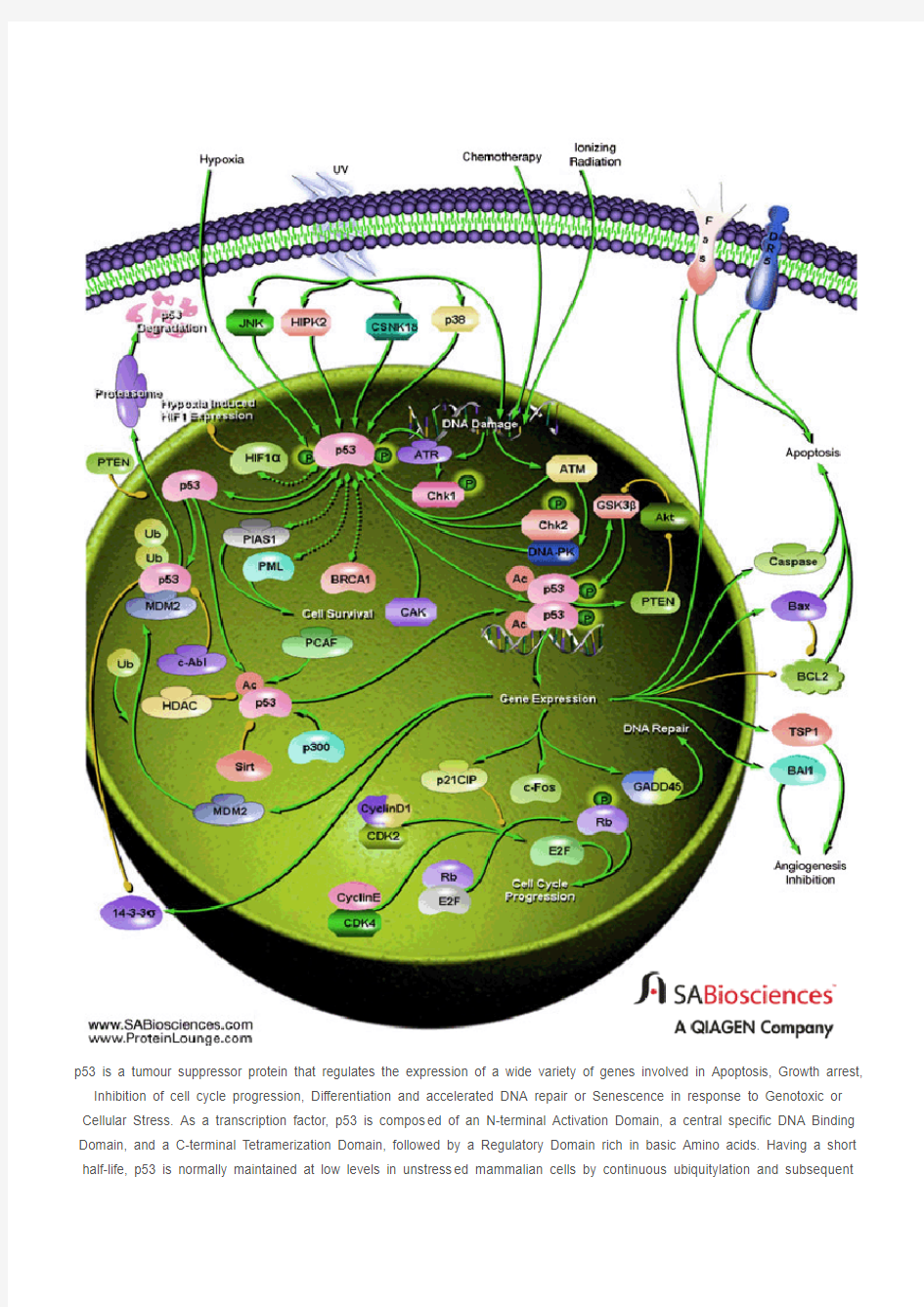
p53 is a tumour suppressor protein that regulates the expression of a wide variety of genes involved in Apoptosis, Growth arrest, Inhibition of cell cycle progression, Differentiation and accelerated DNA repair or Senescence in response to Genotoxic or Cellular Stress. As a transcription factor, p53 is compos ed of an N-terminal Activation Domain, a central specific DNA Binding Domain, and a C-terminal Tetramerization Domain, followed by a Regulatory Domain rich in basic Amino acids. Having a short half-life, p53 is normally maintained at low levels in unstress ed mammalian cells by continuous ubiquitylation and subsequent
degradation by the 26S Proteasome. Nonphosphorylated p53 is ubiquitylated by the MDM2 (Mouse Double Minute-2) ubiquitin ligase. MDM2 binding inactivates p53 by two mechanisms. First, MDM2 binds to the transactivation domain of p53, precluding interaction with the transcriptional machinery. Second, this binding mediates the covalent attachment of ubiquitin to p53. Ubiquitylated p53 is then degraded by the Proteasome. Thus MDM2 acts as a major reg ulator of the tumor suppressor p53 by targeting its destruction. When the cell is confronted with stress like DNA damage, Hypoxia, Cytokines, Metabolic changes, Vi ral infection, or Oncogenes, however, p53 ubiquitylation is suppressed and p53 accumulates in the nucleus, where it is activated and stabilized by undergoing multiple covalent modifications including Phosphorylation and Acetylation (Ref.1 & 2).
Phosphorylation of p53 mostly occurs in the N-terminal activation domain at the Ser6, Ser9, Ser15, Thr18, Ser20, Ser33, Ser37, Ser46, Thr55, and Thr81 residues, with some phosphorylation occurring in the C-terminal linker and basic regions at Ser315, Ser371, Ser376, Ser378, and Ser392. Phosphorylation on most of these sites is induced by DNA damage, with som e, such as Thr55 and Ser376, being repressed upon genotoxic stress. p53 phosphorylation is mediated by several cellular kinases including Chks (Checkpoint Kinases), CSNK1-Delta (Casein Kinase-1-Delta), CSNK2 (Casein Kinase-2), PKA (Protein Kinase A), CDK7 (Cyclin-Dependent Kinase-7), DNA-PK (DNA-Activated- Protein Kinase), HIPK2 (Homeodomain-Interacting Protein Kinase-2), CAK (CDK-Activating Kinase), p38 and JNK (Jun NH2-terminal kinase). Notably, phosphorylation at Ser15 by ATM (Ataxia Telangiectasia Mutated Gene)/ATR (Ataxia-Telangiectasia and Rad3 Related), either directly or through Chk1 (Cell Cycle Checkpoint Kinase-1)/Chk2 (Cell Cycle Checkpoint Kinase-2), or at Ser20 by Chk1/Chk2 has been shown to alleviate the inhibition or degradation of p53, leading to p53 stabilization and activation. The phosphorylation-induced p53 stabilization and activation are mediated through multiple mechanisms and may vary according to the cellular context or microenvironment. HIF-1Alpha (Hypoxia-Inducible Factor-1-Alpha) has been implicated to be involved in p53 stabilization, the precise mechanism by which HIF-1Alpha regulates p53-mediated function remains unknown. Recently, the interaction between p53 and HIF-1Alpha was reported to evoke HIF-1Alpha degradation. Members of the PIAS (Protein Inhibitor of Activated STAT) protein family have also been found to interact with p53. PIAS1 and PIAS-Gamma function as SUMO (Small Ubiquitin Related Modifier-1) ligases for p53. Moreover, the RING finger domain of PIAS1 binds to the C-terminus of the tumor suppressor p53 and catalyzes its sumoylation, a modification which represses p53 activity on a reporter plasmid containing consensus p53 DNA binding sites. PML (Promyelocytic Leukemia) also activates p53 by recruiting it to multiprotein complexes termed PML-nuclear bodies. PML is a tumor suppressor protein and the major component of multiprotein nuclear complexes that have been variably termed Kremer bodies, ND10, PODs (for PML Oncogenic Domains), and PML-NBs (PML-Nuclear Bodies). PML binds directly with p53 and recruits it to PML-NBs. Recruitment to PML-NBs activate p53 by bringing it in close proximity with CBP (CREB-Binding Protein) /p300. BRCA1 (Breast Cancer-1 Gene) and p53 can also physically associate, both in vitro and in vivo and function in a common pathway of tumor suppression. The ability of BRCA1 to biochemically modulate p53 function suggests that this may be a
fundamental role of BRCA1 in tumor suppression (Ref.3, 4 & 5).
Another important modification of p53 is acetylation. p53 is specifically acetylated at Lys370, Lys372, Lys373, Lys381, and Lys382 by p300/CBP and at Lys320 by PCAF (p300/CBP-associated factor). Acetylation has been shown to augment p53 DNA binding, and to stimulate p53-mediated transactivation of its downstream target genes through the recruitment of coactivators. Acetylation may also regulate the stability of p53 by inhibiting its ubiquitination by MDM2. In vivo, acetylation at Lys320, Lys373, and Lys382 is induced by many genotoxic agents, including UV-radiation, IR (Ionizing Radiation), hypoxia, oxidative stress, and even depletion of ribonucleotide pools. p53 can also be deacetylated by HDAC1 (Histone Deacetylase-1) and SIRT1. Human SIRT1 is an enzyme that deacetylates the p53 tumor suppressor protein and has been suggested to modulate p53-dependent functions including DNA damage-induced cell death. p53 deacetylation has been suggested to down-regulate the activation of genes such as Bax and p21WAF1. Phosphorylation and acetylation are interdependent. In deed, phosphorylation at the p53
N-terminus has been shown to enhance its interaction with acetylase p300/CBP and to potentiate p53 acetylation. Activated p53 functions effectively as a transcription factor and induces transcription of several genes. The D NA targets of p53 are consensus
sequences consisting of a 10-base pair repeat of 5'-PuPuPu-C(A/T)(T/A)GPyPyPy-3' (where Pu is a purine and Py is a pyrimidine). It also can bind to a palindromic site having a four or five-base pair inverted repeat of a similar sequence. Complete p53 is inactive for specific DNA binding unless activated by covalent and noncovalent modifications of the basic C-terminal domain. After p53 is activated it can be involved in cell-cycle inhibition, apoptosis, genetic repair, and inhibition of blood-vessel
formation (Ref.5, 6 & 7).
Cell cycle inhibition takes place when there is a block in cell-cycle division. p53 does this by stimulating the expression of p21 WAF1/CIP1 (Cyclin Dependent Kinase Inhibitor-p21). This protein is an inhibitor of CDKs (Cyclin-Dependent Kinases) that regulate the cell cycle via perturbation of their partner cyclin. Cyclins are involved to ensure successful transitions from S phase to G1. Since p21 WAF1/CIP1 inhibits CDKs it results in inhibition of both G1-to-S and G2-to-mitosis transitions by causing hypophosphorylation of Rb (Retinoblastoma) and preventing the release of E2F. Additionally p53 can stimulate 14-3-3, a protein that sequesters Cyclin B1-CDK1 complexes out of the nucleus. This results in a G2 block. Activated p53 may also initiate apoptosis and stop cell proliferation. p53 stimulates a wide network of signals that act through two major apoptotic pathways: Extrinsic Pathways and Intrinsic Pathways. The extrinsic pathway involves engagement of pa rticular `death' receptors that belong to the TNFR (Tumor Necrosis Factor Receptor) family and, through the formation of the DISC
(Death-Inducing-Signaling-Complex), leads to a cascade of activation of Caspases, including Caspase8 and Caspase3, which in turn induce apoptosis. Most common death receptors involved in extrinsic apoptosis Fas, DR5 (Death Receptor-5) and PERP. The intrinsic apoptotic pathway is dominated by the Bcl2 (B-Cell CLL/Lymphoma-2) family of proteins, which governs the release of CytoC (Cytochrome-C) from the mitochondria. The Bcl2 family comprises anti-apoptotic (pro-survival) and pro-apoptotic members. The Bcl2 family is divisible into three classes: pro-survival proteins, whose members are most structurally similar to Bcl2, such as BclXL; pro-apoptotic proteins, BAX (Bcl2 Associated-X Protein) and BAK (Bcl2 Antagonist Killer-1), which are structurally similar to Bcl2 and BclXL and antagonize their pro-survival functions; and the pro-apoptotic `BH3-only' proteins. Intriguingly, a key subset of the Bcl2 family genes are p53 targets, including BAX, Noxa, PUMA (p53-Upregulated Modulator of Apoptosis) and the most recently identified, BID (BH3 Interacting Domain Death Agonist). p53 may also inhibit Bcl2 that is an inhibitor of apoptosis. p53 may also have a role in maintaining genetic stability by 'nucleotide-excision' repair of DNA, chromosomal recombination and chromosome segmentation. GADD45 (Growth Arrest- and DNA Damage-Inducible Gene-45) is a multifunctional protein that is regulated by p53 and that may play a role in DNA repair and cell cycle checkpoints. p53 can play
a role in the inhibition of blood-vessel formation. In order for tumours to reach a large size, they must initiate the growth of nutrient-bringing blood vessels in their vicinity, the process of angiogenesis. p53 stimulates the production of genes that prevent this process from happening. p53 activates the expression of the Tsp1 (Thrombospondin-1), an anti-angiogenic factor, along with other angiogenesis inhibitor BAI1 (Brain-specific Angiogenesis Inhibitor-1) (Ref. 8, 9 & 10).
In addition, p53 regulates MDM2 function in a negative feedback loop, because the MDM2 gene is a target for p53. Therefore, activation of p53 eventually leads to its own inactivation by switching on a pathway that leads to its destruction. MDM2 is subject to further regulation by direct binding of the ARF (Active Response Factor) protein, which prevents MDM2-mediated p53 proteolysis. PTEN (Phosphatase and Tensin Homolog), on the other hand inhibits MDM2-mediated p53 degradation. p53 can transcriptionally activate PTEN, which may further inhibit Akt activity. Therefore, inhibition of Akt by the inhibitors may trigger a positive feedback with perhaps additional anti-tumor effects. The c-Fos proto-oncogene is also a target for transactivation by the p53 tumor suppressor. Mutations in p53 are associated with genomic instability and increased susceptibility to cancer. It is the most frequently mutated protein in all cancer with an estimated 60% of all cancers having mutated forms that affect its growth suppressing activities. However some common tumours have a higher incidence such that 90% of cervical and 70% of colorectal are found to have p53 mutations. The p53 protein can be inactivated in several ways, in cluding inherited mutations that result in a higher incidence of certain familial cancers such as Li-Fraumeni syndrome. Certain DNA tumour viruses, such as the human adenovirus and the papilomavirus, bind to and inactivate the protein. Functional p53 is th ought to provide a protective effect
against tumorigenesis (Ref.2, 11 & 12).
免疫与炎症相关信号通路 一、Jak/Stat Signaling:IL-6 Receptor Family Jak和Stat是许多调节细胞生长、分化、存活和病原体抵抗信号通路中的关 键部分。就有这样一个通路涉及到IL-6(gp130)受体家族,它帮助调节B 细胞的分化,浆细胞生成和急性期反应。细胞因子结合引起受体的二聚化同 时激活受体结合的Jak蛋白,活化的Jak蛋白对受体和自身进行磷酸化。这些磷酸化的位点成为带有SH2结构的Stat蛋白和接头蛋白的结合位臵,接头蛋白将受体和MAP激酶,PI3激酶/Akt还有其他的通路联系在一起。受体结合的Stat蛋白被Jak磷酸化后形成二聚体,转移进入细胞核调节目的基因的 表达。细胞因子信号传导抑制分子(SOCS)家族的成员通过同源或异源的 反馈减弱受体传递的信号。Jak或Stat参与其他受体蛋白的信号传导,在下 面Jak/Stat使用表格中有这方面的列举。研究人员已经发现Stat3和Stat5在一些实体肿瘤中被酪氨酸激酶而不是Jaks组成性激活。 JAK/STAT途径介导细胞因子的效应,如促红细胞生成素,血小板生成素, G-CSF,这些细胞因子分别是用于治疗贫血,血小板减少症和中性粒细胞减 少症的蛋白质类药物。该途径也通过干扰素介导信号通路,干扰素可以用来 作为抗病毒和抗增殖剂。研究人员发现,失调的细胞因子信号有助于癌症的 发生。异常的IL-6的信号或导致自身免疫性疾病,炎症,癌症,如前列腺癌 和多发性骨髓瘤的发生。Jak抑制剂目前正在多发性骨髓瘤模型中进行测试。Stat3具有潜在促癌性(原癌基因),在许多癌症中持续的表达。在一些癌细 胞中,细胞因子信号传导和表皮生长因子受体(EGFR)家族成员之间存在交流。 Jak激活突变是恶性血液病中主要的分子机制。研究人员已经在Jak2假激酶域中发现一个特有的体细胞突变(V617F),这个突变常常发生于真性红细 胞增多症,原发性血小板增多症和骨髓纤维化症患者。这个突变导致Jak2的病理激活,同时激活控制红细胞,巨核细胞和粒细胞增殖分化的促红细胞 生成素(EPO),血小板生成素(TPO)和G-CSF等的受体。而Jak1的功能获得性体细胞突变已发现存在于成人急性淋巴细胞性白血病当中。体细胞激活突变已经证明存在于小儿急性淋巴细胞白血病(ALL)患者中。此外,
Hippo信号通路 一、Hippo信号通路概述 Hippo 信号通路,也称为Salvador / Warts / Hippo(SWH)通路,命名主要源于果蝇中的蛋白激酶Hippo(Hpo),是通路中的关键调控因子。该通路由一系列保守激酶组成,主要是通过调控细胞增殖和凋亡来控制器官大小。 Hippo信号通路是一条抑制细胞生长的通路。哺乳动物中,Hippo信号通路上游膜蛋白受体作为胞外生长抑制信号的感受器,一旦感受到胞外生长抑制信号,就会激活一系列激酶级联磷酸化反应,最终磷酸化下游效应因子YAP和TAZ。而细胞骨架蛋白会与磷酸化后的YAP和TAZ结合,使它滞留在细胞质内,降低其细胞核活性,从而实现对器官大小和体积的调控。 二、Hippo信号通路家族成员 虽然Hippo信号通路在各个物种中保守性很高,但是相同功能的调控因子或效应因子在不同物种间还是存在着差异,下表中我们对比了果蝇与哺乳动物中Hippo信号通路相同功能的关键因子[1]。
Expanded(Ex) FRMD6/Willin 含有FERM结构域的蛋白,能与Kibra及Mer结合,调控Hippo信号通路的上游信号 Dachs(Dachs) 肌浆球蛋白myosin的一种,能结合Wts 并促进其降解 Kibra(Kibra) WWC1 含有WW结构域的蛋白,能与Ex及Mer 结合,调控Hippo信号通路的上游信号 Merlin(Mer) NF2 含有FERM结构域的蛋白,能与Kibra及Ex结合,调控Hippo信号通路的上游信号 Hippo(Hpo) MST1,MST2 Sterile-20-样激酶,磷酸化并激活Wts Salvador(Sav) WW45(SAV1) 含有WW结构域的蛋白,能起到一个脚手架蛋白的作用,易化Hippo对Warts的磷酸化 Warts(Wts)LATS1,LATS2 核内DBF-2相关激酶,能磷酸化Yki并使之失活 Mob as tumor suppressor(Mats) MOBKL1A,MOBKL1B 能与Wts结合的激酶,与Wts结合后能 促进Wts的催化活性 Yorkie(YKi) YAP,TAZ 转录共激活因子,能在非磷酸化的激活状态下与转录因子Sd结合,并激活下游靶基因的转录。这些受调控的下游靶基因主要参与了细胞的增殖、生长并抑制凋亡的发生 Scalloped(Sd) TEAD1,TEAD2,TEAD3, TEAD4 能与Yki结合的转录因子,与Yki共同 作用,调控靶基因的转录 三、Hippo信号通路的功能 近十年相关研究结果表明,无论是果蝇还是哺乳动物,Hippo信号通路都可以通过调节细胞增殖、凋亡和干细胞自我更新能力实现对器官大小的调控。Hippo信号通路异常会导致大量组织过度生长。此外,大量研究证实,Hippo信号通路在癌症发生、组织再生以及干细胞功能调控上发挥着重要功能[2][3][4]。 a.Hippo信号通路在器官大小控制中的作用 起初,关于Hippo信号通路的研究主要集中在器官大小的调控。大量研究表明,Hippo 途径主要通过抑制细胞增殖并促进细胞凋亡,继而实现对器官大小的调控。激酶级联反应是该信号传导的关键。Mst1/2激酶与SA V1形成复合物,然后磷酸化LATS1/2;活化后的LATS1/2激酶随即磷酸化Hippo信号通路下游关键效应分子——Y AP和TAZ,同时抑制了
224 中国医药生物技术 2009年6月第4卷第3期Chin Med Biotechnol, June 2009, V ol. 4, No. 3 DOI:10.3969/cmba.j.issn.1673-713X.2009.03.012 · 综述·Notch信号通路研究进展 王利祥,华子春 1917 年,Morgan 及其同事在果蝇体内发现一种基因,因其功能部分缺失可导致果蝇翅缘出现缺口,故命名该基因为 Notch。随后的研究发现,Notch 从无脊椎动物到脊椎动物的多个物种中表达,其家族成员的结构具有高度保守性,在细胞分化、发育中起着关键作用。迄今研究已阐明 Notch 信号通路的主要成员及核心转导过程,然而随着研究的深入,人们逐渐认识到该通路实际上处于十分复杂的调控网络之中,而这与其在发育过程中功能的多样性相符合。本文结合最新进展,系统阐述 Notch 信号通路的组成,功能,作用机制及调控,并揭示该通路异常与疾病的联系。 1 Notch 受体 Notch 受体是一个相对分子量约为 30 000 的 I 型膜蛋白,由胞外亚基和跨膜亚基组成,2 亚基之间通过 Ca2+ 依赖的非共价键结合形成异源二聚体。胞外亚基包含一组串联排列的 EGFR 和 3 个家族特异性的 LNR 重复序列。EGFR 在 Notch 受体与配体的结合中起关键作用,在果蝇中,Notch 受体的第 11 位和 12 位 EGFR 介导了其与配体的结合。LNR 位于 EGFR 的下游,富含半胱氨酸,介导了 2 亚基之间 Ca2+ 依赖的相互作用。跨膜亚基包括跨膜区、RAM 序列、锚蛋白重复序列、核定位序列、多聚谷氨酰胺序列以及 PEST 序列。RAM 结构域是 Notch 信号效应分子 CBF1/RBPJk 主要的结合部位。ANK 重复序列结构域是 Deltex、Mastermind 等的结合部位,这些蛋白对Notch 信号通路有修饰作用。PEST 结构域与泛素介导的Notch 胞内段降解有关[1]。 2 Notch 配体 Notch 配体与受体一样为 I 型跨膜蛋白。果蝇 Notch 配体有 2 个同源物 Delta 和 Serrate,线虫的 Notch 配体为 Lag 2,故又称 Notch 配体为 DSL 蛋白。脊椎动物体内也发现了多个 Notch 配体,与 Delta 同源性高的称为Delta 样分子,与 Serate 同源性高的被称作 Jagged。目前,发现人的 Notch 配体有 D ll l、3、4和 Jagged l、2。配体胞外 DSL 结构域在进化中高度保守,是配体与受体结合、激活 Notch 信号所必需的。Notch 配体的胞内域较短,仅70 个左右氨基酸残基,功能尚未阐明。近来研究发现,Delta 1 的胞内域能够诱导细胞的生长抑制[2]。有人推测,配体胞内段可能类似与受体胞内段,具有信号转导功能,但具体机制有待进一步研究。3 Notch 信号传递与效应因子 迄今研究发现主要有 6 种信号通路在多细胞生物的生长中发挥关键作用,分别是刺猬、骨形态发生蛋白、无翅、类固醇激素受体、Notch 和受体酪氨酸激酶。Notch 相对于其他信号通路结构较简单,没有第二信使的参与。现有研究提出了 Notch 信号活化的“三步蛋白水解模型”[3]。首先,Notch 以单链前体模式在内质网合成,经分泌运输途径,在高尔基体内被 Furin 样转化酶切割成相对分子质量为180 000 含胞外区的大片段和 120 000 含跨膜区和胞内区的小片段。两者通过 Ca2+依赖性的非共价键结合为异源二聚体,然后被转运到细胞膜。当 Notch 配体与受体结合,Notch 受体相继发生 2 次蛋白水解。第一次由 ADAM 金属蛋白酶家族的 ADAM 10/Kuz 或 ADAM 17/TACE 切割为 2 个片段。N 端裂解产物(胞外区)被配体表达细胞内吞,而 C 端裂解产物随后由早老素 1/2,Pen-2,Aph1 和Nicastrin 组成的γ-促分泌酶复合体酶切释放 Notch 受体的活化形式 NICD。 经典的 Notch 信号通路又称为 CBF-1/RBP-Jκ依赖途径。CBF-1/RBP-Jκ本身是 1 个转录抑制因子,能够特异性地与 DNA 序列“CGTGGGAA”相结合,并招募 SMRT,SKIP,I/II 型组蛋白去乙酰化酶等蛋白形成共抑制复合物,抑制下游基因的转录。当 Notch 信号激活后,NICD 通过上述酶切反应被释放进入胞核,通过 RAM 结构域及 ANK 重复序列与 CBF-1/RBP-Jκ结合使共抑制复合物解离,并募集 SKIP,MAML 1 组成共激活复合体,激活下游基因的转录。Notch 信号的靶基因多为碱性螺旋-环-螺旋转录抑制因子家族成员,如哺乳动物中的 HES、非洲爪蟾中的XHey-1,以及近来发现的 BLBP [3]。此外,存在非CBF-1/RBP-Jκ依赖的 Notch 信号转导途径。最近有研究报道,果蝇 Notch 结合蛋白 Deltex 是某些组织特异性非 Su (H)依赖性信号所必需的,同时发现 Deltex 也具有拮抗Notch 的功能 [4]。 4 Notch 信号途径功能 Notch 信号途径的功能最初是在果蝇神经系统发育的 基金项目:国家自然科学基金(30425009,30730030);江苏省自然科学基金(BK2007715) 作者单位:210093 南京大学医药生物技术国家重点实验室 通讯作者:华子春,Email:zchua@https://www.doczj.com/doc/455043602.html, 收稿日期:2009-02-01
ATM Ataxia telangiectasia mutated (ATM) is a serine/threonine protein kinase that is recruited and activated by DNA double-strand breaks. It phosphorylates several key proteins that initiate activation of the DNA damage checkpoint, leading to cell cycle arrest, DNA repair or apoptosis. Several of these targets, including p53, CHK2 and H2AX are tumor suppressors. The protein is named for the disorder Ataxia telangiectasia caused by mutations of ATM.[1] Contents 1 Introduction 2 Structure 3 Function 4 Regulation 5 Role in cancer 6 Interactions 7 See also 8 References 9 Further reading 10 External links Introduction[edit] Throughout the cell cycle the DNA is monitored for damage. Damages result from errors during replication, by-products of metabolism, general toxic drugs or ionizing radiation. The cell cycle has different DNA damage checkpoints, which inhibit the next or maintain the current cell cycle step. There are two main checkpoints, the G1/S and the G2/M, during the cell cycle, which preserve correct progression. ATM plays a role in cell cycle delay after DNA damage, especially after double-strand breaks (DSBs).[2] ATM together with NBS1 act as primary DSB sensor proteins. Different mediators, such as Mre11 and MDC1, acquire post-translational modifications which are generated by the sensor proteins. These modified mediator proteins then amplify the DNA damage signal, and transduce the signals to downstream effectors such as CHK2 and p53. Structure[edit] The ATM gene codes for a 350 kDa protein consisting of 3056 amino acids.[3] ATM belongs to the superfamily of Phosphatidylinositol 3-kinase-related kinases (PIKKs). The PIKK superfamily comprises six Ser/Thr-protein kinases that show a sequence similarity to phosphatidylinositol 3-kinases (PI3Ks). This protein kinase family includes amongst others ATR (ATM- and RAD3-related), DNA-PKcs (DNA-dependent protein kinase catalytic subunit) and mTOR (mammalian target of rapamycin). Characteristic for ATM are five domains. These are from N-Terminus to C-Terminus the HEAT repeat domain, the FRAP-ATM-TRRAP (FAT) domain, the kinase domain (KD), the PIKK-regulatory domain (PRD) and the FAT-C-terminal (FATC) domain. The
World Journal of Cancer Research 世界肿瘤研究, 2014, 4, 41-46 Published Online October 2014 in Hans. https://www.doczj.com/doc/455043602.html,/journal/wjcr https://www.doczj.com/doc/455043602.html,/10.12677/wjcr.2014.44008 Review of the ERK5 Signaling Pathway Research Song Luo*, Shengfa Su, Weiwei Ouyang#, Bing Lu# Teaching and Research Section of Oncology, Guiyang Medical University, Guiyang Email: 4567436@https://www.doczj.com/doc/455043602.html,, #ouyangww103173@https://www.doczj.com/doc/455043602.html,, #lbgymaaaa@https://www.doczj.com/doc/455043602.html, Received: Sep. 25th, 2014; revised: Oct. 16th, 2014; accepted: Oct. 20th, 2014 Copyright ? 2014 by authors and Hans Publishers Inc. This work is licensed under the Creative Commons Attribution International License (CC BY). https://www.doczj.com/doc/455043602.html,/licenses/by/4.0/ Abstract Extracellular signal regulated kinase 5 (ERK5) is an important part of mitogen activated protein kinase (MAPK) system, and also is a new signal transduction pathway of MAPK signaling system, which has attracted much attention in recent years. ERK5 can be activated by many stimulating factors and plays an important role in cell survival, proliferation and differentiation. Furthermore, ERK5 is closely related to vascular development and proliferation, and other critical functions. This paper focuses on the origin, structure, property, physiological features of ERK5, and the relation-ship between ERK5 and tumor and non-oncologic diseases, and reviews the research direction in the future. Keywords ERK5, Signaling Pathways, MAPK ERK5信号通路研究现状 罗松*,苏胜发,欧阳伟炜#,卢冰# 贵阳医学院肿瘤学教研室,贵阳 Email: 4567436@https://www.doczj.com/doc/455043602.html,, #ouyangww103173@https://www.doczj.com/doc/455043602.html,, #lbgymaaaa@https://www.doczj.com/doc/455043602.html, 收稿日期:2014年9月25日;修回日期:2014年10月16日;录用日期:2014年10月20日 *第一作者。 #通讯作者。
1JAK-STAT信号通路 1)JAK与STAT蛋白 JAK-STAT信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程。与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT。(1)酪氨酸激酶相关受体(tyrosinekinaseassociatedreceptor) 许多细胞因子和生长因子通过JAK-STAT信号通路来传导信号,这包括白介素2?7(IL-2?7)、GM-CSF(粒细胞/巨噬细胞集落刺激因子)、GH(生 长激素)、EGF(表皮生长因子)、PDGF(血小板衍生因子)以及IFN(干扰素)等等。这些细胞因子和生长因子在细胞膜上有相应的受体。这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK 的结合位点。受体与配体结合后,通过与之相结合的JAK的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。 (2)酪氨酸激酶JAK(Januskinase) 很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体(receptor tyrosinekinase,RTK),而JAK却是一类非跨膜型的酪氨酸激酶。JAK是英文Januskinase的缩写,Janus在罗马神话中是掌管开始和终结的两面神。之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸、JAK1个成员:4蛋白家族共包括JAK结构域的信号分子。SH2化多个含特定
JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAKhomologydomain,JH),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域。 (3)转录因子STAT(signaltransducerandactivatoroftranscription)STAT被称为“信号转导子和转录激活子”。顾名思义,STAT在信号转导和转录激活上发挥了关键性的作用。目前已发现STAT家族的六个成员,即STAT1-STAT6。STAT蛋白在结构上可分为以下几个功能区段:N-端保守序列、DNA结合区、SH3结构域、SH2结构域及C-端的转录激活区。其中,序列上最保守和功能上最重要的区段是SH2结构域,它具有与酪氨酸激酶Src的SH2结构域完全相同的核心序列“GTFLLRFSS”。 2)JAK-STAT信号通路 与其它信号通路相比,JAK-STAT信号通路的传递过程相对简单。信号传 递过程如下:细胞因子与相应的受体结合后引起受体分子的二聚化,这使得与受体偶联的JAK激酶相互接近并通过交互的酪氨酸磷酸化作用而活化。JAK激活后催化受体上的酪氨酸残基发生磷酸化修饰,继而这些磷酸化的酪氨酸位点与周围的氨基酸序列形成“停泊位点”(dockingsite),同时含有SH2结构域的STAT蛋白被招募到这个“停泊位点”。最后,激酶JAK 催化结合在受体上的STAT蛋白发生磷酸化修饰,活化的STAT蛋白以二 聚体的形式进入细胞核内与靶基因结合,调控基因的转录。值得一提的是,一种JAK激酶可以参与多种细胞因子的信号转导过程,一种细胞因子的信号通路也可以激活多个JAK激酶,但细胞因子对激活的STAT分子却具有一定的选择性。例如IL-4激活STAT6,而IL-12 。STAT4却特异性激活
9 简述TLR4信号通路在炎症中的功能。 TLR4 recognizes lipopolysaccharide (LPS) together with myeloid differentiation factor 2 (MD2) on the cell surface. LPS is a component derived from the outer membrane of Gram-negative bacteria and is known to be a cause of septic shock. The crystal structure of a complex comprising TLR4, MD2, and LPS revealed that two complexes of TLR4-MD2-LPS interact symmetrically to form a TLR4 homodimer (Park et al., 2009). TLR4 is also involved in the recognition of viruses by binding to viral envelope proteins. In addition, TLR4 modulates the patho-genesis of H5N1 avian in?uenza virus infection by recognizing a DAMP rather than the virus itself (Imai et al., 2008). Acutelung injury caused by avian in?uenza virus infection produces endogenous oxidized phospholipids, which stimulate TLR4. Mice lacking TLR4 were found to be resistant to avian ?u-induced lethality. 1TL R4的结构分布及信号通路 TLR4是人类发现的第一个TLR 相关蛋白,几乎分布于所有的细胞系,主要表达在参与宿主
P53 信号通路 P53是一个肿瘤抑制蛋白,调节各种各样基因的表达,包括细胞凋亡,生长抑制,抑制细胞周期进程,分化和加速DNA修复,基因毒性和细胞应激后的衰老。作为一个转录因子,p53是N端激活域、DNA中央特定结合域和C-端四聚体化域的组成部分,而且其调控域富含碱性氨基酸。p53半衰期很短,在26S蛋白酶体作用下,通过持续的泛素化和后期降解,p53在无刺激的哺乳类动物细胞中维持较低的含量。 去磷酸化的p53在MDM2(鼠双微体基因-2)泛素连接酶作用下被泛素化。MDM2结合p53使其无活性是通过两种途径: 第一,MDM2结合p53的转录激活域,阻止转录元件的相互作用。 第二,介导p53共价结合泛素蛋白,泛素化的p53被蛋白水解酶降解。 通过使p53的失活,MDM2扮演着p53抑癌基因的主要监视者。当细胞面临着DNA损伤、缺氧、细胞因子、代谢改变、病毒感染或者致癌基因等刺激时,导致p53泛素化被抑制和p53在细胞核内积累,通过多个共价修饰包括磷酸化和乙酰化,p53被激活并稳定存在。 p53的磷酸化大多数出现在N-末端激活域的Ser6,Ser9,Ser15,Thr18,Ser20,Ser33,Ser37,Ser46,Thr55和Thr81残基上,另外还有一些p53磷酸化出现在C-末端连接处和碱性区域的Ser315, Ser371, Ser376, Ser378, 和Ser392残基上。大多数位点上的磷酸化是由DNA损伤诱发的,还有一些例如Thr55 and Ser376在基因毒性应激下被压抑。P53磷酸化是由几个细胞激酶所介导,包括Chks,CSNK1-Delta,CSNK2,PKA,CDK7,DNA-PK,HIPK2,CAK, p38和JNK。显然,由ATM/ATR作用在Ser15上磷酸化,直接作用或者通过Chk1/Chk2,在Chk1/Chk2作用下的Ser20磷酸化已经证实能够减缓抑制或减慢降解p53,导致p53稳定并活化。磷酸化诱导的p53稳定和活化是通过多种机制介导以及很多细胞环境或微环境的改变所致。HIF-1Alpha参与p53的稳定,HIF-1Alpha调节p53介导作用的详细机制依然是一个未知数。最近,p53和HIF-1Alpha之间的作用已被报道能够引起HIF-1Alpha的降解。PIAS 蛋白家族也被发现能与p53发生相互作用。PIAS1 和PIAS-Gamma作用可作为p53的SUMO 连接酶。另外,PIAS1的环指域结合p53抑癌基因的C-末端催化其类泛素化(sumoylation),这个修饰,能抑制报告质粒p53的活性,包含共同序列p53DNA的结合位点。PLM通过吸收p53到多蛋白复合体(称为PLM核体)来激活p53。PLM是一个肿瘤抑制蛋白,也是曾发生变异称为Kremer bodies蛋白核配合物(ND10, PODs and PML-NBs)的主要组成成分。PLM直接和p53结合并进入PLM-NBs。补充到PLM-NBs激活p53,通过把它引进到与CBP/p300极为贴近。BRCA1和p53发生联合,在体内和体外是相同的肿瘤抑制途径。BRCA1对于生化调节p53作用的能力暗示着在肿瘤抑制过程,这个可能是BRCA1一个基本的角色。 P53另一个重要的修饰是乙酰化作用。在Lys370,Lys372, Lys373, Lys381, and Lys382 by p300/CBP and at Lys320残基上通过PCAF,p53发生特殊的乙酰化。已证明,乙酰化可以增强p53DNA的结合、通过更新辅激活因子刺激p53介导的下游靶基因转录激活。乙酰化可以通过MDM2抑制p53的泛素化来调节p53的稳定性。在机体内,发生Lys320, Lys373, and Lys382残基上的乙酰化是通过许多基因毒性介质所诱导的,包括紫外线、电离辐射、缺氧、氧化应激,甚至耗尽的核苷酸池。P53同样也可以被HDAC1和SIRT1去乙酰化。人类的SIRT1是一种酶,属于去乙酰化p53肿瘤抑制蛋白,能够被显示调节p53依赖作用,包括DNA诱导损伤的细胞死亡。P53去乙酰化能够被应用到下调Bax and p21WAF1等基因的活性。磷酸化和乙酰化是相互依赖的。的确,p53N-末端的磷酸化被证实能够增强与乙酰化酶p300/CBP的相互作用和加强p53乙酰化。激活p53功能实际上是一个转录因子和诱导一些基因的转录。DNA靶基因p53是一个有十个5'-PuPuPu-C(A/T)(T/A)GPyPyPy-3'重复出现的共有序列。它也可以结合到一个含有四到五个重复出现碱基对的相同序列的回文位点。完整
资深PI最新文章解析信号通路 ------------------------------------------------------------------------------------------------------------------------------------ 摘要:来自新加坡分子与细胞生物学研究院,癌症与发育细胞生物学部的研究人员获得了YAP-TEAD4复合物在YAP因子N端结构域相互作用,以及在TEAD4 C端结构域与YAP相互作用的晶体结构,从中研究人员认为YAP中的PXXΦP片段是与TEAD4相互作用的关键结构,这为研究Hippo信号通路提供了重要的分子机理线索。这一研究成果公布在《Genes Development》杂志上。 生物通报道:来自新加坡分子与细胞生物学研究院,癌症与发育细胞生物学部的研究人员获得了YAP-TEAD4复合物在YAP因子N端结构域相互作用,以及在TEAD4 C端结构域与YAP相互作用的晶体结构,从中研究人员认为YAP中的PXXΦP片段是与TEAD4相互作用的关键结构,这为研究Hippo信号通路提供了重要的分子机理线索。这一研究成果公布在《Genes Development》杂志上。 领导这一研究的是新加坡分子与细胞生物学研究院宋海卫博士,其早年毕业于河南大学化学系,之后进入中科院生物物理研究院进行分子生物学方面的学习,1998年获得利兹大学(The University of Leeds)分子生物学专业博士学位。目前任新加坡分子与细胞生物学研究所资深研究员。 Hippo信号转导通路是几年前发现的一个信号转导通路。研究发现Hippo信号通路是参与调控器官大小发育的关键信号通路,这一观点首先在果蝇中被发现,后来的研究发现在哺乳动物的发育过程中Hippo有相同的功能。06年Cell发表的一篇文章证实Hippo 是一种细胞分裂和死亡的控制开关。Hippo信号转导通路通过促进细胞调亡和限制细胞
信号通路研究思路
证明一个药物能通过抑制P38表达而发挥保护细胞的作用,需要做的是: 要证明你的药物是通过抑制P38表达而发挥保护作用,首先要证明P38表达增加会导致损伤。 其次,要证明你的药物存在保护作用。 再次,证明你的药物可以抑制P38表达。 最后,证明你的药物是由于抑制了P38表达而发挥保护作用。 首先证明P38表达增加会导致损伤。 这里需要建立一个损伤模型。正如你提到的,钙离子导致P38mapk的增高,如果某种损伤可以通过钙离子导致P38mapk的增高,那么你就建立起了一个损伤模型。这时,对P38做个RNA干扰,使其表达下降,再来损伤刺激,如果这时损伤刺激不会导致损伤,那么可以说P38mapk的增高会导致损伤。 这里最好不要用P38的抑制剂SB来处理,因为这个抑制剂是针对P38活性的抑制剂,抑制的是P38的磷酸化,而不是表达量。 如果说明的问题是p38磷酸化水平增加而导致损伤,那么我建议用抑制剂。这时还可以用Dominant-negative。抑制剂的实验证实该药物不影响P38表达,而影响其活化。(应该首先考虑选用抑制剂,因为目前一些药物的作用机制不是抑制靶点的表达,而是抑制靶点的激活。如果在此应用RNAi的话,很可能会漏掉这个机制或增加实验步骤。) 其次,要证明你的药物存在保护作用。
当然就是用你的药物先处理一下,再来损伤刺激,如果这时损伤刺激不会导致损伤,那么可以说你的药物存在保护作用。 再次,证明你的药物可以抑制P38表达。 用你的药物先处理一下,再来损伤刺激,再检测P38表达,如果用药组相对于没有用药组P38表达下降,那么可以说你的药物可以抑制P38表达。 最后,证明你的药物是由于抑制了P38表达而发挥保护作用。 这一步看似不必要,其实是最重要的步骤,而国内的文章往往忽略了这一关键环节。 这里建议还是用RNA干扰P38表达,再用你的药物处理,再进行损伤刺激,如果用药组与没有用药组的损伤程度一致,那么才可以说你的药物是由于抑制了P38表达而发挥保护作用。 抑制剂也有其局限性,有时是“致命”的,主要原因是抑制剂缺乏特异性。虽然我们在文章里看到用抑制剂的时候都说是什么什么的特异性抑制剂,但真的那么特异吗?其实往往是作者为了写文章发文章的需要而夸大了抑制剂的特异性。细胞里无数的信号通路,谁也不能保证抑制剂在作用于靶分子时不会影响其他信号通路。其实无论什么抑制剂,对剂量的要求都相对比较苛刻,为什么?就是因为一旦浓度高了,就不知道会干扰到其他哪些信号通路,从而产生很多说不清道不明的现象。 PI3K的抑制剂---LY294002和wortmannin,它们都能抑制PI3K和相关的激酶,但LY294002的浓度达到200μM常用来抑制DNA依赖的蛋白激酶(DNA-PK);wortmannin在浓度超过3μM常用来抑制运动失调性毛细血管扩张基因
细胞信号转导途径研究方法 一、蛋白质表达水平和细胞内定位研究 1、信号蛋白分子表达水平及分子量检测: Western blot analysis. 蛋白质印迹法是将蛋白质混合样品经SDS-PAGE后,分离为不同条带,其中含有能与特异性抗体(或McAb)相应的待检测的蛋白质(抗原蛋白),将PAGE胶上的蛋白条带转移到NC 膜上此过程称为blotting,以利于随后的检测能够的进行,随后,将NC膜与抗血清一起孵育,使第一抗体与待检的抗原决定簇结合(特异大蛋白条带),再与酶标的第二抗体反应,即检测样品的待测抗原并可对其定量。 基本流程: 检测示意图: 2、免疫荧光技术Immunofluorescence (IF) 免疫荧光技术是根据抗原抗体反应的原理,先将已知的抗原或抗体标记上荧光素制成荧光标记物,再用这种荧光抗体(或抗原)作为分子探针检查细胞或组织内的相应抗原(或抗体)。在细胞或组织中形成的抗原抗体复合物上含有荧光素,利用荧光显微镜观察标本,
荧光素受激发光的照射而发出明亮的荧光(黄绿色或桔红色),可以看见荧光所在的细胞或组织,从而确定抗原或抗体的性质、定位,以及利用定量技术测定含量。 采用流式细胞免疫荧光技术(FCM)可从单细胞水平检测不同细胞亚群中的蛋白质分子,用两种不同的荧光素分别标记抗不同蛋白质分子的抗体,可在同一细胞内同时检测两种不同的分子(Double IF),也可用多参数流式细胞术对胞内多种分子进行检测。 二、蛋白质与蛋白质相互作用的研究技术 1、免疫共沉淀(Co- Immunoprecipitation, Co-IP) Co-IP是利用抗原蛋白质和抗体的特异性结合以及细菌蛋白质的“protein A”能特异性地结合到免疫球蛋白的FC片段的现象而开发出来的方法。目前多用精制的protein A预先结合固化在agarose的beads上,使之与含有抗原的溶液及抗体反应后,beads上的prorein A 就能吸附抗原抗体达到沉淀抗原的目的。 当细胞在非变性条件下被裂解时,完整细胞内存在的许多蛋白质-蛋白质间的相互作用被保留了下来。如果用蛋白质X的抗体免疫沉淀X,那么与X在体内结合的蛋白质Y也能沉淀下来。进一步进行Western Blot和质谱分析。这种方法常用于测定两种目标蛋白质是否在体内结合,也可用于确定一种特定蛋白质的新的作用搭档。缺点:可能检测不到低亲和力和瞬间的蛋白质-蛋白质相互作用。 2、G ST pull-down assay GST pull-down assay是将谷胱甘肽巯基转移酶(GST)融合蛋白(标记蛋白或者饵蛋白,GST, His6, Flag, biotin …)作为探针,与溶液中的特异性搭档蛋白(test protein或者prey被扑获蛋白)结合,然后根据谷胱甘肽琼脂糖球珠能够沉淀GST融合蛋白的能力来确定相互作用的蛋白。一般在发现抗体干扰蛋白质-蛋白质之间的相互作用时,可以启用GST沉降技术。该方法只是用于确定体外的相互作用。 示意图:
P53信号通路 P53是一个肿瘤抑制蛋白,调节各种各样基因的表达,包括细胞凋亡,生长抑制,抑制细胞周期进程,分化和加速DNA修复,基因毒性和细胞应激后的衰老。作为一个转录因子,p53是N端激活域、DNA中央特定结合域和C-端四聚体化域的组成部分,而且其调控域富含碱性氨基酸。p53半衰期很短,在26S蛋白酶体作用下,通过持续的泛素化和后期降解,p53在无刺激的哺乳类动物细胞中维持较低的含量。 去磷酸化的p53在MDM2(鼠双微体基因-2)泛素连接酶作用下被泛素化。MDM2结合p53使其无活性是通过两种途径: 第一,MDM2结合p53的转录激活域,阻止转录元件的相互作用。 第二,介导p53共价结合泛素蛋白,泛素化的p53被蛋白水解酶降解。 通过使p53的失活,MDM2扮演着p53抑癌基因的主要监视者。当细胞面临着DNA损伤、缺氧、细胞因子、代谢改变、病毒感染或者致癌基因等刺激时,导致p53泛素化被抑制和p53在细胞核内积累,通过多个共价修饰包括磷酸化和乙酰化,p53被激活并稳定存在。 p53的磷酸化大多数出现在N-末端激活域的Ser6,Ser9,Ser15,Thr18,Ser20,Ser33,Ser37,Ser46,Thr55和Thr81残基上,另外还有一些p53磷酸化出现在C-末端连接处和碱性区域的Ser315,Ser371,Ser376,Ser378,和Ser392残基上。大多数位点上的磷酸化是由DNA损伤诱发的,还有一些例如Thr55 and Ser376在基因毒性应激下被压抑。P53磷酸化是由几个细胞激酶所介导,包括Chks,CSNK1-Delta,CSNK2,PKA,CDK7,DNA-PK,HIPK2,CAK,p38和JNK。显然,由ATM/ATR作用在Ser15上磷酸化,直接作用或者通过Chk1/Chk2,在Chk1/Chk2作用下的Ser20磷酸化已经证实能够减缓抑制或减慢降解p53,导致p53稳定并活化。磷酸化诱导的p53稳定和活化是通过多种机制介导以及很多细胞环境或微环境的改变所致。HIF-1Alpha参与p53的稳定,HIF-1Alpha调节p53介导作用的详细机制依然是一个未知数。最近,p53和HIF-1Alpha之间的作用已被报道能够引起HIF-1Alpha的降解。PIAS蛋白家族也被发现能与p53发生相互作用。PIAS1和PIAS-Gamma作用可作为p53的SUMO连接酶。另外,PIAS1的环指域
目录 actin肌丝 (5) Wnt/LRP6 信号 (7) WNT信号转导 (7) West Nile 西尼罗河病毒 (8) Vitamin C 维生素C在大脑中的作用 (10) 视觉信号转导 (11) VEGF,低氧 (13) TSP-1诱导细胞凋亡 (15) Trka信号转导 (16) dbpb调节mRNA (17) CARM1甲基化 (19) CREB转录因子 (20) TPO信号通路 (21) Toll-Like 受体 (22) TNFR2 信号通路 (24) TNFR1信号通路 (25) IGF-1受体 (26) TNF/Stress相关信号 (27) 共刺激信号 (29) Th1/Th2 细胞分化 (30) TGF beta 信号转导 (32) 端粒、端粒酶与衰老 (33) TACI和BCMA调节B细胞免疫 (35) T辅助细胞的表面受体 (36) T细胞受体信号通路 (37) T细胞受体和CD3复合物 (38) Cardiolipin的合成 (40) Synaptic突触连接中的蛋白 (42) HSP在应激中的调节的作用 (43) Stat3 信号通路 (45) SREBP控制脂质合成 (46) 酪氨酸激酶的调节 (48) Sonic Hedgehog (SHH)受体ptc1调节细胞周期 (51) Sonic Hedgehog (Shh) 信号 (53) SODD/TNFR1信号 (56) AKT/mTOR在骨骼肌肥大中的作用 (58) G蛋白信号转导 (59) IL1受体信号转导 (60) acetyl从线粒体到胞浆过程 (62) 趋化因子chemokine在T细胞极化中的选择性表达 (63) SARS冠状病毒蛋白酶 (65) SARS冠状病毒蛋白酶 (67) Parkin在泛素-蛋白酶体中的作用 (69)
Rheumatol Int DOI 10.1007/s00296-014-3137-5 Role of integrins and their ligands in osteoarthritic cartilage Jian Tian · Fang?Jie Zhang · Guang?Hua Lei Received: 25 May 2014 / Accepted: 17 September 2014 ? Springer-Verlag Berlin Heidelberg 2014 [1]. Radiographic evidence of OA occurs in the majority of people by 65 years of age, and among them about 80 % in people who aged over 75 years [2]. However, the pathogen-esis of this disease is not fully elucidated. Cartilage damage is one of the major pathological changes in OA. Articular cartilage is an avascular, a neu-ral, alymphatic, and viscoelastic connective tissue that functions autonomously to bear loads and provide almost friction-free movement of diarthrodial joints [3]. Chondro-cytes, the only cell population of adult articular cartilage, are strongly involved in maintaining the dynamic equi-librium between synthesis and degradation of the extra-cellular matrix (ECM) [4]. Collagens represent the major structural components of the articular cartilage. Cartilage is made up of two main ECM macromolecules: type II collagen and aggrecan, a large aggregating proteoglycan [5, 6]. Cartilage destruction is thought to be mediated by two main enzyme families: the matrix metalloproteinases (MMPs) are responsible for the cartilage collagen break-down, whereas enzymes from disintegrin and metallopro-teinase domain with thrombospondin motifs (ADAMTS) family mediate cartilage aggrecan loss [7]. Activation of biochemical pathways involves the production of proin-flammatory cytokines, inflammation, degradation of the ECM by MMPs and ADAMTS, and cessation of ECM syn-thesis via dedifferentiation and apoptosis of chondrocytes [8, 9]. Therefore, the ECM is a vital cellular environment, and interactions between the cell and ECM are important in regulating many biological processes, which include cell growth, differentiation, and survival [10, 11]. Cell–matrix interactions control cell function and behav-ior by signal transduction through a variety of cell sur-face receptors. The integrins are the major family of ECM receptors, which can transmit information from the matrix to the cell. Integrin binding of ECM ligands results in the Abstract Osteoarthritis (OA) is a degenerative disease, which is characterized by articular cartilage destruction, and mainly affects the older people. The extracellular matrix (ECM) provides a vital cellular environment, and interactions between the cell and ECM are important in reg-ulating many biological processes, including cell growth, differentiation, and survival. However, the pathogenesis of this disease is not fully elucidated, and it cannot be cured totally. Integrins are one of the major receptors in chondro-cytes. A number of studies confirmed that the chondrocytes express several integrins including α5β1, αV β3, αV β5, α6β1, α1β1, α2β1, α10β1, and α3β1, and some integrins ligands might act as the OA progression biomarkers. This review focuses on the functional role of integrins and their extracellular ligands in OA progression, especially OA car-tilage. Clear understanding of the role of integrins and their ligands in OA cartilage may have impact on future develop-ment of successful therapeutic approaches to OA.Keywords Chondrocyte · Integrin · Fibronectin · Tenascin C · Osteopontin · Osteoarthritis · Cartilage Introduction Osteoarthritis (OA) is a degenerative disease and is char-acterized by articular cartilage destruction along with changes occurring in other joint components including bone, menisci, synovium, ligaments, capsule, and muscles Rheumatology INTERNATIONAL J. Tian · F.-J. Zhang · G.-H. Lei (*) Department of Orthopaedics, Xiangya Hospital, Central South University, No. 87 Xiangya Road, Changsha 410008, Hunan, China e-mail: gh.lei9640@https://www.doczj.com/doc/455043602.html,; lgh9640@https://www.doczj.com/doc/455043602.html,