丙醇高斯程序应用
- 格式:doc
- 大小:457.50 KB
- 文档页数:40
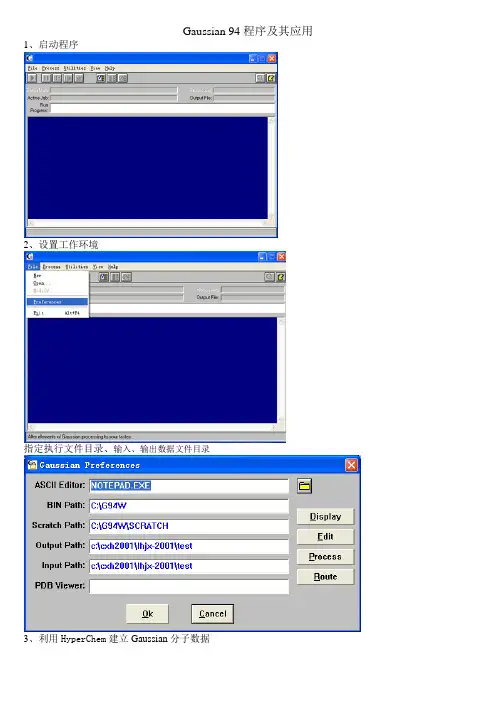
Gaussian 94程序及其应用1、启动程序2、设置工作环境指定执行文件目录、输入、输出数据文件目录3、利用HyperChem建立Gaussian分子数据(Ⅰ)打开高斯程序,File------open,打开刚才保存的1-氯乙烯的数据,并对该分子进行sto-3g构型优化单击图标单击即可运行。
Molecule Specification运行结果为:-- Stationary point found.----------------------------! Optimized Parameters !! (Angstroms and Degrees) !------------------------ -------------------------! Name Definition Value Derivative Info. !-----------------------------------------------------------------------------! R1 R(2,1) 1.3075 -DE/DX = -0.0002 ! ! R2 R(3,1) 1.082 -DE/DX = 0.0001 ! ! R3 R(4,1) 1.0827 -DE/DX = 0. ! ! R4 R(5,2) 1.0849 -DE/DX = 0. ! ! R5 R(6,1) 2.7154 -DE/DX = 0. ! ! R6 R(6,2) 1.7776 -DE/DX = 0.0002 ! ! A1 A(2,1,3) 122.6964 -DE/DX = 0. ! ! A2 A(2,1,4) 120.3199 -DE/DX = 0. ! ! A3 A(3,1,4) 116.9837 -DE/DX = 0. ! ! A4 A(1,2,5) 124.643 -DE/DX = 0. ! ! A5 A(2,1,6) 33.4715 -DE/DX = 0.0001 ! ! A6 A(3,1,6) 89.2249 -DE/DX = 0. ! ! A7 A(4,1,6) 153.7914 -DE/DX = 0. ! ! A8 A(1,2,6) 122.5954 -DE/DX = 0. ! ! A9 A(5,2,6) 112.7616 -DE/DX = 0. ! ! A10 A(1,6,2) 23.9331 -DE/DX = 0. ! ! D1 D(5,2,1,3) 180. -DE/DX = 0. ! ! D2 D(5,2,1,4) 0. -DE/DX = 0. ! ! D3 D(5,2,1,6) 180. -DE/DX = 0. ! ! D4 D(6,2,1,3) 0. -DE/DX = 0. ! ! D5 D(6,2,1,4) 180. -DE/DX = 0. ! ! D6 D(2,6,1,3) 180. -DE/DX = 0. ! ! D7 D(2,6,1,4) 0. -DE/DX = 0. ! ! D8 D(1,6,2,5) 180. -DE/DX = 0. ! ----------------------------------------------------------------------------- GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad------------------------------------------------------------------------------------------------------------------- Final structure in terms of initial Z-matrix:CC,1,R2H,1,R3,2,A3H,1,R4,2,A4,3,180.,0H,2,R5,1,A5,3,180.,0Cl,2,R6,1,A6,3,0.,0Variables:R2=1.30747709R3=1.08204192R4=1.08272704R5=1.08487191R6=1.77756291A3=122.69641181A4=120.31991643A5=124.64301574A6=122.59542361|1|GINC-UNK|FOpt|RHF|STO-3G|C2H3Cl1|PCUSER|18-Dec-1911|0||# HF/STO-3GOPT||1-氯乙烯sto-3g构型优化||0,1|C,-0.6957009082,0.,-1.5082013006|C,-0.7581567701,0.,-0.2022167693|H,0.2417669563,0.,-2.0485426002|H,-1.6031560746,0.,-2.0988136568|H,-1.6791317147,0.,0.3711524101|Cl,0.691980406,0.,0.8258066039||Version=x86-Win32-G94RevE.1|State=1-A'|HF=-531.0786764|RMSD=2.822e-009|RMSF=1.175e-004|Dipole=-0.6686084,0.,-0.5817464|PG=CS [SG(C2H3Cl1)]||@(Ⅱ)对优化结果进行单点计算,首先复制输出文件的结果,然后粘贴到数据输入文件中去单击进行运行运行结果为:*********************************************Gaussian 94: x86-Win32-G94RevE.1 23-Nov-199627-Dec-1911*********************************************--------------# HF/sto-3g sp--------------1/38=1/1;2/12=2,17=6,18=5/2;3/11=9,25=1,30=1/1,2,3;4//1;5/5=2,32=1,38=4/2;6/7=2,8=2,9=2,10=2,19=1,28=1/1;99/5=1,9=1/99;----------------------1-氯乙烯sto-3g单点计算----------------------Symbolic Z-matrix:Charge = 0 Multiplicity = 1CC 1 R2H 1 R3 2 A3H 1 R4 2 A4 3 180. 0H 2 R5 1 A5 3 180. 0Cl 2 R6 1 A6 3 0. 0Variables:R2 1.34R3 1.07973R4 1.07973R5 1.07973R6 1.07973A3 120.00816A4 120.00816A5 120.00816A6 120.00816------------------------------------------------------------------------Z-MATRIX (ANGSTROMS AND DEGREES)CD Cent Atom N1 Length/X N2 Alpha/Y N3 Beta/Z J ------------------------------------------------------------------------1 1 C2 2 C 1 1.340000( 1)3 3 H 1 1.079730( 2) 2 120.008( 6)4 4 H 1 1.079730( 3) 2 120.008( 7) 3 180.000( 10) 05 5 H 2 1.079730( 4) 1 120.008( 8) 3 180.000( 11) 06 6 Cl 2 1.079730( 5) 1 120.008( 9) 3 .000( 12) 0------------------------------------------------------------------------Z-Matrix orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 .000000 .000000 .0000002 6 .000000 .000000 1.3400003 1 .934997 .000000 -.5399984 1 -.934997 .000000 -.5399985 1 -.934997 .000000 1.8799986 17 .934997 .000000 1.879998----------------------------------------------------------Distance matrix (angstroms):1 2 3 4 51 C .0000002 C 1.340000 .0000003 H 1.079730 2.099669 .0000004 H 1.079730 2.099669 1.869993 .0000005 H 2.099669 1.079730 3.058310 2.419996 .0000006 Cl 2.099669 1.079730 2.419996 3.058310 1.86999366 Cl .000000Interatomic angles:C2-C1-H3=120.0082 C2-C1-H4=120.0082 H3-C1-H4=119.9837 C1-C2-H5=120.0082 C1-C2-Cl6=120.0082 H5-C2-Cl6=119.9837Stoichiometry C2H3ClFramework group CS[SG(C2H3Cl)]Deg. of freedom 9Full point group CS NOp 2Largest Abelian subgroup CS NOp 2Largest concise Abelian subgroup C1 NOp 1Standard orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 1.327232 .287457 .0000002 6 .000000 .471996 .0000003 1 1.733322 -.712996 .0000004 1 1.990848 1.139179 .0000005 1 -.406090 1.472449 .0000006 17 -.663616 -.379726 .000000----------------------------------------------------------Rotational constants (GHZ): 67.9749568 10.3831325 9.0072766 Isotopes: C-12,C-12,H-1,H-1,H-1,Cl-35Standard basis: STO-3G (5D, 7F)There are 18 symmetry adapted basis functions of A' symmetry.There are 4 symmetry adapted basis functions of A" symmetry.Crude estimate of integral set expansion from redundant integrals=1.000.Integral buffers will be 262144 words long.Raffenetti 1 integral format.Two-electron integral symmetry is turned on.22 basis functions 66 primitive gaussians16 alpha electrons 16 beta electronsnuclear repulsion energy 115.4166028311 Hartrees.One-electron integrals computed using PRISM.The smallest eigenvalue of the overlap matrix is 5.614E-02Projected CNDO Guess.Initial guess orbital symmetries:Occupied (A') (A') (A') (A') (A') (A') (A") (A') (A') (A')(A") (A') (A') (A') (A') (A")Virtual (A") (A') (A') (A') (A') (A')Warning! Cutoffs for single-point calculations used.Requested convergence on RMS density matrix=1.00E-04 within 64 cycles. Requested convergence on MAX density matrix=1.00E-02.Requested convergence on energy=5.00E-05.Keep R1 integrals in memory in canonical form, NReq= 450705.SCF Done: E(RHF) = -530.067323383 A.U. after 6 cyclesConvg = .2684E-04 -V/T = 2.0096S**2 = .0000********************************************************************** Population analysis using the SCF density.********************************************************************** Orbital Symmetries:Occupied (A') (A') (A') (A') (A') (A") (A') (A') (A') (A')(A") (A') (A') (A') (A') (A")Virtual (A") (A') (A') (A') (A') (A')The electronic state is 1-A'.Alpha occ. eigenvalues -- -103.91000 -11.20069 -10.97041 -10.61756 -8.07206 Alpha occ. eigenvalues -- -8.04379 -8.04306 -1.43047 -.93263 -.80151 Alpha occ. eigenvalues -- -.70761 -.69994 -.59994 -.49157 -.36604 Alpha occ. eigenvalues -- -.23382Alpha virt. eigenvalues -- .31471 .63974 .71238 .85138 .93482 Alpha virt. eigenvalues -- 1.04677Condensed to atoms (all electrons):1 2 3 4 5 61 C 4.900028 .595825 .402833 .395532 -.025331 -.1731992 C .595825 5.732803 -.032981 -.030424 .404607 -.0257843 H .402833 -.032981 .614347 -.022367 .001621 -.0053884 H .395532 -.030424 -.022367 .616178 -.003463 .0042305 H -.025331 .404607 .001621 -.003463 .590164 -.0887226 Cl -.173199 -.025784 -.005388 .004230 -.088722 16.752508 Total atomic charges:11 C -.0956882 C -.6440453 H .0419364 H .0403145 H .1211266 Cl .536356Sum of Mulliken charges= .00000Atomic charges with hydrogens summed into heavy atoms:11 C -.0134372 C -.5229193 H .0000004 H .0000005 H .0000006 Cl .536356Sum of Mulliken charges= .00000Electronic spatial extent (au): <R**2>= 172.1980Charge= .0000 electronsDipole moment (Debye):X= -.8342 Y= .0253 Z= .0000 Tot= .8345 Quadrupole moment (Debye-Ang):XX= -24.2345 YY= -23.1281 ZZ= -23.5995XY= -1.3849 XZ= .0000 YZ= .0000Octapole moment (Debye-Ang**2):XXX= -5.1092 YYY= -1.4749 ZZZ= .0000 XYY= -1.4845 XXY= -.4300 XXZ= .0000 XZZ= -2.4727 YZZ= -1.1777 YYZ= .0000 XYZ= .0000Hexadecapole moment (Debye-Ang**3):XXXX= -136.6205 YYYY= -54.0979 ZZZZ= -19.0554 XXXY= -15.6858 XXXZ= .0000 YYYX= -16.1524 YYYZ= .0000 ZZZX= .0000 ZZZY= .0000 XXYY= -29.3803 XXZZ= -26.6547 YYZZ= -13.0041 XXYZ= .0000 YYXZ= .0000 ZZXY= -5.5929N-N= 1.154166028311E+02 E-N=-1.481757123171E+03 KE= 5.250305341478E+02 Symmetry A' KE= 4.795223533234E+02Symmetry A" KE= 4.550818082437E+011|1|GINC-UNK|SP|RHF|STO-3G|C2H3Cl1|PCUSER|27-Dec-1911|0||# HF/STO-3G S P||1-氯乙烯sto-3g单点计算||0,1|C|C,1,1.34|H,1,1.07973,2,120.00816|H,1,1.07973,2,120.00816,3,180.,0|H,2,1.07973,1,120.00816,3,180.,0|Cl,2,1.07973,1,120.00816,3,0.,0||Version=x86-Win32-G94RevE.1|State=1-A'|HF=-530.0673234|RMSD=2.684e-005|Dipole=0.0353257,0.,0.3264277|PG=CS [SG(C2H3Cl1)]||@94(Ⅲ)对结果进行相关能的计算,首先复制输出文件的结果,然后粘贴到数据输入文件中去运行结果为:*********************************************Gaussian 94: x86-Win32-G94RevE.1 23-Nov-199621-Dec-1911*********************************************---------------# HF/STO-3G MP21-氯乙烯的相关能的计算----------------------Symbolic Z-matrix:Charge = 0 Multiplicity = 1CC 1 R2H 1 R3 2 A3H 1 R4 2 A4 3 180. 0H 2 R5 1 A5 3 180. 0Cl 2 R6 1 A6 3 0. 0Variables:R2 1.30748R3 1.08204R4 1.08273R5 1.08487R6 1.77756A3 122.69641A4 120.31992A5 124.64302A6 122.59542Standard orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 1.277970 1.060880 .0000002 6 .000000 .784660 .0000003 1 2.041653 .294331 .0000004 1 1.614773 2.089893 .0000005 1 -.791346 1.526761 .0000006 17 -.619582 -.881425 .000000********************************************************************** Population analysis using the SCF density.********************************************************************** Orbital Symmetries:Occupied (A') (A') (A') (A') (A') (A") (A') (A') (A') (A')(A') (A') (A') (A") (A') (A")Virtual (A") (A') (A') (A') (A') (A')The electronic state is 1-A'.Alpha occ. eigenvalues -- -103.69833 -11.10838 -11.06348 -10.38048 -7.82460 Alpha occ. eigenvalues -- -7.81915 -7.81906 -1.08405 -.95938 -.75052 Alpha occ. eigenvalues -- -.62638 -.56978 -.46613 -.45464 -.39226 Alpha occ. eigenvalues -- -.33409Alpha virt. eigenvalues -- .28706 .35950 .60378 .66744 .86552 Alpha virt. eigenvalues -- .97887Condensed to atoms (all electrons):1 2 3 4 5 61 C 4.775666 .611616 .392437 .391154 -.025797 -.0283222 C .611616 4.815767 -.024760 -.024584 .392986 .2421413 H .392437 -.024760 .563313 -.021350 .001803 -.0032304 H .391154 -.024584 -.021350 .570233 -.004029 .0010555 H -.025797 .392986 .001803 -.004029 .558724 -.0254696 Cl -.028322 .242141 -.003230 .001055 -.025469 16.964994 Total atomic charges:11 C -.1167542 C -.0131663 H .0917874 H .0875215 H .1017816 Cl -.151168Sum of Mulliken charges= .00000Atomic charges with hydrogens summed into heavy atoms:11 C .0625542 C .0886153 H .0000004 H .0000005 H .0000006 Cl -.151168Sum of Mulliken charges= .00000Electronic spatial extent (au): <R**2>= 244.8248Charge= .0000 electronsDipole moment (Debye):X= .9907 Y= 2.0231 Z= .0000 Tot= 2.2527Quadrupole moment (Debye-Ang):XX= -22.3101 YY= -22.5444 ZZ= -24.2687XY= -.5141 XZ= .0000 YZ= .0000Octapole moment (Debye-Ang**2):XXX= .0302 YYY= -.5479 ZZZ= .0000 XYY= -.0783XXY= .4325 XXZ= .0000 XZZ= -1.4613 YZZ= -2.0260YYZ= .0000 XYZ= .0000Hexadecapole moment (Debye-Ang**3):XXXX= -121.1758 YYYY= -141.9518 ZZZZ= -19.9152 XXXY= -44.5432XXXZ= .0000 YYYX= -40.0365 YYYZ= .0000 ZZZX= .0000ZZZY= .0000 XXYY= -42.1538 XXZZ= -25.1760 YYZZ= -30.1162XXYZ= .0000 YYXZ= .0000 ZZXY= -15.6039N-N= 8.805534228086E+01 E-N=-1.429996710208E+03 KE= 5.224301068131E+02Symmetry A' KE= 4.769281466379E+02Symmetry A" KE= 4.550196017524E+011|1|GINC-UNK|SP|RMP2-FC|STO-3G|C2H3Cl1|PCUSER|21-Dec-1911|0||# HF/STO-3G MP2||1-氯乙烯的相关能的计算||0,1|C|C,1,1.30748|H,1,1.08204,2,122.69641|H,1,1.08273,2,120.31992,3,180.,0|H,2,1.08487,1,124.64302,3,180.,0|Cl,2,1.77756,1,122.59542,3,0.,0||Version=x86-Win32-G94RevE.1|State=1-A'|HF=-531.0786764|MP2=-531.2060553|RMSD=2.919e-009|PG=CS [SG(C2H3Cl1)]||@讨论:1、优化分子几何构型即寻找能量极小值所对应的分子几何构型。


昆明理工大学理学院上机实验报告课程名称:计算化学实验名称:专业班级:学生姓名:学号:上机作业4:Gaussian程序使用二:频率和热力学性质计算对乙烯酮( H2C=C=O)分子的振动频率和热力学性质进行计算。
1)写出乙烯酮( H2C=C=O)分子的内坐标及高斯输入文件。
,其中C=C: 1.35 C=O: 1.20 C-H: 1.09,H-C=C:120.0°。
(提示:设虚原子)%CHK=YIXITONG.CHK%rwf=yixitong.rwf#p B3LYP /6-31G* spYixitong0 1CC 1 R1X 2 R2 1 A1O 2 R3 3 A1 1 180.0H 1 R4 2 A2 3 0.0H 1 R4 2 A2 3 180.0R1=1.35R2=1.0R3=1.20R4=1.09A1=90.0A2=120.02)对H2C=C=O分子进行结构优化,给出结构的对称性,优化后的结构数据(键长、键角、二面角),能量值,并通过GaussView或ChemOffice将输入的结构图形以球棍形式列出。
计算方法:B3LYP基组设定:6-31G*优化:OPT对称性:C2V能量= -152.5984712921 C 0.0000002 C 1.314839 0.0000003 O 2.486220 1.171381 0.0000004 H 1.082743 2.077840 3.167280 0.0000005 H 1.082743 2.077840 3.167280 1.878582 0.0000003)对优化后的结构进行频率计算。
(提示:保持*.chk文件不变),找出计算的红外频率、振动模式及对称性,对频率进行矫正,并将校正后的数据和实验值相对比(填写表1)。
给出主要振动模式的图形、对应的峰值和红外光谱图(要求用origin作图)。
表1. 乙烯酮的红外分析振动模式实验值(cm-1) 计算值(cm-1) 校正值(cm-1) 红外强度对称性面内振动438 440.7177 423.662 2.9343 B2面外摇摆528 538.7049 517.8573 74.4313 B1面外摇摆588 590.7611 567.899 74.0799 B1面内扭曲977 1003.0527 964.22341 8.3269 B2伸缩振动1118 1179.8738 1134.2135 6.9638 A1面内伸缩1388 1434.7138 1379.1909 16.2242 A1面内拉伸2152 2239.9335 2153.2481 526.0268 A1面内拉伸3071 3209.1869 3084.9915 23.7501 A1面外拉伸3166 3298.0569 3170.4221 5.7145 B2计算方法:B3LYP基组设定:6-31G*频率计算:Freq geom=check guess=read频率矫正因子:0.9613(2)(1)(3)(4)(5) (6(7)(8)(9)-10010020030040050060070080005010015020025030010.7036549533175.0751177974257.3788746988E p s i l o nFrequency (cm-1)4)列表给出H2C=C=O分子的热力学数据。

高斯伪谱法程序
高斯伪谱法是一种数值计算方法,常用于求解偏微分方程。
该方法以傅里叶级数为基础,将函数的离散值通过傅里叶变换转化为频域,再进行计算。
高斯伪谱法具有高精度、高效率和较好的稳定性,广泛应用于科学和工程领域。
实现高斯伪谱法的程序一般包括以下步骤:
1. 离散化:将求解区域离散化,将偏微分方程转化为离散化方程。
2. 选取基函数:选择一组正交的基函数,一般选择正交的勒让
德多项式或柯西-罗尔多项式。
3. 计算系数:通过求解离散化方程,计算出系数。
4. 重构解:使用基函数和系数重构出解。
5. 检验误差:检验求解结果的误差,可以使用残差法或拉格朗
日插值法。
高斯伪谱法的程序实现需要较高的数学和编程能力,但是随着计算机技术的发展,现在已经有许多高斯伪谱法的计算软件和包可供使用。
- 1 -。


下面是Link功能一览表:9Link功能一览表(续):10Link功能一览表(续):11开始作业暂停当前作业当前link后暂停终止当前作业和批处理恢复当前作业在当前作业完成后终止批处理12设定多步任务的开始任务放弃任务15创建Gaussian输入文件电荷与多重度分子说明部分,段后通常加空行19Gaussian程序能完成的任务类型:25逆时针,为正顺时针,为负•当没有构建分子的图像软件可用时,与直角坐标相比使用内坐标要方便得多。
•对具有较高对称性的分子,输入内坐标少优化变量、节省计算时间并保证优化结果与分子的对称性严格相符。
%chk=h2o2.chk#p hf/6-31g optH2O2 energy calculation 0 1HO 1 0.9O 2 1.4 1 105.0H 3 0.9 2 105.0 1 120.0%chk=h2o2.chk#p hf/6-31g optH2O2 energy calculation0 1H 0.000 0.0000.000O 0.000 0.9000.000O 1.350 1.2620.000H 1.464 1.742-0.752上述两个作业将对H2O2分子的结构进行完全优化,包括所有的键长、键角和二面角。
例1:使用HF方法,优化H2O2分子内坐标表示笛卡尔坐标表示41✓通过同一个变量控制C-H✓把二面角定义为180和0来控制分子的平面构型对称性44使用一个虚原子来定位Td对称性Bq1、画结构•单击工具,在工作窗口中画苯环。
•单击元素工具,选氧元素,再点,替换苯环上H7和H8形成两个OH。
•选碳元素,再点,替换一个OH上的H形成OCH3。
•单击,出现如下窗口,从中选择羧基,替换苯环上的H9.52,531.构建分子2.选择6个碳原子例:用GaussView在某结构的中心添加鬼原子54554. 点击鼠标右键出现此窗口5. 选择place fragment at centroid of selected Atoms3.选择鬼原子程序,编辑或按钮,弹出对话以及保存56Entering Link 1 = C:\program1\G09WD1\G09W\l1.exe PID= 6620.写出分子的内坐标其中: 1.20 ,C-C: 1.54写出呋喃分子的内坐标,使其具有构建二茂铁分子,并存成Gaussian输入文件。

白酒贮存过程中主要成分间氢键相互作用的量子化学计算黄张君1,张文华1,曾运航1,*,石碧1,王松涛2,3,沈才洪2,3(1.四川大学轻工科学与工程学院,四川成都610065;2.国家固态酿造工程技术研究中心,四川泸州646000;3.泸州老窖股份有限公司,四川泸州646000)摘 要:用量子化学的方法研究乙醇-水、乙醇-风味成分、水-风味成分以及乙醇-水-风味成分之间的氢键相互作用,以期更深入地认识白酒的老熟机理。
用密度泛函理论计算结果表明:在乙醇、水、风味成分的二元复合物和三元复合物中,结构稳定性由高到低的排序分别为乙醇/水-酸>乙醇/水-醇>乙醇/水-酯,以及乙醇-水-酸>乙醇-水-醇>乙醇-水-酯。
酸类物质的羰基氧原子和羟基氢原子能分别与乙醇、水的羟基氢原子和氧原子生成氢键,使乙醇/ 水-酸和乙醇-水-酸复合物形成环状结构,故结构稳定性高。
这应该是白酒在贮存过程中风味成分的质量浓度呈现“酸增酯减”的原因之一,同时也是贮存可以增强酒体中氢键相互作用、减少游离分子的原因。
关键词:白酒贮存;量子化学;风味成分;氢键;相互作用Quantum Chemistry Calculation of Hydrogen Bond Interactions between Major Compounds in Baijiu during Storage HUANG Zhangjun1, ZHANG Wenhua1, ZENG Yunhang1,*, SHI Bi1, WANG Songtao2,3, SHEN Caihong2,3(1. College of Biomass Science and Engineering, Sichuan University, Chengdu 610065, China;2. National Engineering Research Center of Solid-State Brewing, Luzhou 646000, China;3. Luzhou Laojiao Co. Ltd., Luzhou 646000, China)Abstract: In this work, the hydrogen bond interactions between ethanol and water, between ethanol and flavor compounds, between water and flavor compounds, and among ethanol, water and flavor compounds were investigated by quantum chemistry calculation to provide insights into the mechanism of Baijiu aging. The results density functional theory showed that the structural stability of the binary and ternary complexes was in the decreasing order of ethanol/water-acid > ethanol/ water-alcohol > ethanol/water-ester, and ethanol-water-acid > ethanol-water-alcohol > ethanol-water-ester, respectively. The carbonyl oxygen atom and hydroxyl hydrogen atom in the organic acids could form hydrogen bonds with the hydrogen atom and oxygen atom in the hydroxyl group of ethanol or water, separately, causing the ethanol/water-acid and ethanol-water-acid complexes to form ring structures and consequently improving the structural stability. This may partially explain why the flavor components of Baijiu showed an increase in acids and a decrease in esters during storage and may also explain why storage could strengthen the hydrogen bond interactions and reduce free molecules in Baijiu.Keywords: Baijiu storage; quantum chemistry; flavor compound; hydrogen bond; interactionDOI:10.7506/spkx1002-6630-20200531-375中图分类号:TS261;O641 文献标志码:A 文章编号:1002-6630(2021)12-0045-07引文格式:黄张君, 张文华, 曾运航, 等. 白酒贮存过程中主要成分间氢键相互作用的量子化学计算[J]. 食品科学, 2021, 42(12): 45-51. DOI:10.7506/spkx1002-6630-20200531-375. HUANG Zhangjun, ZHANG Wenhua, ZENG Yunhang, et al. Quantum chemistry calculation of hydrogen bond interactions between major compounds in Baijiu during storage[J]. Food Science, 2021, 42(12): 45-51. (in Chinese with English abstract) DOI:10.7506/spkx1002-6630-20200531-375. 收稿日期:2020-05-31基金项目:国家固态酿造工程技术研究中心基金项目(K2019-243);四川大学技术开发项目(19H0393)第一作者简介:黄张君(1987—)(ORCID: 0000-0003-4594-2491),女,工程师,博士研究生,研究方向为酒类新产品的研究和开发。

高斯过程应用随机过程高斯过程是一种概率模型,常常被用于建模连续型的随机变量。
它的特点是可以通过有限个数据点来描述整个函数的概率分布,因此在机器学习领域被广泛应用。
在实际应用中,高斯过程可以用来进行回归分析、分类、聚类等任务。
随机过程是一种随时间变化的随机变量序列,常用来描述动态系统的行为。
随机过程的性质通常可以通过其均值函数和协方差函数来描述。
高斯过程是一种特殊的随机过程,其均值函数和协方差函数均为高斯函数,因此其性质更容易进行分析和计算。
在机器学习领域,高斯过程常用于回归分析。
给定一组输入输出的数据点,我们希望通过高斯过程模型来预测输入输出之间的关系。
通过学习数据点之间的相关性和噪声水平,我们可以得到一个高斯过程模型,从而可以对新的输入数据进行预测。
这种方法不仅可以用于预测,也可以用于对不确定性进行建模。
除了回归分析,高斯过程还可以用于分类问题。
在分类问题中,我们需要将数据点分为不同的类别。
通过将高斯过程模型与逻辑回归等分类算法相结合,可以得到一种更加准确和稳健的分类器。
这种方法在文本分类、图像分类等领域有着广泛的应用。
此外,高斯过程还可以用于聚类分析。
聚类分析是一种无监督学习方法,旨在将数据点划分为不同的簇。
通过将高斯过程模型与聚类算法相结合,可以得到一种更加灵活和自适应的聚类方法。
这种方法在社交网络分析、生物信息学等领域有着广泛的应用。
总的来说,高斯过程是一种强大且灵活的工具,可以应用于各种机器学习任务中。
通过深入理解高斯过程的原理和性质,我们可以更好地应用它来解决实际问题,从而推动机器学习领域的发展和进步。
希望未来能够看到更多基于高斯过程的创新应用,为人类社会带来更多的福祉。

Gaussian软件应用——研究化学反应和反应性讲解Gaussian软件应用——研究化学反应和反应性第八章研究化学反应和反应性本章讨论应用电子结构理论研究化学反应.我们将从电子密度开始,然后回顾第四章中有关反应势垒的讨论,再讨论反应研究中的更复杂的技术,最后,通过对相应反应的计算,来研究未知体系的反应热.本章将引入两种新的计算方法* 势能面* 反应路径分析8.1 预测电子密度将电子密度或静电势可视化是研究一个分子体系的反应性的重要的第一步.例8.1 文件e8_01a, e8_01b 取代苯的电子密度在有机化学中,亲电芳香取代反应的定位效应是已经被深入研究的课题.在这里,我们采用电子密度对这一现象进行研究.已经知道氯苯和硝基苯的硝化是基于同样的反应机理:苯环首先受NO2+的攻击,产生各种异构体的阳离子异构体.当硝化完成后,产物分布如下.邻位间位对位氯硝基苯29% 1% 70%二硝基苯7% 88% 1%我们在这里检验间位和对位异构体的中间体.分子采用B3LYP/6-31G(d)进行优化,电子密度在HF/6-31G(d)等级计算.将电子密度按照平行苯环平面的方向切片,得到不同厚度位置的电子密度图. 间位的氯硝基苯和对位的二硝基苯的电子密度分布显示,其保留了有较大共振范围的电子结构,相反,另两个构型的电子密度分布显示其电子分布相对局域化,并且向苯环外的方向集中.通过电子密度的图形,可以定性的理解电子密度和反应性的关系,在得到结论之前,检查这个体积的电子密度是必要的.关于这方面的进一步资料可以参见Gaussian出版的白皮书Visualizing Results from Gaussian. 8.2 计算反应焓变例8.2 文件e8_02 水解反应现在分析水解反应H+ + H2O --> H3O+目的是计算标准反应焓变dH298.其计算方法可以表示为dH298 = dE298 + d(PV)dE298 = dEe0 + d(dEe)298 + dEv0 + d(dEv)298 + dEr298 + dEt298其中dEe0: 0K时产物与反应物的能量差;d(dEe)298: 0K到298K电子能量的变化.对于这个反应,这一项可以忽略;dEv0: 0K时反应物和产物的零点能之差;d(dEv)298: 0K到298K振动能量的变化;dEr298: 产物和反应物的旋转能之差;dEt298: 产物和反应物的平动能之差;d(PV): 由于有一摩尔分子消失,PV=-RT.dEe0由单点能得到,本例采用的计算方法是B3LYP/6-311+G(2df,2p).其他的各项都要考虑内能校正,通过频率分析得到.这样,所要做的工作就是进行优化然后进行频率分析得到所需数值.采用B3LYP/6-31G(d)就能够得到足够精确的结果.这里注意我们不用计算H+,由于没有电子,它的电子能量显然是0;由于只有一个原子,其振动,转动能显然也是零,这样,其只有平动能,其值为1.5RT = 0.889kcal.mol.(详见统计热力学).最终计算得到dH298=-163.3kcal.mol.实验值为-165.3+-1.8kcal/mol. 两者符合的相当好.8.3 研究势能面对于势能面的研究对反应路径分析来讲,可能产生出人意料的好的结果.本节中, 我们通过实例研究势能面的应用方法.考虑丙烯基正离子的旋转异构体的变化,H1 H1| |H2a C H2b H2a C H3b\ / \ / <---> \ / \ /C C C C| | | |H3a H3b H3a H2b(I) (I')曾经认为两个异构体之间的变化是通过一个具有Cs对称性的过渡态完成的,在该构型中,H2b-C-H3b组成的平面垂直与碳原子平面.采用HF/6-311++G(d,p)能够找到这样的过渡态,但是进一步的采用MP2和QCISD 以及同样基组的研究却没有得到过渡态,而得到了极小值!这个新的具有Cs对称性的结构中,H1迁移到了端位的碳原子上.这个新结构的能量比势能面中平衡结构的能量高10kcal/mol.这样就有了另一条反应路线:中间碳上的氢迁移到端位的碳原子上;新形成的甲基旋转;旋转后的甲基上的一个氢原子迁移回中间碳原子.在这个例子中,应用了IRC计算来确定过渡态的确是连接产物与反应物的.本章后面将对这一方法进行讨论.HF方法的研究得到了假的过渡态,原因是,由于HF方法本身的限制,其计算的亚甲基旋转的势垒要低于氢原子迁移的势垒.8.4 势能面扫描势能面扫描可以研究一个区域内的势能面.一般的扫描都是由一系列的在不同结构上的单点能计算组成的.当进行势能面扫描时,要设置分子结构的变量,设置需要变化的结构的范围和步长.在Gaussian中,势能面扫描是自动进行的,下面是一个进行势能面扫描的算例.#T UMP4/6-311+G(d,p) Scan TestCH PES Scan0 2CH 1 RR 0.5 40 0.04该算例要求一个对于CH的势能面扫描,所用的关键词是scan,变量的设置格式是:名称初始值[点数步长]当只有一个参数时,变量在整个扫描中是不变的,当三个参数都设定时,变量将在一定范围内变化.当有多个变量时,所有的可能构型都要计算.所有等级的计算结果都在输出文件中列出,比如进行的MP2的势能面扫描也将列出HF方法的结果.根据得到的扫描结果,可以得到所要的势能面,通过它,可能得到极小值的可能位置.势能面扫描过程中不进行几何优化.8.5 反应路径分析在第四章中我们提到,得到一个过渡态机构不能说明它就是连接产物和反应物的结构.分析其是不是所需过渡态的一个方法是分析虚频的简正振动状态.有时,对振动的分析也不能够确定.本节讨论更为精确的方法.IRC方法检验过渡态分子的趋势.计算从过渡态开始,根据能量降低的方向来寻找极小值,就是说,寻找过渡态所连接的两个极小值. 反应路径是连接反应物与产物的,但是连接反应物和产物可以有不止一条路径, 通过不同的过渡态连接,通过IRC计算,我们可以寻找真正的反应路径,也就是能量最低的反应路径.反应路径计算可以确认得到的过渡态就是连接反应物和产物的过渡态,一旦确认,还可以计算活化能(注意零点能校正).运行IRC在Gaussian中,运行IRC的关键词是IRC.需要注意的是,IRC计算是从过渡态开始的,在两个反应方向上各进行固定步骤的计算(默认是6步).IRC计算的方法是这样的:* 优化过渡态* 进行频率分析,确认所得到的是过渡态,计算零点能,生成进行IRC 计算的力矩阵* 运行IRC,在鞍点的能量下降方向,寻找极小值.一般的,需要增大寻找的次数,从而尽可能的接近极小值.方法是设置MaxPoints.确定反应势垒,一般还要进行更多的工作,* 对过渡态的高等级的能量计算* 对反应物和产物进行优化和频率分析,得到零点能,进行高等级能量计算8.6 势能面研究实例我们现在用Gaussian的反应路径分析来研究甲醛的势能面.这个势能面上有很多极小值,包括甲醛,羟基卡宾,以及H2和CO.每一组之间都可以组合成不同的反应物产物对.这里研究两个反应H2CO <--> CO + H2H2CO <--> HCOH甲醛的解离我们要确定反应过渡态的结构,预测反应的活化能.为此,我们需要以下信息:* 甲醛,氢分子,一氧化碳分子的考虑零点能的能量.* 过渡态的几何构型和零点能校正的能量.计算在HF/6-31G(d)水平进行,结果如下SCF能量零点能总能量H2 -1.12683 0.00968 -1.11716CO -112.73788 0.00508 -112.7280H2 + CO -113.84996H2CO -113.86633 0.02668 -113.83966计算过渡态的能量,方法是* 过渡态几何构型优化,计算SCF能量* 频率分析,计算零点能* IRC计算,确认过渡态优化过渡态例8.3 文件e8_03 CH2O --> H2 + CO IRC首先考虑氧原子垂直于CHH平面的构型,同时增大OCH夹角.计算中设置Opt=(TS,CalcFC). CalcFC一般对于过渡态的优化是有帮助的.得到的该点几何构型与猜测的结构接近,SCF能量-113.69352频率分析频率分析表明其有一个虚频,零点能0.01774(校正后),总能量-113.68578IRC计算IRC计算需要优化好的过渡态和相应的力矩阵,得到的方法是* 从临时文件中获得(IRC=RCFC),或* 在IRC计算的初始进行计算(IRC=CalcFC)IRC计算在输出文件末尾对计算进行总结,列出能量和优化的变量的值.第一个值和最后一个值是整条路径的起点和终点. 在起点上,我们得到了一个类似甲醛分子的结构,可以认定该反应路线是通向甲醛的,在终点上,得到了一个C-H键伸长的结构,C-O键略微缩短,也表明这条反应路线是通向解离分子的.计算活化能IRC计算确认了所得到的就是我们所要的过渡态,下面就可以计算活化能了.能量活化能(kcal/mol)过渡态-113.67578反应物-113.83966 102.8(正向)产物-113.84996 109.3(反向)计算表明两个反应方向的势垒相似.注意IRC得到的产物的能量不一定等于两个单独的体系的和,因为当IRC计算得到分子配合物的极小值,与两个分离体系的能量和有些差别1,2氢迁移反应现在用同样的步骤研究第二个反应.反式氢基卡宾的包含零点能的总能量是-113.75709,计算方法是RHF/6-31G(d).寻找过渡态猜测过渡态在碳原子上的一个氢原子象氧原子方向迁移,处于同时与碳原子和氧原子作用的位置,对其进行的频率分析表明其位一阶鞍点,包含零点能的总能量为-113.67941反应路径分析IRC分析得到的两个结构,一个类似于HCOH,一个类似于H2CO,说明该结构为该反应的过渡态.活化能预测计算得到的活化能为100.6(正向)和48.7(反向)kcal/molIRC的注意事项虽然实际的反应结构如极小点,极大点,鞍点等在势能面上存在几何的和数学的意义,但不能简单推广到物理的和化学的意义.实际的分子是有动能的,这样它就可以不遵循反应路径.当然,计算得到的结果提供了最经济的反应途径.8.7 等构反应(Isodesmic Reactions)等构反应是指反应前后各种键的数量不变的反应,比如乙醛与乙烷生成丙酮和甲烷的反应,反应前后,各种价键的数量都没有变化. 由于这一特点,对这样体系的研究可以得到相当精确的结果.例8.5 文件e8_05 等构反应的反应焓变化本例计算上面提到的反应的反应焓变,步骤如下:* 在HF/6-31G(d)水平优化结构* 进行频率分析计算零点能* 在B3LYP/6-311+G(3df,2p)水平计算单点能结果得到反应焓变-9.95kcal/mol,实验值-9.9+-0.3kcal/mol.例8.6 文件e8_06 通过等构反应确定二氧化碳分子生成焓本例中利用等构反应CO2 + CH4 --> 2H2CO 来确定二氧化碳分子的生成焓.原理如下:dHcalc = 2 E0(H2CO) - (E0(CO2) + E0(CH4))dHf(CO2) = -(dHcalc - dHf(CH4) + 2dHf(H2CO))甲烷和甲醛的生成焓实验值分别为-16.0和-25.0kcal/mol(0K)计算方法同上例,所得到的反应焓变为60.64kcal/mol,这样计算得到的二氧化碳的生成焓为-94.64kcal/mol.实验值为-93.96kcal/mol.等构反应的局限等构反应的局限等构反应对于反应体系和生成焓的研究非常重要,但其也有明显的缺点,如下* 必须依赖良好的实验数据,实验数据不准确,得到的生成焓自然也不可信* 该技术不能推广到活化势垒方面* 该技术不能用于实际上发生不了的等构反应* 不同的等构反应可以得到不同的生成焓数值.例8.7 文件e8_07 等构反应的局限本例通过两个不同的等构反应计算乙烷的生成焓,研究理论MP2/6-31G(d)//HF/6-31G(d)反应一丙烷+ 氢--> 乙烷+ 甲烷反应二乙烷+ 氢--> 2 甲烷另外通过6个反应计算SiF4的生成焓,研究方法MP2/6-31G(d,p)//HF/6-31G(d) SiH4 + F2 --> 2H2 + SiF4 (i)SiH4 + 4HF --> 4H2 + SiF4 (ii)SiF3H + HF --> SiF4 (iii)SiF2H2 + 2HF --> 2H2 + SiF4 (iv)SiFH3 + 3HF --> 3H2 + SiF4 (v)SiH4 + 4F2 --> SiF4 + 4HF (vi)结果如下乙烷生成焓实验值-20.0+-0.1 kcal/mol反应一-17.361反应二-22.258SiF4 实验值-386.0+-0.3 kcal/mol(i) -375.983(ii) -366.588(iii) -380.506(iv) N/A(v) -376.112(vi) -385.379虽然得到的乙烷生成焓和实验值比较吻合,但两个结果之间竟然有高达5kcal/mol 的差距,对于这样的简单的烃的体系仍然有这样大的差异,在使用这一方法是就不得不小心了.对于SiF4,不同反应得到的数据差别也很明显,虽然也有很符合的结果,但最大的差距竟然达到20kcal/mol.同时注意这些硅化合物的生成焓本身的可靠性不高.8.8 练习练习8.1 文件8_01a~c 水和反应计算两个反应的反应焓Li+ + H2O --> H2OLi+ at 298.15KH2O + H2O --> (H2O)2 at 373K两个反应的实验值分别是-34.0+-0.2kcal/mol和-3.6+-.5kcal/mol计算方法是B3LYP/6-311+G(2df,2p)//B3LYP/6-31G(d)由于本例中要计算298.15K和373K两个温度的水分子的热力学数据,所以,可以在一个计算中完成,输入文件如下%Chk=water#T RHF/6-31G(d) Freq=ReadIso...--Link1--%Chk=water%Nosave#T RHF/6-31G(d) Geom=Check Freq=(ReacFC, ReadIso) Guess=Read Test...373.0 1.0 0.9135161611计算得到的第一个反应反应焓变为-34.0kcal/mol.注意对Li+不能进行频率分析,其焓的校正为1.5RT(只有平动能)对于第二个反应,注意由于DFT方法在处理弱作用体系上有些困难,要确认所得到的分子是真正的极小值,而不是过渡态.本例中需要采用Opt=CalcAll来进行优化.计算结果为反应焓-2.9kcal/mol练习8.2 文件8_02a, 8_02b 键的解离本例中通过势能面扫描研究键的断裂过程,研究的体系是CH键CH UMP4/6-311+G(d,p) 键长范围0.2-2.5ACH4 RQCISD(T)/6-311++G(d,p) 和键长范围.75-3.15AUQCISD(T,E4T)/6-311++G(d,p)UQCISD的E4T选项进行的是在MP4(SDTQ)水平进行MP4计算,而不是默认的MP4(SDQ). 计算中还需要设置的关键词有IOP(2/16=1)表示在扫描中忽略对称性变化Guess=(Always,Mix)表示将HOMO和LUMO轨道混合来消除自旋对称性的影响,并且在每一个点都重新计算新的猜测波函数.下面是检查势能面扫描数据的方法* 将HF, MP2, MP4, QCISD(T)不同水平计算的键能对键长作图* 利用这些图来分析键解离:* 限制性和非限制性方法的比较* 电子相关* 电子相关* 三重态对QCISD水平的贡献对于CH,UHF曲线在其他曲线之上,相对而言,HF的结果是最差的.MP2的曲线比MP3和MP4(SDTQ)曲线要高些,但三者相差不多.对于CH4,UHF得到的曲线远高于其他结果,MP2的结果也比其他结果要高些,而其他各个计算方法得到的曲线基本相似.每一次提高微扰的次数,能量曲线就向低的方向移动.QCISD曲线与MP4曲线很接近,在对于整个曲线的描述上,QCISD(T)比MP4(SDTQ)的结果要好很多.限制性方法和非限制性方法得到的结果只在键分裂之后才显现出来,RHF方法得到的曲线趋势就是错误的,因为其得到的结果是,当原子进一步远离时,能量继续上升,实际上当两个原子距离达到一定程度后,能量将基本保持不变.而对于QCISD(T)方法,两者的差别不大,限制性方法得到的曲线在键解离后要略高于非限制性方法的结果.这说明,在描述键解离的过程时,非限制性方法的结果要比限制性方法好,特别是在采取低等级计算模型时.练习8.3 文件8_03 H2CO的势能面我们以前研究过H2CO的两个反应的势能面,得到了五个稳定点:其中三个极小值,甲醛,反式羟基卡宾以及一氧化碳和氢分子;两个过渡态分别连接甲醛和另外两个产物.现在要做的,就是研究这两个产物之间的变化途径.反应分两步: 反式羟基卡宾<--> 顺式羟基卡宾<--> 一氧化碳+ 氢在HF/6-31G(d)水平研究这一问题,研究步骤是:* 寻找过渡态* 确定得到的稳定点是过渡态,计算零点能* 确定这一过渡态连接的两个极小值一个可能的连接顺式和反式结构的过渡态就是HCO平面与COH 平面成90度的二面角.我们采用Opt=QST3,分别给出两个异构体和这一猜测的构型.然后进行IRC计算,得到的是接近顺式和反式异构体的构型,说明这个过渡态结构就是所要找的过渡态.用同样的方法,可以得到有顺式异构体到一氧化碳和氢的反应的过渡态.反应的活化能为正向反向反式<--> 顺式25.8 20.5顺式<--> 解离68.0 131.6这样就得到了关于H2CO的新的势能面.练习8.4 文件8_04 原子电荷分析这个练习采用非Mulliken布局分析方法计算原子电荷,考察这些结果.原子电荷计算并不是有量子化学理论推导出来的,所有的计算原子电荷的方法都是武断的.练习采用的方法有* Muliken布局分析(默认)* Natural布局分析(Pop=NPA)* 采用CHelpG方法的Breneman静电势衍生电荷* 采用Merz-Kollman-Singh方法的静电势衍生电荷计算方法为MP2/6-31G(d).为了节省计算时间,在计算第二个以及后面的几个算例时可以采用在临时文件中的电子密度,Geom=CHeckpointDensity=(Checkpoint,MP2),同时注意在第一个算例中加入Density=MP2从而采用MP2方法得到的电子密度来计算原子电荷,默认方法为HF方法.计算的分子是丙烯阳离子.Mulliken方法得到的原子电荷,三个碳原子上都有一定的负电荷;Natural方法的结果是,中间碳原子上是负电荷,其余原子上均为正电荷;CHelpG和MKS方法得到的结果,也只有中间的碳原子上有负电荷.后面三个方法得到的中间碳原子上负电荷的值越来越少.练习8.5 文件8_04 基团电荷计算上例中的CH2和CH基团的电荷,结果如下Mulliken NPA CHelpG MKSCH +0.18 -0.05 -0.10 +0.09CH2 +0.41 +0.52 +0.47 +0.45练习8.6 文件8_06 分子中的原子电荷和键级分子中的原子理论(theory of atoms in molecules),提供了另外的更加复杂的计算原子电荷和相关性质的方法.这一理论利用图论和电子密度函数来定义化学性质,如键和原子电荷.这样的理论有清楚的量子化学意义.Gassian中的AIM关键词涉及到这一算法.这里用AIM=BondOrders来计算丙烯正离子的原子电荷和键级.计算的碳碳键键级为1.4,碳氢键键级为0.9.所有原子上都是正电荷.练习8.7 文件8_07 Si+ 和硅烷的势能面硅簇反应是一个新的领域,非常值得用电子结构方法研究.这个练习检验硅正离子进攻硅烷(SiH4)的势能面.这个反应是硅簇反应的中心:Si+ + SiH4 --> Si2H2+ --> Si3H4+ --> Si4H6+ --> Si5H10+ ...每一步反应都生成氢我们只研究第一个反应H Si+| | HSi-H + Si+ --> Si + |/ \ / \ HH H H H在Krishnan Raghavachari的研究中,发现了如下的极小值H H H H H H2\ \ \ / \ / |Si-H...Si Si-Si Si-Si Si--Si/ | / | \ / \ /H H H H H H H SiSiH4...Si+ H3Si...SiH+ H2Si...SiH2+ H2Si-Si+...H2最右边的结构是产物配合物,结合能1kcal/mol,在我们的研究中可以认为是反应的终点.另外还有如下过渡态H H-H\ / \|Si--Si/H确定哪一个是连接过渡态和的极小值就可以确认反应势垒.计算采用HF/6-31G(d)进行频率分析和IRC,采用MP4方法进行能量计算,IRC计算中要包含下列关键词IRC=(RCFC, StepSize=30, MaxPoints=15) SCF=QC这样的设置增加了两端搜索的点的数量.具体的计算方法是* 进行频率分析,计算零点能,准备IRC* IRC计算,确定与过渡态相连接的极小值* UMP4/6-31G(d,p)计算能量最终确认该过渡态连接的是H3Si-SiH+和H2Si-Si+...H2其他的极小值当然与其他的过渡态相连.有兴趣的可以继续寻找其他的过渡态, 完成整个反应的势能面.练习8.8 文件8_08 等构反应这里研究等构反应CH3COX + H3CCH3 <--> CH3COCH3 + CH3XX = H, F, Cl计算反应焓变,方法为B3LYP/6-311+G(3df,2p)//B3LYP/6-31G(d) 结果为-9.95, 16.71, 7.14;实验值-9.9+-0.3, 17.9+-1.3, 6.6+-0.3 采用其他方法,HF方法的结果很糟糕,MP2方法过分考虑了电子相关,MP3方法的结果也比较好.对于AM1方法,结果比HF方法还要差.练习8.9 文件8_09a~b 通过等构反应计算生成焓通过等构反应研究氟代甲烷和苯的生成焓.所用反应为CHF3 + 2CH4 --> 3CH3FC6H6 + 6CH4 --> 3C2H6 + 3C2H4结果为-54.27和23.89,实验值-55.9+-2.0,24.0+-0.2 练习8.10文件8_10 一个SN2反应研究SN2反应Cl- + H3CF --> ClCH3 + F-方法如下* 过渡态结构优化* 过渡态频率分析* 从过渡态的IRC分析* 寻找中间体两个极小值的几何结构* 频率分析计算零点能得到的过渡态是Cl...CH3...F两个极小值是F-...CH3Cl和CH3F...Cl-。


乙烷分子高斯程序应用1、用HyperChem程序画乙烷分子图,保存为*.PDB格式2、设置工作环境3、格式转化:4、分子构型进行优化(opt)5、进行振动分析、单点计算6、计算结果输出:优化结果输出:Final structure in terms of initial Z-matrix: CC,1,R2H,1,R3,2,A3H,1,R4,2,A4,3,D4,0H,1,R4,2,A4,3,-D4,0H,2,R6,1,A6,3,180.,0H,2,R7,1,A7,3,D7,0H,2,R7,1,A7,3,-D7,0V ariables:R2=1.52714912R3=1.0851582R4=1.08516911R6=1.08517148R7=1.08518242A3=111.18808808A4=111.18735176A6=111.18983868A7=111.18910141D4=119.99975739D7=-60.000242381|1|GINC-UNK|FOpt|RHF|6-31G(d)|C2H6|PCUSER|11-Jan-1911|0||# HF/6-31G*OPT||乙烷||0,1|C,0.0000021919,0.,-0.7635805116|C,-0.0000021919,0.,0.7635686082|H,1.0118036956,-0.0000000002,-1.1557871264|H,-0.5059007695,-0.8762601514,-1.155782425|H,-0.5059007693,0.8762601516,-1.155782425|H,-1.0118041011,0.0000000002,1.1558109401|H,0.5059009868,-0.8762605204,1.1558062284|H,0.5059009871,0.8762605202,1.1558062284||V ersion=x86-Win32-G94RevE.1|State=1-A'|HF=-79.2287541|RMSD=5.028e-010|RMSF=1.707e-004|Dipole=0.,0.,-0.0000069|PG=CS [SG(C2H2),X(H4)]||@HE WHO LAUGHS LAST PROBA BLY DIDN'T GET THE JOKE.Job cpu time: 0 days 0 hours 0 minutes 8.0 seconds.File lengths (MBytes): R WF= 5 Int= 0 D2E= 0 Chk= 2 Scr= 1Normal termination of Gaussian 94振动分析结果:1 2 3A" A' A" Frequencies -- 320.9807 887.6569 887.6878Red. masses -- 1.0078 1.0562 1.0562Frc consts -- .0612 .4903 .4904IR Inten -- .0000 2.4997 2.4996Raman Activ -- .0000 .0000 .0000Depolar -- .0000 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .00 .00 -.05 .00 .00 .00 .00 .052 6 .00 .00 .00 -.05 .00 .00 .00 .00 .053 1 .00 .00 .41 .16 .51 .00 .00 .00 -.224 1 .35 .00 -.20 .20 -.26 -.03 .03 .44 -.175 1 -.35 .00 -.20 .20 -.26 .03 -.03 -.44 -.176 1 .00 .00 .41 .16 .51 .00 .00 .00 -.227 1 -.35 .00 -.20 .20 -.26 .03 -.03 -.44 -.178 1 .35 .00 -.20 .20 -.26 -.03 .03 .44 -.174 5 6A' A' A" Frequencies -- 1062.1629 1337.0508 1337.0597Red. masses -- 3.1308 1.4544 1.4544Frc consts -- 2.0811 1.5319 1.5320IR Inten -- .0000 .0000 .0000Raman Activ -- 13.9063 3.1051 3.1046Depolar -- .3025 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .31 .00 .14 .00 .00 .00 .00 .142 6 .00 -.31 .00 -.14 .00 .00 .00 .00 -.143 1 .02 .37 .00 -.06 -.51 .00 .00 .00 -.254 1 -.01 .37 -.01 -.20 .25 .08 .08 .44 -.115 1 -.01 .37 .01 -.20 .25 -.08 -.08 -.44 -.116 1 -.02 -.37 .00 .06 .51 .00 .00 .00 .257 1 .01 -.37 -.01 .20 -.25 .08 .08 .44 .118 1 .01 -.37 .01 .20 -.25 -.08 -.08 -.44 .117 8 9A' A' A' Frequencies -- 1546.7873 1578.7780 1643.8192Red. masses -- 1.1984 1.2740 1.0212Frc consts -- 1.6893 1.8709 1.6259IR Inten -- .1468 .0000 .0000Raman Activ -- .0000 4.1660 38.8356Depolar -- .6187 .7157 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .09 .00 .00 -.11 .00 -.02 .00 .002 6 .00 .09 .00 .00 .11 .00 .02 .00 .003 1 -.17 -.37 .00 .18 .36 .00 -.12 -.28 .004 1 .08 -.37 .14 -.09 .36 -.15 .34 .14 -.265 1 .08 -.37 -.14 -.09 .36 .15 .34 .14 .266 1 -.16 -.37 .00 -.18 -.36 .00 .12 .28 .007 1 .08 -.37 -.14 .09 -.36 -.15 -.34 -.14 -.268 1 .08 -.37 .14 .09 -.36 .15 -.34 -.14 .2610 11 12A" A" A' Frequencies -- 1643.8212 1649.7178 1649.7193Red. masses -- 1.0212 1.0628 1.0628Frc consts -- 1.6259 1.7041 1.7041IR Inten -- .0000 5.7101 5.7108Raman Activ -- 38.8364 .0001 .0001Depolar -- .7500 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .00 -.02 .00 .00 -.05 .05 .00 .002 6 .00 .00 .02 .00 .00 -.05 .05 .00 .003 1 .00 .00 .49 .00 .00 .52 .12 .22 .004 1 -.26 .24 .04 -.28 .19 .04 -.36 -.11 .285 1 .26 -.24 .04 .28 -.19 .04 -.36 -.11 -.286 1 .00 .00 -.49 .00 .00 .52 .12 .22 .007 1 -.26 .24 -.04 .28 -.19 .04 -.36 -.11 -.288 1 .26 -.24 -.04 -.28 .19 .04 -.36 -.11 .2813 14 15A' A' A" Frequencies -- 3203.2175 3209.3460 3253.3299Red. masses -- 1.0340 1.0379 1.1032Frc consts -- 6.2510 6.2986 6.8798IR Inten -- 72.4201 .0057 .0008Raman Activ -- .0175 221.0308 150.1856Depolar -- .0191 .0193 .7500Atom AN X Y Z X Y Z X Y Z1 6 .00 .03 .00 .00 -.04 .00 .00 .00 -.072 6 .00 .03 .00 .00 .04 .00 .00 .00 .073 1 .38 -.14 .00 -.39 .14 .00 .00 .00 -.014 1 -.19 -.14 -.33 .19 .14 .33 .24 .18 .405 1 -.19 -.14 .33 .19 .14 -.33 -.24 -.18 .406 1 .39 -.14 .00 .38 -.14 .00 .00 .00 .017 1 -.19 -.14 .34 -.19 -.14 .33 .24 .18 -.408 1 -.19 -.14 -.34 -.19 -.14 -.33 -.24 -.18 -.4016 17 18A' A" A' Frequencies -- 3253.3981 3278.4462 3278.5130Red. masses -- 1.1032 1.1037 1.1037Frc consts -- 6.8801 6.9891 6.9894IR Inten -- .0008 101.4189 101.4133Raman Activ -- 150.1842 .0011 .0011Depolar -- .7500 .7500 .7500Atom AN X Y Z X Y Z X Y Z1 6 -.07 .00 .00 .00 .00 .07 .07 .00 .002 6 .07 .00 .00 .00 .00 .07 .07 .00 .003 1 .54 -.20 .00 .00 .00 .01 -.54 .21 .004 1 .12 .10 .24 -.24 -.18 -.40 -.13 -.10 -.245 1 .12 .10 -.24 .24 .18 -.40 -.13 -.10 .246 1 -.54 .20 .00 .00 .00 .01 -.54 .21 .007 1 -.12 -.10 .24 .24 .18 -.40 -.12 -.10 .248 1 -.12 -.10 -.24 -.24 -.18 -.40 -.12 -.10 -.24Zero-point vibrational energy 209475.1 (Joules/Mol)50.06574 (Kcal/Mol)WARNING-- EXPLICIT CONSIDERATION OF 1 DEGREES OF FREEDOM AS VIBRATIONS MAY CAUSE SIGNIFICANT ERRORVIBRATIONAL TEMPERA TURES: 461.82 1277.13 1277.18 1528.21 1923.71 (KELVIN) 1923.72 2225.47 2271.50 2365.08 2365.082373.56 2373.56 4608.69 4617.51 4680.794680.89 4716.93 4717.02Zero-point correction= .079785 (Hartree/Particle)Thermal correction to Energy= .083192Thermal correction to Enthalpy= .084136Thermal correction to Gibbs Free Energy= .056714Sum of electronic and zero-point Energies= -79.148969Sum of electronic and thermal Energies= -79.145563Sum of electronic and thermal Enthalpies= -79.144618 Sum of electronic and thermal Free Energies= -79.172040。
有关高斯过程的实际应用
1、贝叶斯优化:贝叶斯优化是一种基于概率模型的机器学习方法,它使用高斯过程(GP)来估计函数的未知参数,以找到具有最佳性能的参数。
贝叶斯优化可以用于自动化机器学习模型的超参数调优,使得模型可以更好地拟合数据。
2、计算机视觉:高斯过程可以用于计算机视觉中的目标检测和识别,其中,GP可以用于模型拟合和估计,以获得更准确的结果。
GP也可以用于模式识别,以改善分类器的性能。
3、系统调控:高斯过程也可以用于系统调控,例如,GP可以用于模型拟合以改善系统性能,以及用于控制系统以达到期望的输出。
此外,GP还可以用于预测系统的输出,以及模拟系统的行为。
对乙醇结构优化高斯模拟介绍乙醇(C2H5OH)是一种常见的有机化合物,也是酒精的主要成分之一。
乙醇的结构优化是研究乙醇分子内原子的相对位置和键长的过程,通过计算和模拟,可以得到乙醇分子的最稳定结构。
高斯模拟是一种常用的计算化学方法,可以用于模拟和优化分子的结构。
乙醇的结构乙醇分子由两个碳原子、六个氢原子和一个氧原子组成。
乙醇的结构可以用分子式C2H5OH表示,其中C表示碳原子,H表示氢原子,O表示氧原子。
乙醇的结构可以通过高斯模拟进行优化,得到最稳定的构型。
高斯模拟的原理高斯模拟是一种基于量子力学原理的计算方法,可以用于计算分子的结构、能量和性质等。
在高斯模拟中,分子的电子结构通过解薛定谔方程来描述,通过求解方程得到分子的波函数和能量。
优化分子的结构是通过调整分子内原子的相对位置和键长,使得分子的总能量达到最低。
乙醇结构优化的步骤乙醇的结构优化可以通过以下步骤进行:1.构建初始结构:根据乙醇的化学式,可以构建一个初始结构,确定碳原子、氢原子和氧原子的相对位置。
2.选择合适的计算方法:在高斯模拟中,需要选择适合的计算方法和基组。
常用的计算方法包括密度泛函理论(DFT)和哈特里-福克(HF)方法。
3.设置计算参数:在进行高斯模拟之前,需要设置一些计算参数,如收敛准则、基组的选择和计算的精度等。
4.进行结构优化:通过改变原子的位置和键长,使得乙醇分子的总能量达到最低。
可以使用梯度下降法或者分子动力学模拟等方法进行结构优化。
5.分析结果:优化完成后,可以分析乙醇分子的最稳定结构和键长,比较不同结构的能量差异,进一步理解乙醇的性质。
结果与讨论通过高斯模拟对乙醇进行结构优化,可以得到乙醇分子的最稳定结构。
乙醇的最稳定结构是通过调整碳原子、氢原子和氧原子的相对位置和键长得到的。
根据优化后的结构,可以计算乙醇分子的能量和其他性质,如键长、键角和电荷分布等。
优化后的乙醇结构可以用于进一步研究乙醇的性质和反应。
例如,可以通过计算乙醇的电子亲和能和电离能来研究其与其他分子的相互作用。
Gaussian 和GaussView 软件在化学教学中应用一、研究背景随着计算机技术的飞速发展以及高考改革的全面推行,越来越多的计算软件和绘图软件应用于高中化学的教学中,例如Photoshop、ChenDraw、Multiwfn、Gaussian 和GaussView 程序等。
计算机辅助教学可以将抽象的化学知识生动形象的表现出来,从而弥补了传统的化学教学手段难以生动的表现抽象的化学知识的缺点,这样不仅有助于学生对复杂的结构和现象的理解,还可以增强他们对化学学习的兴趣。
近年来,全国多省进行高考改革,辽宁省于2018 年也加入其中。
改革下的化学学科随之也有新的变化。
化学学科作为高中理科生教育的重要科目之一,这些年来越来越得到学生的喜爱。
自从有学者国的高中教育进入新高考改革时代以来,高中化学老师对学生的课堂教学的重视程度也得到了进一步加深。
高中化学老师不但需要完成新课改所要求的教学目标,还需要从有效提升高中化学教学效率方面进行创新,让高中生能够在学习中更加热爱化学课程,并且能够在化学课程的学习过程中更加努力,这样才可以提升高中生对化学课程的学习兴趣和老师对学生的教学效率,从而成功地实现新高考改革背景下的高中化学教学创新和常识重构。
高中化学的内容比较繁杂,尤其是选修五有机化学部分,需要学生极强的空间想象能力,部分学生由于空间想象能力有限,因此对于有机化学的学习产生抵触心理。
而Gaussian 计算软件能够以图形化的方式更加清晰、直观地描述有机化学反应中分子的变化过程。
以人教版《有机化学基础》(选修5)中同分异构体教学为例, Gaussian 和GaussView 软件在辅助课堂教学方面的重要作用,可以直观地展示了如何构筑有机物分子的同分异构体。
Gaussian 和GaussView软件极大丰富了课堂教学的形式,也为以后大学化学的学习奠定基础。
Gaussian 是一个功能强大的量子化综合软件包,它可以在不同型号的大型计算机,工作站和个人计算机上运行。
用gaussian软件研究丙酮异构化机理
1 打开gaussian view
Vie w→builder
画出丙酮的分子式:
单击打开元素周期表,选C原子,再选价态。
画完后点击clean
画出后计算优化:
Calculat e→gaussian
Jobtype :opt+freq
Link0中将6改为12
计算过程中可将丙酮改为丙烯醇,最好不要重新画
修改键,点击,再点击键两端原子,修改,改完后点clean。
用丙酮同样的方法优化
2
优化后将两结构优化的结果提交到gaussian生成两个gjf文件(修改一下文件名)打开gaussian03w
File→open,将丙酮优化后生成的gjf文件打开
Route section后opt后加上=qst2
再用写字板打开丙烯醇优化后生成的gjf文件
从文件名开始到最后拷到
数字10后面,空一格:
点击OK,→再点
也可以直接点run
打开计算结果
在gaussian view的result查看中间体性质。
不足之处请包涵。
高斯程序应用用HyperChem程序画丙醇分子图:设置工作环境:格式转化:分子构型进行优化(opt):优化结果输出:Entering Link 1 = F:\G94W\l1.exe PID= 5608.Copyright (c) 1988,1990,1992,1993,1995 Gaussian, Inc.All Rights Reserved.This is part of the Gaussian 94(TM) system of programs. It is based on the the Gaussian 92(TM) system (copyright 1992 Gaussian, Inc.), the Gaussian 90(TM) system (copyright 1990 Gaussian, Inc.), the Gaussian 88(TM) system (copyright 1988 Gaussian, Inc.), the Gaussian 86(TM) system (copyright 1986 Carnegie Mellon University), and the Gaussian 82(TM) system (copyright 1983 Carnegie Mellon University). Gaussian is a federally registered trademark of Gaussian, Inc.This software is provided under written license and may be used, copied, transmitted, or stored only in accord with that written license.The following legend is applicable only to US Government contracts under DFARS:RESTRICTED RIGHTS LEGENDUse, duplication or disclosure by the US Government is subjectto restrictions as set forth in subparagraph (c)(1)(ii) of theRights in Technical Data and Computer Software clause at DFARS 252.227-7013.Gaussian, Inc.Carnegie Office Park, Building 6, Pittsburgh, PA 15106 USAThe following legend is applicable only to US Government contracts under FAR:RESTRICTED RIGHTS LEGENDUse, reproduction and disclosure by the US Government is subject to restrictions as set forth in subparagraph (c) of the Commercial Computer Software - Restricted Rights clause at FAR 52.227-19.Gaussian, Inc.Carnegie Office Park, Building 6, Pittsburgh, PA 15106 USA---------------------------------------------------------------Warning -- This program may not be used in any manner that competes with the business of Gaussian, Inc. or will provide assistance to any competitor of Gaussian, Inc. The licenseeof this program is prohibited from giving any competitor of Gaussian, Inc. access to this program. By using this program, the user acknowledges that Gaussian, Inc. is engaged in the business of creating and licensing software in the field of computational chemistry and represents and warrants to the licensee that it is not a competitor of Gaussian, Inc. and thatit will not use this program in any manner prohibited above.---------------------------------------------------------------Cite this work as:Gaussian 94, Revision E.1,M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M. W. Gill,B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith,G. A. Petersson, J. A. Montgomery, K. Raghavachari,M. A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. B. Foresman,J. Cioslowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y. Ayala, W. Chen, M. W. Wong, J. L. Andres,E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox,J. S. Binkley, D. J. Defrees, J. Baker, J. P. Stewart,M. Head-Gordon, C. Gonzalez, and J. A. Pople,Gaussian, Inc., Pittsburgh PA, 1995.*********************************************Gaussian 94: x86-Win32-G94RevE.1 23-Nov-199612-Jan-1911*********************************************---------------# HF/6-31G* opt---------------1/18=20,38=1/1,3;2/9=110,12=2,17=6,18=5/2;3/5=1,6=6,7=1,11=9,25=1,30=1/1,2,3;4//1;5/5=2,38=4/2;6/7=2,8=2,9=2,10=2,28=1/1;7//1,2,3,16;1//3(1);99//99;2/9=110/2;3/5=1,6=6,7=1,11=9,25=1,30=1/1,2,3;4/5=5,16=2/1;5/5=2,38=4/2;7//1,2,3,16;1//3(-5);2/9=110/2;3/5=1,6=6,7=1,11=9,25=1,30=1,39=1/1,3;6/7=2,8=2,9=2,10=2,28=1/1;99/9=1/99;----丙醇----Symbolic Z-matrix:Charge = 0 Multiplicity = 1CC 1 R2H 1 R3 2 A3H 1 R4 2 A4 3 D4 0 H 1 R4 2 A4 3 -D4 0 C 2 R6 1 A6 3 180. 0H 2 R7 1 A7 3 D7 0H 2 R7 1 A7 3 -D7 0O 6 R9 2 A9 1 180. 0H 6 R10 2 A10 1 D10 0H 6 R10 2 A10 1 -D10 0H 9 R12 6 A12 2 180. 0Variables:R2 1.54R3 1.09021R4 1.08951R6 1.54029R7 1.09032R9 1.43R10 1.08985R12 0.95991A3 109.44878A4 109.46169A6 109.49368A7 109.50249A9 109.49368A10 109.40677A12 109.47314D4 119.95934D7 59.99236D10 59.9767GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad Berny optimization.Initialization pass.----------------------------! Initial Parameters !! (Angstroms and Degrees) !------------------------ -------------------------! Name Definition Value Derivative Info. !-----------------------------------------------------------------------------! R1 R(2,1) 1.54 estimate D2E/DX2 !! R2 R(3,1) 1.0902 estimate D2E/DX2 !! R3 R(4,1) 1.0895 estimate D2E/DX2 !! R4 R(5,1) 1.0895 estimate D2E/DX2 !! R5 R(6,2) 1.5403 estimate D2E/DX2 !! R6 R(7,2) 1.0903 estimate D2E/DX2 !! R7 R(8,2) 1.0903 estimate D2E/DX2 !! R8 R(9,6) 1.43 estimate D2E/DX2 !! R9 R(10,6) 1.0898 estimate D2E/DX2 !! R10 R(11,6) 1.0898 estimate D2E/DX2 ! ! R11 R(12,9) 0.9599 estimate D2E/DX2 ! ! A1 A(2,1,3) 109.4488 estimate D2E/DX2 ! ! A2 A(2,1,4) 109.4617 estimate D2E/DX2 ! ! A3 A(3,1,4) 109.454 estimate D2E/DX2 ! ! A4 A(2,1,5) 109.4617 estimate D2E/DX2 ! ! A5 A(3,1,5) 109.454 estimate D2E/DX2 ! ! A6 A(4,1,5) 109.5471 estimate D2E/DX2 ! ! A7 A(1,2,6) 109.4937 estimate D2E/DX2 ! ! A8 A(1,2,7) 109.5025 estimate D2E/DX2 ! ! A9 A(6,2,7) 109.4506 estimate D2E/DX2 ! ! A10 A(1,2,8) 109.5025 estimate D2E/DX2 ! ! A11 A(6,2,8) 109.4506 estimate D2E/DX2 ! ! A12 A(7,2,8) 109.4275 estimate D2E/DX2 ! ! A13 A(2,6,9) 109.4937 estimate D2E/DX2 ! ! A14 A(2,6,10) 109.4068 estimate D2E/DX2 ! ! A15 A(9,6,10) 109.5113 estimate D2E/DX2 ! ! A16 A(2,6,11) 109.4068 estimate D2E/DX2 ! ! A17 A(9,6,11) 109.5113 estimate D2E/DX2 ! ! A18 A(10,6,11) 109.4975 estimate D2E/DX2 ! ! A19 A(6,9,12) 109.4731 estimate D2E/DX2 ! ! D1 D(6,2,1,3) 180. estimate D2E/DX2 ! ! D2 D(6,2,1,4) -60.0407 estimate D2E/DX2 ! ! D3 D(6,2,1,5) 60.0407 estimate D2E/DX2 ! ! D4 D(7,2,1,3) 59.9924 estimate D2E/DX2 ! ! D5 D(7,2,1,4) 179.9517 estimate D2E/DX2 ! ! D6 D(7,2,1,5) -59.967 estimate D2E/DX2 ! ! D7 D(8,2,1,3) -59.9924 estimate D2E/DX2 ! ! D8 D(8,2,1,4) 59.967 estimate D2E/DX2 ! ! D9 D(8,2,1,5) 179.9517 estimate D2E/DX2 ! ! D10 D(9,6,2,1) 180. estimate D2E/DX2 ! ! D11 D(9,6,2,7) -59.9606 estimate D2E/DX2 ! ! D12 D(9,6,2,8) 59.9606 estimate D2E/DX2 ! ! D13 D(10,6,2,1) 59.9767 estimate D2E/DX2 ! ! D14 D(10,6,2,7) 179.9839 estimate D2E/DX2 ! ! D15 D(10,6,2,8) -60.0627 estimate D2E/DX2 ! ! D16 D(11,6,2,1) -59.9767 estimate D2E/DX2 ! ! D17 D(11,6,2,7) 60.0627 estimate D2E/DX2 ! ! D18 D(11,6,2,8) 179.9839 estimate D2E/DX2 ! ! D19 D(12,9,6,2) 180. estimate D2E/DX2 ! ! D20 D(12,9,6,10) -60.0407 estimate D2E/DX2 ! ! D21 D(12,9,6,11) 60.0407 estimate D2E/DX2 ! -----------------------------------------------------------------------------Trust Radius=3.00E-01 FncErr=1.00E-07 GrdErr=1.00E-07Number of steps in this run= 61 maximum allowed number of steps= 100. GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradInput orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 .000000 .000000 .0000002 6 .000000 .000000 1.5400003 1 1.028000 .000000 -.3630004 1 -.513000 -.890000 -.3630005 1 -.513000 .890000 -.3630006 6 -1.452000 .000000 2.0540007 1 .514000 .890000 1.9040008 1 .514000 -.890000 1.9040009 8 -1.452000 .000000 3.48400010 1 -1.965000 -.890000 1.69000011 1 -1.965000 .890000 1.69000012 1 -2.357000 .000000 3.804000----------------------------------------------------------Distance matrix (angstroms):1 2 3 4 51 C .0000002 C 1.540000 .0000003 H 1.090208 2.162913 .0000004 H 1.089513 2.162563 1.779545 .0000005 H 1.089513 2.162563 1.779545 1.780000 .0000006 C 2.515397 1.540292 3.462988 2.741480 2.7414807 H 2.163680 1.090317 2.489093 3.059807 2.4887788 H 2.163680 1.090317 2.489093 2.488778 3.0598079 O 3.774462 2.426405 4.577096 4.058723 4.05872310 H 2.740333 2.162366 3.736972 2.514580 3.08083011 H 2.740333 2.162366 3.736972 3.080830 2.51458012 H 4.475027 3.268202 5.368623 4.642879 4.6428796 7 8 9 106 C .0000007 H 2.163274 .0000008 H 2.163274 1.780000 .0000009 O 1.430000 2.674632 2.674632 .00000010 H 1.089846 3.059352 2.488220 2.067294 .00000011 H 1.089846 2.488220 3.059352 2.067294 1.78000012 H 1.970159 3.555944 3.555944 .959909 2.32696411 1211 H .00000012 H 2.326964 .000000Stoichiometry C3H8OFramework group CS[SG(C3H2O),X(H6)]Deg. of freedom 18Full point group CS NOp 2Largest Abelian subgroup CS NOp 2Largest concise Abelian subgroup CS NOp 2Standard orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 -.752500 1.745265 .0000002 6 -.752500 .205265 .0000003 1 -1.780500 2.108265 .0000004 1 -.239500 2.108265 .8900005 1 -.239500 2.108265 -.8900006 6 .699500 -.308735 .0000007 1 -1.266500 -.158735 -.8900008 1 -1.266500 -.158735 .8900009 8 .699500 -1.738735 .00000010 1 1.212500 .055265 .89000011 1 1.212500 .055265 -.89000012 1 1.604500 -2.058735 .000000----------------------------------------------------------Rotational constants (GHZ): 26.1932011 3.7793692 3.5233968 Isotopes: C-12,C-12,H-1,H-1,H-1,C-12,H-1,H-1,O-16,H-1,H-1,H-1Standard basis: 6-31G(d) (6D, 7F)There are 54 symmetry adapted basis functions of A' symmetry.There are 22 symmetry adapted basis functions of A" symmetry.Crude estimate of integral set expansion from redundant integrals=1.505. Integral buffers will be 262144 words long.Raffenetti 1 integral format.Two-electron integral symmetry is turned on.76 basis functions 144 primitive gaussians17 alpha electrons 17 beta electronsnuclear repulsion energy 129.9815628224 Hartrees.One-electron integrals computed using PRISM.The smallest eigenvalue of the overlap matrix is 4.568E-03Projected INDO Guess.Initial guess orbital symmetries:Occupied (A') (A') (A') (A') (A') (A') (A') (A') (A") (A')(A') (A") (A') (A") (A') (A') (A")Virtual (A') (A') (A") (A') (A') (A") (A') (A') (A") (A')(A') (A') (A') (A') (A") (A') (A') (A') (A") (A')(A') (A') (A') (A") (A") (A') (A') (A') (A") (A')(A') (A') (A") (A') (A') (A') (A') (A") (A") (A')(A') (A") (A') (A') (A') (A") (A') (A') (A') (A")(A') (A') (A') (A") (A") (A') (A") (A') (A")Requested convergence on RMS density matrix=1.00E-08 within 64 cycles. Requested convergence on MAX density matrix=1.00E-06.Integral accuracy reduced to 1.0E-05 until final iterations.Initial convergence to 1.0E-05 achieved. Increase integral accuracy.SCF Done: E(RHF) = -193.108299804 A.U. after 13 cyclesConvg = .4210E-08 -V/T = 2.0019S**2 = .0000********************************************************************** Population analysis using the SCF density.**********************************************************************Orbital Symmetries:Occupied (A') (A') (A') (A') (A') (A') (A') (A') (A') (A")(A') (A") (A') (A') (A") (A') (A")Virtual (A') (A') (A") (A') (A') (A') (A") (A") (A') (A')(A') (A') (A') (A") (A') (A") (A') (A') (A') (A")(A') (A") (A') (A") (A') (A') (A") (A') (A') (A')(A") (A') (A') (A") (A') (A') (A') (A') (A") (A")(A') (A') (A') (A") (A') (A") (A') (A") (A") (A")(A') (A') (A') (A') (A') (A') (A') (A') (A')The electronic state is 1-A'.Alpha occ. eigenvalues -- -20.55156 -11.27158 -11.21870 -11.21602 -1.34488 Alpha occ. eigenvalues -- -1.04902 -.91138 -.80335 -.68097 -.65514 Alpha occ. eigenvalues -- -.59587 -.57563 -.52470 -.49437 -.49018 Alpha occ. eigenvalues -- -.46479 -.43719Alpha virt. eigenvalues -- .22690 .23466 .29032 .30348 .31236 Alpha virt. eigenvalues -- .33005 .34373 .36169 .39842 .41533 Alpha virt. eigenvalues -- .43838 .73018 .74626 .75456 .78471 Alpha virt. eigenvalues -- .81502 .90319 .94052 .94858 1.02025 Alpha virt. eigenvalues -- 1.11199 1.12830 1.15746 1.17541 1.17793 Alpha virt. eigenvalues -- 1.19291 1.22184 1.22823 1.25013 1.28552 Alpha virt. eigenvalues -- 1.33387 1.48874 1.61956 1.69077 1.69771 Alpha virt. eigenvalues -- 1.73921 1.91863 1.99185 2.04423 2.14662 Alpha virt. eigenvalues -- 2.21819 2.27026 2.29587 2.31811 2.40990 Alpha virt. eigenvalues -- 2.52234 2.59474 2.61339 2.70118 2.75726Alpha virt. eigenvalues -- 2.75727 2.82709 2.90232 3.06969 3.24236 Alpha virt. eigenvalues -- 4.18505 4.58468 4.72822 4.90741Condensed to atoms (all electrons):1 2 3 4 5 61 C 5.128832 .322241 .394698 .390228 .390228 -.0590002 C .322241 5.090717 -.035129 -.039559 -.039559 .3624573 H .394698 -.035129 .522716 -.025265 -.025265 .0044074 H .390228 -.039559 -.025265 .544804 -.028204 -.0042455 H .390228 -.039559 -.025265 -.028204 .544804 -.0042456 C -.059000 .362457 .004407 -.004245 -.004245 4.7685107 H -.038013 .395663 -.002616 .004153 -.002754 -.0381678 H -.038013 .395663 -.002616 -.002754 .004153 -.0381679 O .002259 -.048330 -.000039 -.000027 -.000027 .21376410 H -.000236 -.045730 -.000125 .002919 .000049 .39660511 H -.000236 -.045730 -.000125 .000049 .002919 .39660512 H -.000204 .005358 .000003 -.000001 -.000001 -.0211077 8 9 10 11 121 C -.038013 -.038013 .002259 -.000236 -.000236 -.0002042 C .395663 .395663 -.048330 -.045730 -.045730 .0053583 H -.002616 -.002616 -.000039 -.000125 -.000125 .0000034 H .004153 -.002754 -.000027 .002919 .000049 -.0000015 H -.002754 .004153 -.000027 .000049 .002919 -.0000016 C -.038167 -.038167 .213764 .396605 .396605 -.0211077 H .533571 -.026250 .002198 .004674 -.004474 -.0001548 H -.026250 .533571 .002198 -.004474 .004674 -.0001549 O .002198 .002198 8.401143 -.039213 -.039213 .25794510 H .004674 -.004474 -.039213 .595713 -.045189 -.00320511 H -.004474 .004674 -.039213 -.045189 .595713 -.00320512 H -.000154 -.000154 .257945 -.003205 -.003205 .329724 Total atomic charges:11 C -.4927832 C -.3180623 H .1693554 H .1579025 H .1579026 C .0225837 H .1721708 H .1721709 O -.75265710 H .13821111 H .13821112 H .435000Sum of Mulliken charges= .00000Atomic charges with hydrogens summed into heavy atoms:11 C -.0076242 C .0262773 H .0000004 H .0000005 H .0000006 C .2990047 H .0000008 H .0000009 O -.31765710 H .00000011 H .00000012 H .000000Sum of Mulliken charges= .00000Electronic spatial extent (au): <R**2>= 384.4487Charge= .0000 electronsDipole moment (Debye):X= 1.3469 Y= 1.0634 Z= .0000 Tot= 1.7161 Quadrupole moment (Debye-Ang):XX= -22.6184 YY= -29.3407 ZZ= -26.5057XY= -2.9045 XZ= .0000 YZ= .0000Octapole moment (Debye-Ang**2):XXX= 10.4824 YYY= -8.5464 ZZZ= .0000 XYY= 8.4059 XXY= -10.3430 XXZ= .0000 XZZ= 1.3520 YZZ= -1.7820 YYZ= .0000 XYZ= .0000Hexadecapole moment (Debye-Ang**3):XXXX= -127.3697 YYYY= -308.8705 ZZZZ= -45.7961 XXXY= 42.6892 XXXZ= .0000 YYYX= 45.8838 YYYZ= .0000 ZZZX= .0000 ZZZY= .0000 XXYY= -55.7957 XXZZ= -30.7554 YYZZ= -58.9171 XXYZ= .0000 YYXZ= .0000 ZZXY= 21.9484N-N= 1.299815628224E+02 E-N=-7.123599860541E+02 KE= 1.927509295435E+02 Symmetry A' KE= 1.822597192938E+02Symmetry A" KE= 1.049121024977E+01***** Axes restored to original set *****-------------------------------------------------------------------Center Atomic Forces (Hartrees/Bohr)Number Number X Y Z-------------------------------------------------------------------1 6 .003252623 .000000000 .0117895022 6 -.013276735 .000000000 -.0035267613 1 -.005730690 .000000000 -.0029142724 1 .002449622 .003805382 -.0036693385 1 .002449622 -.003805382 -.0036693386 6 .010218108 .000000000 .0176709017 1 -.001146847 -.005346019 .0020749968 1 -.001146847 .005346019 .0020749969 8 -.009358321 .000000000 -.01121855610 1 -.000527977 .003279543 -.00124806911 1 -.000527977 -.003279543 -.00124806912 1 .013345418 .000000000 -.006115992-------------------------------------------------------------------Cartesian Forces: Max .017670901 RMS .006206408Internal Forces: Max .017334548 RMS .004599720GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad Berny optimization.Search for a local minimum.Step number 1 out of a maximum of 61All quantities printed in internal units (Hartrees-Bohrs-Radians)Second derivative matrix not updated -- first step.Eigenvalues --- .00233 .00237 .01295 .03834 .04893Eigenvalues --- .05137 .05719 .05725 .05824 .07659Eigenvalues --- .11233 .11705 .13705 .16000 .16000Eigenvalues --- .16000 .16000 .21946 .22074 .28493Eigenvalues --- .28519 .34776 .34776 .34789 .34830Eigenvalues --- .34830 .34869 .34869 .40989 .55493Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.00000RFO step: Lambda=-4.09258586E-03.Linear search not attempted -- first point.Iteration 1 RMS(Cart)= .03711253 RMS(Int)= .00086830Iteration 2 RMS(Cart)= .00094285 RMS(Int)= .00028427Iteration 3 RMS(Cart)= .00003007 RMS(Int)= .00028184Iteration 4 RMS(Cart)= .00000218 RMS(Int)= .00028186TrRot= .000000 .000000 .000000 .000000 .000000 .000000Variable Old X -DE/DX Delta X Delta X Delta X New X(Linear) (Quad) (Total)R1 2.91018 -.00154 .00000 -.00531 -.00531 2.90487R2 2.06019 -.00443 .00000 -.01260 -.01260 2.04760R3 2.05888 -.00304 .00000 -.00862 -.00862 2.05027R4 2.05888 -.00304 .00000 -.00862 -.00862 2.05027R5 2.91073 -.01312 .00000 -.04538 -.04538 2.86535R6 2.06040 -.00421 .00000 -.01197 -.01197 2.04843R7 2.06040 -.00421 .00000 -.01197 -.01197 2.04843R8 2.70231 -.01733 .00000 -.04187 -.04187 2.66044 R9 2.05951 -.00201 .00000 -.00571 -.00571 2.05380 R10 2.05951 -.00201 .00000 -.00571 -.00571 2.05380 R11 1.81396 -.01462 .00000 -.02615 -.02615 1.78781 A1 1.91024 .00459 .00000 .02702 .02662 1.93686 A2 1.91047 .00522 .00000 .03228 .03178 1.94224 A3 1.91033 -.00501 .00000 -.03147 -.03188 1.87846 A4 1.91047 .00522 .00000 .03228 .03178 1.94224 A5 1.91033 -.00501 .00000 -.03147 -.03188 1.87846 A6 1.91196 -.00500 .00000 -.02860 -.02920 1.88276 A7 1.91103 .00672 .00000 .04060 .04032 1.95134 A8 1.91118 -.00017 .00000 .01551 .01566 1.92684 A9 1.91027 -.00282 .00000 -.01722 -.01805 1.89222 A10 1.91118 -.00017 .00000 .01551 .01566 1.92684 A11 1.91027 -.00282 .00000 -.01722 -.01805 1.89222 A12 1.90987 -.00075 .00000 -.03726 -.03778 1.87209 A13 1.91103 -.00588 .00000 -.02266 -.02258 1.88845 A14 1.90951 .00168 .00000 .00641 .00648 1.91599 A15 1.91133 .00249 .00000 .01629 .01631 1.92764 A16 1.90951 .00168 .00000 .00641 .00648 1.91599 A17 1.91133 .00249 .00000 .01629 .01631 1.92764 A18 1.91109 -.00246 .00000 -.02276 -.02280 1.88829 A19 1.91067 -.00239 .00000 -.01456 -.01456 1.89611 D1 3.14159 .00000 .00000 .00000 .00000 3.14159 D2 -1.04791 -.00014 .00000 -.00227 -.00232 -1.05023 D3 1.04791 .00014 .00000 .00227 .00232 1.05023 D4 1.04706 -.00056 .00000 -.01332 -.01356 1.03351 D5 3.14075 -.00070 .00000 -.01559 -.01587 3.12488 D6 -1.04662 -.00042 .00000 -.01105 -.01124 -1.05786 D7 -1.04706 .00056 .00000 .01332 .01356 -1.03351 D8 1.04662 .00042 .00000 .01105 .01124 1.05786 D9 3.14075 .00070 .00000 .01559 .01756 3.15831 D10 3.14159 .00000 .00000 .00000 .00000 3.14159 D11 -1.04651 .00218 .00000 .03332 .03302 -1.01349 D12 1.04651 -.00218 .00000 -.03332 -.03302 1.01349 D13 1.04679 -.00048 .00000 -.01001 -.01001 1.03678 D14 3.14131 .00170 .00000 .02331 .02357 3.16488 D15 -1.04829 -.00266 .00000 -.04333 -.04302 -1.09132 D16 -1.04679 .00048 .00000 .01001 .01001 -1.03678 D17 1.04829 .00266 .00000 .04333 .04302 1.09132 D18 3.14131 -.00170 .00000 -.02331 -.02301 3.11830 D19 3.14159 .00000 .00000 .00000 .00000 3.14159 D20 -1.04791 -.00002 .00000 .00394 .00385 -1.04406 D21 1.04791 .00002 .00000 -.00394 -.00385 1.04406Item Value Threshold Converged?Maximum Force .017335 .000450 NORMS Force .004600 .000300 NOMaximum Displacement .112943 .001800 NORMS Displacement .036822 .001200 NOPredicted change in Energy=-2.000500E-03 GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradInput orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 .759831 .000000 -1.7418112 6 .715876 .000000 -.2052503 1 1.782259 .000000 -2.1005654 1 .268469 -.877046 -2.1498285 1 .268469 .877046 -2.1498286 6 -.707458 .000000 .3174537 1 1.224854 .872844 .1873108 1 1.224854 -.872844 .1873109 8 -.659180 .000000 1.72446710 1 -1.232120 -.880322 -.04441511 1 -1.232120 .880322 -.04441512 1 -1.544733 .000000 2.057396----------------------------------------------------------Distance matrix (angstroms):1 2 3 4 51 C .0000002 C 1.537189 .0000003 H 1.083543 2.174717 .0000004 H 1.084954 2.179625 1.750199 .0000005 H 1.084954 2.179625 1.750199 1.754092 .0000006 C 2.528538 1.516277 3.470663 2.794480 2.7944807 H 2.167858 1.083983 2.511360 3.072296 2.5252538 H 2.167858 1.083983 2.511360 2.525253 3.0722969 O 3.745487 2.369512 4.537786 4.079204 4.07920410 H 2.761157 2.143717 3.753559 2.585448 3.12616011 H 2.761157 2.143717 3.753559 3.126160 2.58544812 H 4.443533 3.198424 5.325178 4.664509 4.6645096 7 8 9 106 C .0000007 H 2.124294 .0000008 H 2.124294 1.745688 .0000009 O 1.407842 2.583466 2.583466 .00000010 H 1.086824 3.027212 2.467889 2.057224 .00000011 H 1.086824 2.467889 3.027212 2.057224 1.76064412 H 1.930914 3.453939 3.453939 .946069 2.30006511 1211 H .00000012 H 2.300065 .000000Stoichiometry C3H8OFramework group CS[SG(C3H2O),X(H6)]Deg. of freedom 18Full point group CS NOp 2Largest Abelian subgroup CS NOp 2Largest concise Abelian subgroup CS NOp 2Standard orientation:----------------------------------------------------------Center Atomic Coordinates (Angstroms)Number Number X Y Z----------------------------------------------------------1 6 1.465138 1.206792 .0000002 6 .000000 .741689 .0000003 1 1.528681 2.288469 .0000004 1 1.992620 .846683 .8770465 1 1.992620 .846683 -.8770466 6 -.110836 -.770532 .0000007 1 -.517468 1.122988 -.8728448 1 -.517468 1.122988 .8728449 8 -1.476811 -1.111313 .00000010 1 .381442 -1.175355 .88032211 1 .381442 -1.175355 -.88032212 1 -1.553191 -2.054293 .000000----------------------------------------------------------Rotational constants (GHZ): 26.8991374 3.8313903 3.5741980 Isotopes: C-12,C-12,H-1,H-1,H-1,C-12,H-1,H-1,O-16,H-1,H-1,H-1Standard basis: 6-31G(d) (6D, 7F)There are 54 symmetry adapted basis functions of A' symmetry.There are 22 symmetry adapted basis functions of A" symmetry.Crude estimate of integral set expansion from redundant integrals=1.505. Integral buffers will be 262144 words long.Raffenetti 1 integral format.Two-electron integral symmetry is turned on.76 basis functions 144 primitive gaussians17 alpha electrons 17 beta electronsnuclear repulsion energy 131.0838942242 Hartrees.One-electron integrals computed using PRISM.The smallest eigenvalue of the overlap matrix is 4.389E-03Initial guess read from the read-write file:Initial guess orbital symmetries:Occupied (A') (A') (A') (A') (A') (A') (A') (A') (A') (A")(A') (A") (A') (A') (A") (A') (A")Virtual (A') (A') (A") (A') (A') (A') (A") (A") (A') (A')(A') (A') (A') (A") (A') (A") (A') (A') (A') (A")(A') (A") (A') (A") (A') (A') (A") (A') (A') (A')(A") (A') (A') (A") (A') (A') (A') (A') (A") (A")(A') (A') (A') (A") (A') (A") (A') (A") (A") (A")(A') (A') (A') (A') (A') (A') (A') (A') (A')Requested convergence on RMS density matrix=1.00E-08 within 64 cycles.Requested convergence on MAX density matrix=1.00E-06.Integral accuracy reduced to 1.0E-05 until final iterations.Initial convergence to 1.0E-05 achieved. Increase integral accuracy.SCF Done: E(RHF) = -193.110364246 A.U. after 13 cyclesConvg = .9144E-08 -V/T = 2.0010S**2 = .0000***** Axes restored to original set *****-------------------------------------------------------------------Center Atomic Forces (Hartrees/Bohr)Number Number X Y Z-------------------------------------------------------------------1 6 .001367252 .000000000 .0056818312 6 -.001959179 .000000000 -.0068386883 1 .000908610 .000000000 -.0002302364 1 -.000542056 -.000764258 -.0002911225 1 -.000542056 .000764258 -.0002911226 6 .003389929 .000000000 .0045017137 1 .001109824 .000541223 .0001944628 1 .001109824 -.000541223 .0001944629 8 -.001538434 .000000000 -.00331756710 1 -.001595127 -.000439380 -.00084975511 1 -.001595127 .000439380 -.00084975512 1 -.000113459 .000000000 .002095775-------------------------------------------------------------------Cartesian Forces: Max .006838688 RMS .002021700Internal Forces: Max .004833275 RMS .001081579GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad Berny optimization.Search for a local minimum.Step number 2 out of a maximum of 61All quantities printed in internal units (Hartrees-Bohrs-Radians)Update second derivatives using information from points 1 2Trust test= 1.03E+00 RLast= 1.59E-01 DXMaxT set to 4.24E-01Eigenvalues --- .00233 .00237 .01295 .03532 .04586Eigenvalues --- .05212 .05382 .05428 .05741 .08200Eigenvalues --- .11061 .12049 .13627 .15136 .16000Eigenvalues --- .16000 .17131 .20946 .22144 .27556Eigenvalues --- .30432 .34762 .34776 .34784 .34830Eigenvalues --- .34853 .34869 .36541 .40801 .56424Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.000001000.000001000.000001000.000001000.00000Eigenvalues --- 1000.00000RFO step: Lambda=-2.84994529E-04.Quartic linear search produced a step of -.00115.Iteration 1 RMS(Cart)= .01008000 RMS(Int)= .00008374Iteration 2 RMS(Cart)= .00008982 RMS(Int)= .00001493Iteration 3 RMS(Cart)= .00000157 RMS(Int)= .00001490TrRot= .000002 .000000 -.000001 .000000 -.000001 .000000 Variable Old X -DE/DX Delta X Delta X Delta X New X(Linear) (Quad) (Total) R1 2.90487 -.00483 .00001 -.01727 -.01727 2.88760 R2 2.04760 .00093 .00001 .00182 .00183 2.04943 R3 2.05027 .00097 .00001 .00219 .00220 2.05247 R4 2.05027 .00097 .00001 .00219 .00220 2.05247 R5 2.86535 .00191 .00005 .00358 .00363 2.86898 R6 2.04843 .00103 .00001 .00213 .00214 2.05057 R7 2.04843 .00103 .00001 .00213 .00214 2.05057 R8 2.66044 -.00128 .00005 -.00598 -.00593 2.65451 R9 2.05380 .00141 .00001 .00365 .00366 2.05745 R10 2.05380 .00141 .00001 .00365 .00366 2.05745 R11 1.78781 .00084 .00003 -.00027 -.00024 1.78757 A1 1.93686 -.00005 -.00003 .00179 .00176 1.93861 A2 1.94224 -.00009 -.00004 .00139 .00135 1.94360 A3 1.87846 .00011 .00004 -.00120 -.00116 1.87730 A4 1.94224 -.00009 -.00004 .00139 .00135 1.94360 A5 1.87846 .00011 .00004 -.00120 -.00116 1.87730 A6 1.88276 .00003 .00003 -.00243 -.00242 1.88035 A7 1.95134 .00202 -.00005 .01424 .01417 1.96551 A8 1.92684 -.00081 -.00002 -.00395 -.00402 1.92282 A9 1.89222 -.00026 .00002 .00126 .00128 1.89350A10 1.92684 -.00081 -.00002 -.00395 -.00402 1.92282A11 1.89222 -.00026 .00002 .00126 .00128 1.89350A12 1.87209 .00006 .00004 -.00967 -.00965 1.86244。