
Cell Metabolism
Short Article
Hypoxic Regulation of Glutamine Metabolism through HIF1and SIAH2Supports Lipid Synthesis that Is Necessary for Tumor Growth
Ramon C.Sun1and Nicholas C.Denko1,*
1Department of Radiation Oncology,James Cancer Hospital and Comprehensive Cancer Center,Ohio State University Wexner School of Medicine,Columbus,OH43210,USA
*Correspondence:nicholas.denko@https://www.doczj.com/doc/2518821123.html,
https://www.doczj.com/doc/2518821123.html,/10.1016/j.cmet.2013.11.022
SUMMARY
Recent reports have identi?ed a phenomenon by which hypoxia shifts glutamine metabolism from oxidation to reductive carboxylation.We now identify the mechanism by which HIF-1activation results in a dramatic reduction in the activity of the key mito-chondrial enzyme complex a ketoglutarate dehydro-genase(a KGDH).HIF-1activation promotes SIAH2 targeted ubiquitination and proteolysis of the 48kDa splice variant of the E1subunit of the a KGDH complex(OGDH2).Knockdown of SIAH2or mutation of the ubiquitinated lysine residue on OGDH2 (336KA)reverses the hypoxic drop in a KGDH activity, stimulates glutamine oxidation,and reduces gluta-mine-dependent lipid synthesis.336KA OGDH2-expressing cells require exogenous lipids or citrate for growth in hypoxia in vitro and fail to grow as model tumors in immunode?cient mice.Reversal of hypoxic mitochondrial function may provide a target for the development of next-generation anticancer agents targeting tumor metabolism. INTRODUCTION
Glutamine is the most abundant amino acid in blood and is second only to glucose as a carbon source for energy production and anabolic processes.Recently,glutamine was identi?ed as being essential for fuelling mitochondrial metabolism in rapidly dividing cancer cells transformed with either c-MYC(Gao et al.,2009;Wise et al.,2008)or Kirsten rat sarcoma viral oncogene homolog(Son et al.,2013).Inhibiting glutamine meta-bolism has been proposed as an anticancer therapy(Wise and Thompson,2010).
Mitochondrial glutamine metabolism can follow either oxida-tive or reductive pathways(Holleran et al.,1995).Mitochondrial utilization of glutamine begins with a two-step conversion of glutamine to a ketoglutarate(a KG),typically by glutaminase and glutamate dehydrogenase.a KG can be either oxidized by a KG dehydrogenase(a KGDH)to succinate(standard tricarbox-ylic acid[TCA]cycle reaction)or reductively carboxylated by iso-citrate dehydrogenase(reverse TCA cycle)to isocitrate and then citrate.The reductive cycle has been shown to be favored in cells where HIF-1a is stabilized(Metallo et al.,2012b;Wise et al., 2011)or in cells with compromised electron transport capacity (Mullen et al.,2012).Glutamine-derived citrate can be trans-ported to the cytoplasm in order to generate acetyl CoA for anabolic processes such as fatty acid synthesis(Gameiro et al.,2013b;Metallo et al.,2012b).
a KGDH consists of E1(oxogluterate dehydrogenase,OGDH), E2(dihydrolipoamide S-succinyltransferase,DLST),and E3(di-hydrolipoamide dehydrogenase,DLD)components that collec-tively convert a KG to succinyl-CoA and nicotinamide adenine dinucleotide(Patel and Harris,1995).The three-enzyme(E1-E2-E3)dehydrogenase complex structure is conserved in the pyruvate dehydrogenase(PDH)and branched chain a-ketoacid dehydrogenase complexes.PDH serves as the major entry point for glucose-derived pyruvate into the mitochondrial TCA cycle (Patel and Korotchkina,2001).Recent work has found that HIF1activation inhibits PDH,which blocks glucose oxidation (Kim et al.,2006;Papandreou et al.,2006).In humans, there are three major splice variants of the OGDH gene,V1 (114kDa),V2(48kDa),and V3(114kDa),although the functional differences of these enzymes remain unreported. Environmental hypoxia has a major effect on gene expression, largely by the induction of the HIF a family of transcription factors through protein stabilization(Epstein et al.,2001).However, protein destabilization by hypoxia-regulated ubiquitination (Vin?as-Castells et al.,2010)or SUMOylation(Comerford et al., 2003)has also been reported.The E3ubiquitin ligase SIAH2 destabilizes several critical proteins in hypoxia(Qi et al.,2008). In this work,we connect the crucial HIF-1and SIAH2regulatory systems that control the metabolism of glutamine.Furthermore, we show that this circuit is necessary for the growth of cells in hypoxia and as model tumors.
RESULTS
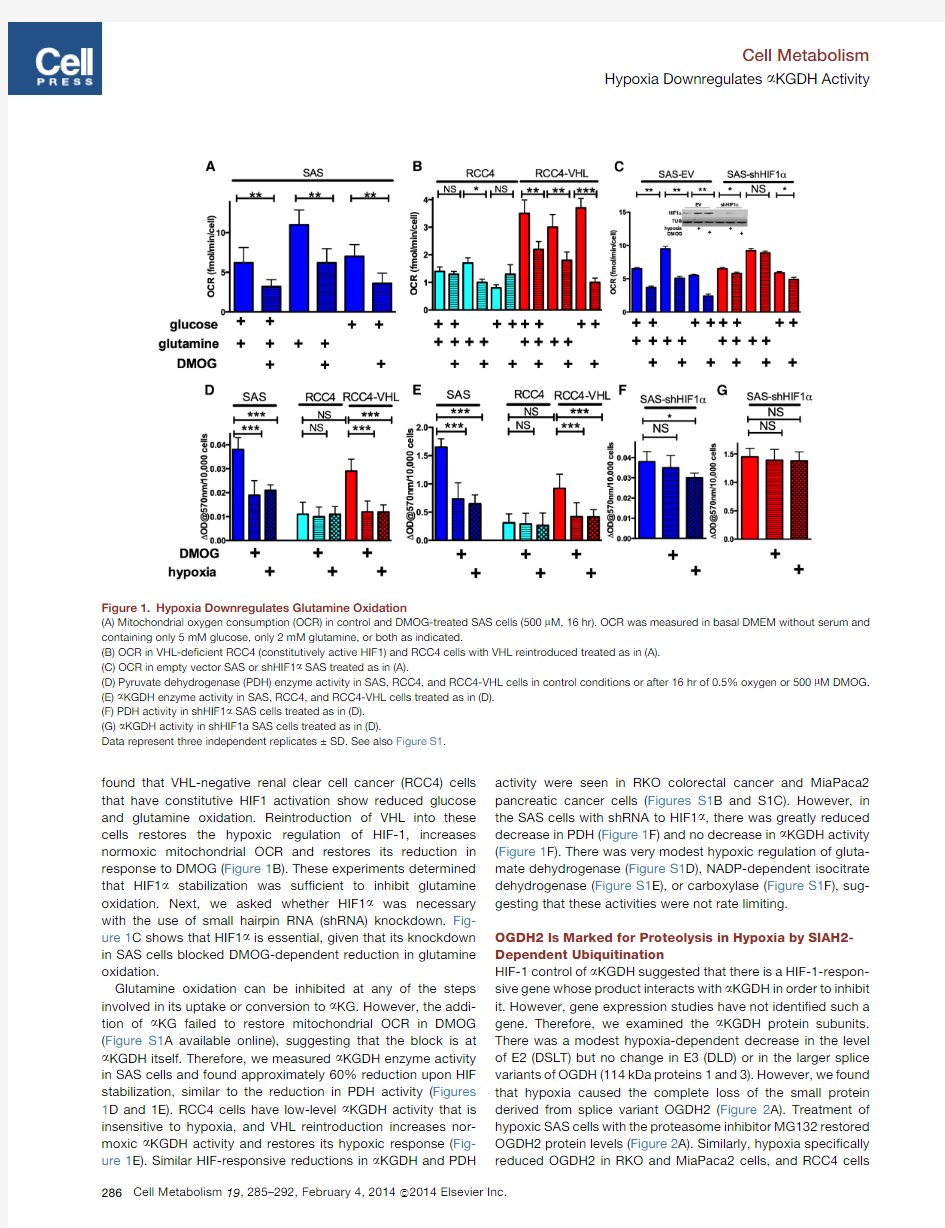
HIF1Stabilization Is Necessary and Suf?cient to Reduce Mitochondrial Glutamine Oxidation and a KGDH Activity Hypoxia inhibits mitochondrial glucose oxidation(Denko,2008; Kim et al.,2006),so we asked whether it could also inhibit glutamine oxidation.We found reduced mitochondrial oxygen consumption(OCR)in the head and neck tumor cell line SAS when they were treated with the HIF-stabilizing PHD inhibitor DMOG.This decrease was detected in complete basal, glucose-only,or glutamine-only media(Figure1A).We also
found that VHL-negative renal clear cell cancer (RCC4)cells that have constitutive HIF1activation show reduced glucose and glutamine oxidation.Reintroduction of VHL into these cells restores the hypoxic regulation of HIF-1,increases normoxic mitochondrial OCR and restores its reduction in response to DMOG (Figure 1B).These experiments determined that HIF1a stabilization was suf?cient to inhibit glutamine oxidation.Next,we asked whether HIF1a was necessary with the use of small hairpin RNA (shRNA)knockdown.Fig-ure 1C shows that HIF1a is essential,given that its knockdown in SAS cells blocked DMOG-dependent reduction in glutamine oxidation.
Glutamine oxidation can be inhibited at any of the steps involved in its uptake or conversion to a KG.However,the addi-tion of a KG failed to restore mitochondrial OCR in DMOG (Figure S1A available online),suggesting that the block is at a KGDH itself.Therefore,we measured a KGDH enzyme activity in SAS cells and found approximately 60%reduction upon HIF stabilization,similar to the reduction in PDH activity (Figures 1D and 1E).RCC4cells have low-level a KGDH activity that is insensitive to hypoxia,and VHL reintroduction increases nor-moxic a KGDH activity and restores its hypoxic response (Fig-ure 1E).Similar HIF-responsive reductions in a KGDH and PDH
activity were seen in RKO colorectal cancer and MiaPaca2pancreatic cancer cells (Figures S1B and S1C).However,in the SAS cells with shRNA to HIF1a ,there was greatly reduced decrease in PDH (Figure 1F)and no decrease in a KGDH activity (Figure 1F).There was very modest hypoxic regulation of gluta-mate dehydrogenase (Figure S1D),NADP-dependent isocitrate dehydrogenase (Figure S1E),or carboxylase (Figure S1F),sug-gesting that these activities were not rate limiting.
OGDH2Is Marked for Proteolysis in Hypoxia by SIAH2-Dependent Ubiquitination
HIF-1control of a KGDH suggested that there is a HIF-1-respon-sive gene whose product interacts with a KGDH in order to inhibit it.However,gene expression studies have not identi?ed such a gene.Therefore,we examined the a KGDH protein subunits.There was a modest hypoxia-dependent decrease in the level of E2(DSLT)but no change in E3(DLD)or in the larger splice variants of OGDH (114kDa proteins 1and 3).However,we found that hypoxia caused the complete loss of the small protein derived from splice variant OGDH2(Figure 2A).Treatment of hypoxic SAS cells with the proteasome inhibitor MG132restored OGDH2protein levels (Figure 2A).Similarly,hypoxia speci?cally reduced OGDH2in RKO and MiaPaca2cells,and RCC4
cells
Figure 1.Hypoxia Downregulates Glutamine Oxidation
(A)Mitochondrial oxygen consumption (OCR)in control and DMOG-treated SAS cells (500m M,16hr).OCR was measured in basal DMEM without serum and containing only 5mM glucose,only 2mM glutamine,or both as indicated.
(B)OCR in VHL-de?cient RCC4(constitutively active HIF1)and RCC4cells with VHL reintroduced treated as in (A).(C)OCR in empty vector SAS or shHIF1a SAS treated as in (A).
(D)Pyruvate dehydrogenase (PDH)enzyme activity in SAS,RCC4,and RCC4-VHL cells in control conditions or after 16hr of 0.5%oxygen or 500m M DMOG.(E)a KGDH enzyme activity in SAS,RCC4,and RCC4-VHL cells treated as in (D).(F)PDH activity in shHIF1a SAS cells treated as in (D).(G)a KGDH activity in shHIF1a SAS cells treated as in (D).
Data represent three independent replicates ±SD.See also Figure S1.
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
have very low levels of OGDH2that could be increased with MG132(Figure S2A).
a KGDH E2has been reported to be marked for proteolysis by the E3ubiquitin ligase SIAH2upon mitochondrial disruption (Ha-belhah et al.,2004);therefore,we examined whether SIAH2could be responsible for the hypoxic destruction of OGDH2.We found that the SIAH2inhibitor Vitamin K3(Shah et al.,2009)could also restore OGDH2protein (Figure 2A).Further-more,treatment of hypoxic cells with both MG132and the deu-biquitinating enzyme inhibitor nsc632839resulted in the appear-ance of variant of OGDH27kDa larger,suggesting the addition of ubiquitin (Figure 2B).Larger,polyubiquitinated species could not be detected because of nonspeci?c antibody reactivity at 60kDa.We also found that MG132or VitK3could restore hypox-ic a KGDH activity (Figure 2B).These treatments had no effect on OGDH2protein levels in normoxic conditions (Figure S2
B).
Figure 2.SIAH2E3Ligase Is Responsible for OGDH2Protein Degradation after HIF1Stabilization
(A)Western blot of SAS protein extracts from control,0.5%O 2conditions,and 0.5%O 2with 10m M MG132,50m M VitK3,or 10m M DUBI and MG132as indicated.
(B)a KGDH activity in SAS cells treated with either 500m M DMOG or DMOG in combination with 10m M MG132or 50m M VitK3.
(C)Western blot analysis of the indicted proteins in SAS,RCC4,and RCC4-VHL cells with and without shRNA-SIAH2as indicated cultured in normoxia or 16hr 0.5%oxygen.
(D)a KGDH activity in control and shRNA-SIAH2SAS,RCC4,and RCC4-VHL cells ±16hr 500m M DMOG.
(E)Mitochondrial OCR in control and shRNA-SIAH2SAS,RCC4,and RCC4-VHL cells ±16hr 500m M DMOG measured in basal,serum-free media containing only glutamine.
Data from (B),(D),and (E)are two independent experiments in triplicate ±SD.See also Figure S2.
In order to genetically connect SIAH2ubiquitination to HIF-dependent decrease of OGDH2protein,we reduced SIAH2protein with stable shRNA in SAS,RCC4,and RCC4VHL cells (Figure 2C,top row).This treatment decreased both the baseline and hypoxia-inducible SIAH2protein and blocked HIF-depen-dent reduction of OGDH2and the SIAH2target protein PHD3(Nakayama et al.,2004).This genetic manipulation also blocked the HIF-dependent decrease in a KGDH activity and mito-chondrial OCR in the SAS cells (Figures 2D and 2E).We also found that stable knockdown of SIAH2in VHL-de?cient RCC4cells resulted in increased nor-moxic OGDH2protein levels,a KGDH activity,and mitochondrial OCR,which were all insensitive to hypoxia or DMOG
(Figures 2C–2E).Finally,SIAH2knockdown in the RCC4VHL cells resulted in no change in normoxic OGDH2protein or activity but blocked the reduction of these measures in DMOG (Figures 2C–2E).
HIF-Dependent Change in Glutamine Metabolism Enhances the Growth of Tumors In Vivo
To determine the functional consequence of hypoxic SIAH2dependent proteolysis of OGDH2,we needed to speci?cally block it while allowing SIAH2to mark other proteins (Nakayama et al.,2009).Therefore,we identi?ed the speci?c ubiquitinated lysine residue on OGDH2using mass spectrometry so we could mutate it and block the SIAH2-dependent destruction.We immunoprecipitated FLAG-OGDH2from hypoxic cells treated with MG132and DUBI and analyzed the precipitate at the Ohio State University Comprehensive Cancer Center proteomic core
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
facility.We identi?ed 24mitochondrial proteins as copurifying with OGDH2,including the E2subunit of a KGDH (DSLT)and the E1b subunit of PDH (Table S1).One peptide of OGDH2was ubiquitinated at residue lysine 336(Figure S2C).We converted lysine 336to alanine by site-directed mutagenesis and stably introduced the wild-type (WT)or mutant proteins into SAS cells.The modi?ed protein showed signi?cant resis-tance to hypoxic degradation (Figure 3B,second and third rows)and did show high-molecular-weight polyubiquitinated species when treated with hypoxia,MG132,and DUBI (Fig-ure S3A).The cells expressing the 336KA OGDH2protein were also resistant to HIF-dependent reduction of a KGDH activity (Figure 3C)and OCR (Figure 3D).336KA OGDH2showed similar function when introduced into RKO and MiaPaca2cells (Figures S3B–S3I).
We could then determine the importance of reductive gluta-mine metabolism for the growth of model tumors.Cells express-ing either WT or hypoxia-resistant OGDH2were injected into immunode?cient mice for growth as tumors.Overexpression of the WT protein had a modest inhibitory effect on tumor growth (Figure 3E),possibly because high-level expression resulted
in
Figure 3.Hypoxia-Resistant a KGDH Activ-ity Is Not Compatible with Tumor Growth
(A)Different splice variants of OGDH.OGDH 1and OGHD3are 99%identical except at amino acid positions 153–169(represented by the inverted triangle).OGDH2is identical to OGDH1until amino acid 403.
(B)Western blot of lysates from SAS cells stably expressing empty vector,FLAG-WT OGDH2,and FLAG-336KA OGDH2exposed to normoxia or 0.5%oxygen for 16hr.Lysates were probed for proteins as indicated.Note that both anti-FLAG and anti-OGDH2show OGDH2336KA that is resistant to hypoxic degradation.
(C)a KGDH activity in SAS cell lines described in (B)treated with either control,16hr 500m M DMOG,or 0.5%oxygen.
(D)Mitochondrial OCR in SAS cell lines described in (B)treated with either control,500m M DMOG,or 0.5%oxygen and measured in glutamine-only media.
(E)Tumor volume of SAS cells expressing empty vector,OGDH-WT,and OGDH-336KA grown in nude mice (n =8–10per group).Statistically signi?cant growth differences exist between all three groups.
(F)Western blot analysis of lysates of tumors harvested after growth as described in (E).Note the decrease in markers of proliferation as tumors grow more slowly.
Data from (C)and (D)are mean ±SD and tumor volumes are mean ±SE.See also Figure S3and Table S1.
incomplete degradation (Figures 3B and 3F).Expression of the hypoxia-resis-tant 336KA protein had a profound inhib-itory effect (Figure 3E).Although both OGDH2-modi?ed cell line tumors grew slower than the parental tumors,the cells
expressing 336KA OGDH2grew signi?cantly slower than the cells expressing WT OGDH2.Similar tumor growth inhibition of the two proteins was seen in RKO and MiaPaca2cells (Figures S3B–S3I).Analysis of lysates from these tumors showed that WT FLAG-OGDH2was expressed at intermediate levels in vivo,whereas 336KA FLAG-OGDH2was expressed at much higher levels (Figures 3F,S3E,and S3I).Furthermore,analysis of the tumor lysates for markers of proliferation (Ki67and cyclin D)indi-cated that there was reduced proliferation in the336KA OGDH2tumors.There was no increase in the apoptotic marker,cleaved caspase 3.These results suggest that reductive carboxylation of glutamine is necessary for the proliferation of tumor cells in vivo.Cellular Growth in Hypoxia Requires Glutamine-Derived Lipid Production
The tumors expressing 336KA OGDH2appeared to have reduced proliferation,so we examined the effect of forced gluta-mine oxidation on the proliferation of tumor cells in vitro.Reduc-tive carboxylation of glutamine in hypoxia has been proposed to be a mechanism for generating citrate for lipid synthesis when glucose entry into the TCA cycle is reduced (Le et al.,2012;
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
Figure 4.OGDH 336KA Inhibits Cell Growth under Hypoxia by Reducing Glutamine-Derived Lipid Production
(A)Relative hypoxic proliferation of SAS,RCC4,and RCC4VHL cells grown for 72hr in the indicated media presented as a percentage of normoxic growth.Hypoxic growth requires either glutamine (2mM)or glutamine derivatives a KG (2mM)or citrate (2mM).
(B)Hexane-soluble lipids derived from a 1hr pulse of 0.5m Ci 14C-glutamine in SAS cells expressing either empty vector or OGDH2-336KA grown for 16hr in normoxia or hypoxia.
(C)Hexane-soluble lipids derived from a 1hr pulse of 0.5m Ci 14C-glucose in SAS cells as described in (B).
(D)Relative hypoxic proliferation of SAS cells expressing empty vector or 336KA-OGDH2after 72hr in basal glutamine-containing media and 10%charcoal-stripped serum supplemented with either 2mM citrate or 2mM a KG.Note that WT cells have maximal hypoxic proliferation with glutamine,but 336KA-OGDH2-expressing cells require citrate.
(legend continued on next page)
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
Metallo et al.,2012a;Wise et al.,2011).Removal of glutamine from the media causes signi?cant growth inhibition in hypoxia,which can be rescued with the addition of a KG (Wise et al.,2011)or citrate (Figure 4A).Therefore,we directly measured the incorporation of either glucose or glutamine into newly synthesized fatty acids with 14C-labeled precursors.Cells expressing either empty vector or 336KA OGDH2were treated with hypoxia and pulsed with radio-labeled glucose or gluta-mine.Glutamine uptake was slightly reduced by hypoxia in all lines,whereas glucose uptake was increased (Figures S4A–S4D).Fatty acid production was measured by determining hexane-extractable counts from the pulsed cells.Both lines showed hypoxic decrease in glucose incorporation into lipids (Figure 4C).However,there was a speci?c reduction in glutamine incorporation into lipids in the 336KA-OGDH2-expressing cells.Glutamine-derived lipids were increased by hypoxia in empty vector cells but were further reduced in 336KA cells (Figure 4B).Consistent results were observed in RCC4and RCC4VHL cells (Figures S4E and S4F).
The 336KA OGDH2cells allowed us to determine whether the inability to generate lipids from glutamine would inhibit hypoxic proliferation in vitro.We found that the 336KA OGDH2-expressing cells grew as fast as the empty vector cells in normoxia but had a reduced growth rate in hypoxia,even in complete media with glutamine and 10%charcoal stripped serum (to remove the serum lipids)(Figure 4D).This 336KA-dependent growth defect was due to an inability to produce mitochondrial metabolites,given that hypoxic growth of these cells could be rescued by addition of exogenous citrate but not exogenous a KG (which is simply oxidized instead of being used to make citrate)(Figure 4D).To establish that the growth defect was due to a lack of citrate to produce lipids,we added the desaturated lysophospholipid (C18:1)that is rapidly taken up by cells (Kamphorst et al.,2013).This lipid could rescue the glutamine-dependent growth of WT OGDH2cells and the cit-rate-dependent growth of the 336KA OGDH2-expressing cells.The saturated lysophospholipid (C24:0)that is poorly taken up by cells could not rescue either cell lines (Figures 4E and S4G).DISCUSSION
Microenvironmental hypoxia is a signi?cant stress that produces compensatory metabolic changes in tumor cells.The cell con-serves oxygen by reducing its use for mitochondrial energy production.However,reduction of TCA cycle reactions leads to a reduced supply of mitochondrial intermediates.Under hyp-oxic pressure,cells use alternative fuels such as glutamine to generate citrate and support proliferation (Figure 4F).Cells can use this compensation until the oxygen level becomes too low,at which point they die (Papandreou et al.,2005).
Recent publications have described how low levels of intracel-lular citrate and high levels of a KG are associated with HIF-dependent reductive carboxylation of glutamine (Fendt et al.,2013;Gameiro et al.,2013a ).These elegant tracer studies are
entirely consistent with our results and complement our current ?ndings.Their conclusions focused on the changes in the me-tabolites during hypoxia but did not identify how these changes occurred.We now propose that hypoxia causes a decrease in glucose-derived citrate because of decreased PDH activity and also increases a KG levels because of decreased a KGDH activity.These changes in substrate concentrations drive the reverse reaction at isocitrate dehydrogenase.
The biochemical regulation of the mammalian a KGDH complex is understudied.The E1component of the related PDH contains two proteins,the a protein with a thiamine pyro-phosphate binding domain (TBD)and the b protein with a trans-ketolase-like domain (TK).In PDH,the E1subunit functions as a heterotetramer of a 2b 2.The full-length a KGDH E1(variants 1or 3)OGDH protein contains both a TBD and TK domain and func-tions as a homodimer.The smaller OGDH variant 2contains just the TBD.However,PDHE1b copuri?ed with OGDH2(Table S1).This suggests that OGDH2can function in the a KGDH complex by adding transketolase activity with the use of the PDH version found in the E1b enzyme.Careful biochemical puri?cation and analysis will be needed to determine whether this is indeed the case.
The mechanism by which OGDH2is speci?cally degraded in hypoxia is interesting,given that lysine 336is conserved on the full-length OGDH1and OGDH3.There may be variable accessibility to the lysine based on the 3D structure of the whole versus half protein or by function of its interactions with PDH E1b .It is also not yet clear how an intramitochondrial protein could be degraded by the cytoplasmic proteasome,although recent work has shown that such a mechanism exists (Azzu and Brand,2010).
Fatty acid desaturation and cholesterol synthesis require molecular oxygen as an enzymatic substrate,indicating why these processes should be regulated under hypoxia.Recent work has shown that hypoxic cells preferentially take up unsatu-rated lysophospholipids from the growth media,and the level of activity of the sterol desaturase is also downregulated by hypox-ia (Kamphorst et al.,2013).Here,we show that de novo lipogen-esis is also required for the growth of cells in hypoxia in vitro and in the hypoxia that exists within the tumor microenvironment.Glutamine appears to play a crucial role in hypoxic lipogenesis because glucose ?ux into the mitochondria is limited by reduced PDH activity.However,it is not apparent why blocking the hypoxic downregulation of PDH (and stimulate glucose ?ux into the mitochondria)can also block the growth of model tumors (Hitosugi et al.,2011;McFate et al.,2008).
EXPERIMENTAL PROCEDURES
Cells
MiaPaca2pancreatic cancer and RKO colorectal cancer cells were from ATCC.SAS head and neck cancer,RCC4renal cancer,and RCC4-VHL cells were gifts from Q.Le and A.Giaccia of Stanford University.All cells were cultured in Dulbecco’s modi?ed Eagle’s medium (DMEM)with 10%fetal bovine serum (FBS),25mM glucose,and 4mM L-glutamine;unless stated
(E)Relative hypoxic proliferation of SAS cells described in (D)in basal media with 10%charcoal-stripped serum.Supplementation with absorbable lysophos-pholipid rescues the glutamine dependence of the WT OGDH2cells and the citrate dependence of the 336KA-OGDH2-expressing cells.(F)A model illustrating how hypoxic degradation of OGDH2shifts the fate of a KG from energy production to the production of lipids.Data from panels (A)–(E)represents mean ±SD.See also Figure S4.
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
otherwise,metabolic experiments were conducted in5mM glucose.Hypoxia (0.5%oxygen)was generated in a HypOxygen H35workstation.Cells were regularly tested for mycoplasma.For lipid-dependent experiments,DMEM was supplemented with10%charcoal-stripped FBS(Invitrogen).For tumor growth,53106cells were injected subcutaneously into the?ank of nude mice,the growth of tumors was monitored with calipers,and volume was calculated by w23l30.52.
Dehydrogenase Assay
For a KGDH activity assay,104treated cells were permeabilized with0.5% Triton X-100and incubated1mM MgCl2,0.1mM CaCl2,0.05mM EDTA, 0.2%Triton X-100,0.3mM ThDP,rotenone,3mM a KG,3mM NAD+,1mM CoA,0.75mM nitroblue tetrazolium,and0.05mM phenazine methosulfate in50mM Tris(pH7.6)at37 C for40min.Then,cells were solubilized in 10%SDS and0.01N HCl overnight.Optical density at570was determined (Molecular Devices).Slight modi?cations were made for PDH,isocitrate dehy-drogenase(IDH),and GDH activities(see the Supplemental Information).
Oxygen Consumption Rate
1–23104cells were plated in96-well Seahorse Assay plates.The next morn-ing,culture media was replaced with bicarbonate-free DMEM with or without glucose or glutamine,and oxygen consumption rate was measured.
Mass Spectrometry
Carboxy-tagged FLAG-OGDH2expression plasmid was purchased from Origene.FLAG-OGDHV2cells were cultured for16hr in0.5%oxygen and Mg132and DUBI for3hr.Precipitated FLAG-OGDH2was analyzed on a Thermo Scienti?c LTQ Orbitrap mass spectrometer equipped with a micro-spray source(Michrom Bioresources),data collected were searched by Mascot Daemon(Matrix Science).Modi?cations identi?ed were methionine oxidation(variable),deamidation(variable),ubiquitination(variable),and car-bamidomethyl cysteine(?xed).
Lipid Synthesis
53104cells were plated in12-well plates in0.5%oxygen or0.5mM DMOG in DMEM(5mM glucose and1mM glutamine),and,16hr later,0.5m Ci14C-L-glutamine(0.5m Ci/mM)or glucose(0.1m Ci/mM)was added for1hr.Cells were rinsed with PBS,and lipids were extracted with500ul of hexane:isopro-panol(3:1)for30min.Total extractable counts are reported.
Statistics
Statistical comparisons were made with a two-tailed Student’s t test, and values are indicated with*p<0.05,>0.01;**p<0.01,>0.001;***p< 0.001.
SUPPLEMENTAL INFORMATION
Supplemental Information contains Supplemental Experimental Procedures, four?gures,and one table and can be found with this article online at http:// https://www.doczj.com/doc/2518821123.html,/10.1016/j.cmet.2013.11.022.
ACKNOWLEDGMENTS
This work was supported by the NCI(NCD).The authors would like to thank the proteomics core at OSU CCC.They would also like to thank Drs.Ioanna Pa-pandreou,Amato Giaccia,Ze’ev Ronai,Naduparambil Jacob,Deliang Guo, and members of the https://www.doczj.com/doc/2518821123.html,b for their helpful discussions.
Received:July12,2013
Revised:September7,2013
Accepted:November1,2013
Published:February4,2014
REFERENCES
Azzu,V.,and Brand,M.D.(2010).Degradation of an intramitochondrial protein by the cytosolic proteasome.J.Cell Sci.123,578–https://www.doczj.com/doc/2518821123.html,erford,K.M.,Leonard,M.O.,Karhausen,J.,Carey,R.,Colgan,S.P.,and Taylor,C.T.(2003).Small ubiquitin-related modi?er-1modi?cation mediates resolution of CREB-dependent responses to hypoxia.Proc.Natl.Acad.Sci. USA100,986–991.
Denko,N.C.(2008).Hypoxia,HIF1and glucose metabolism in the solid tumour.Nat.Rev.Cancer8,705–713.
Epstein,A.C.,Gleadle,J.M.,McNeill,L.A.,Hewitson,K.S.,O’Rourke,J.,Mole, D.R.,Mukherji,M.,Metzen, E.,Wilson,M.I.,Dhanda, A.,et al.(2001).
C.elegans EGL-9and mammalian homologs de?ne a family of dioxygenases that regulate HIF by prolyl hydroxylation.Cell107,43–54.
Fendt,S.M.,Bell, E.L.,Keibler,M.A.,Olenchock, B.A.,Mayers,J.R., Wasylenko,T.M.,Vokes,N.I.,Guarente,L.,Vander Heiden,M.G.,and Stephanopoulos,G.(2013).Reductive glutamine metabolism is a function of the a-ketoglutarate to citrate ratio in cells.Nat Commun4,2236.
Gameiro,P.A.,Laviolette,L.A.,Kelleher,J.K.,Iliopoulos,O.,and Stephanopoulos,G.(2013a).Cofactor balance by nicotinamide nucleotide transhydrogenase(NNT)coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid(TCA)cycle.J.Biol.Chem.288,12967–12977.
Gameiro,P.A.,Yang,J.,Metelo,A.M.,Pe′rez-Carro,R.,Baker,R.,Wang,Z., Arreola,A.,Rathmell,W.K.,Olumi,A.,Lo′pez-Larrubia,P.,et al.(2013b). In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-de?cient cells to glutamine deprivation.Cell Metab.17, 372–385.
Gao,P.,Tchernyshyov,I.,Chang,T.C.,Lee,Y.S.,Kita,K.,Ochi,T.,Zeller,K.I., De Marzo,A.M.,Van Eyk,J.E.,Mendell,J.T.,and Dang,C.V.(2009).c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism.Nature458,762–765.
Habelhah,H.,Laine,A.,Erdjument-Bromage,H.,Tempst,P.,Gershwin,M.E., Bowtell,D.D.,and Ronai,Z.(2004).Regulation of2-oxoglutarate(alpha-keto-glutarate)dehydrogenase stability by the RING?nger ubiquitin ligase Siah. J.Biol.Chem.279,53782–53788.
Hitosugi,T.,Fan,J.,Chung,T.W.,Lythgoe,K.,Wang,X.,Xie,J.,Ge,Q.,Gu, T.L.,Polakiewicz,R.D.,Roesel,J.L.,et al.(2011).Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase1is important for cancer metabolism.Mol.Cell44,864–877.
Holleran,A.L.,Briscoe,D.A.,Fiskum,G.,and Kelleher,J.K.(1995).Glutamine metabolism in AS-30D hepatoma cells.Evidence for its conversion into lipids via reductive carboxylation.Mol.Cell.Biochem.152,95–101.
Kamphorst,J.J.,Cross,J.R.,Fan,J.,de Stanchina,E.,Mathew,R.,White, E.P.,Thompson,C.B.,and Rabinowitz,J.D.(2013).Hypoxic and Ras-trans-formed cells support growth by scavenging unsaturated fatty acids from https://www.doczj.com/doc/2518821123.html,A110,8882–8887.
Kim,J.W.,Tchernyshyov,I.,Semenza,G.L.,and Dang,C.V.(2006).HIF-1-mediated expression of pyruvate dehydrogenase kinase:a metabolic switch required for cellular adaptation to hypoxia.Cell Metab.3,177–185.
Le,A.,Lane,A.N.,Hamaker,M.,Bose,S.,Gouw,A.,Barbi,J.,Tsukamoto,T., Rojas,C.J.,Slusher,B.S.,Zhang,H.X.,et al.(2012).Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab.15,110–121.
McFate,T.,Mohyeldin,A.,Lu,H.,Thakar,J.,Henriques,J.,Halim,N.D.,Wu, H.,Schell,M.J.,Tsang,T.M.,Teahan,O.,et al.(2008).Pyruvate dehydroge-nase complex activity controls metabolic and malignant phenotype in cancer cells.J.Biol.Chem.283,22700–22708.
Metallo,C.M.,Gameiro,P.A.,Bell,E.L.,Mattaini,K.R.,Yang,J.,Hiller,K., Jewell, C.M.,Johnson,Z.R.,Irvine, D.J.,Guarente,L.,et al.(2012a). Reductive glutamine metabolism by IDH1mediates lipogenesis under hypox-ia.Nature481,380–384.
Metallo,C.M.,Gameiro,P.A.,Bell,E.L.,Mattaini,K.R.,Yang,J.J.,Hiller,K., Jewell, C.M.,Johnson,Z.R.,Irvine, D.J.,Guarente,L.,et al.(2012b). Reductive glutamine metabolism by IDH1mediates lipogenesis under hypox-ia.Nature481,380–384.
Mullen,A.R.,Wheaton,W.W.,Jin,E.S.,Chen,P.H.,Sullivan,L.B.,Cheng,T., Yang,Y.,Linehan,W.M.,Chandel,N.S.,and DeBerardinis,R.J.(2012).
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
Reductive carboxylation supports growth in tumour cells with defective mito-chondria.Nature481,385–388.
Nakayama,K.,Frew,I.J.,Hagensen,M.,Skals,M.,Habelhah,H.,Bhoumik,A., Kadoya,T.,Erdjument-Bromage,H.,Tempst,P.,Frappell,P.B.,et al.(2004). Siah2regulates stability of prolyl-hydroxylases,controls HIF1alpha abun-dance,and modulates physiological responses to hypoxia.Cell117,941–952. Nakayama,K.,Qi,J.,and Ronai,Z.(2009).The ubiquitin ligase Siah2and the hypoxia response.Mol.Cancer Res.7,443–451.
Papandreou,I.,Krishna,C.,Kaper,F.,Cai,D.,Giaccia,A.J.,and Denko,N.C. (2005).Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment.Cancer Res.65,3171–3178.
Papandreou,I.,Cairns,R.A.,Fontana,L.,Lim,A.L.,and Denko,N.C.(2006). HIF-1mediates adaptation to hypoxia by actively downregulating mitochon-drial oxygen consumption.Cell Metab.3,187–197.
Patel,M.S.,and Harris,R.A.(1995).Mammalian alpha-keto acid dehydroge-nase complexes:gene regulation and genetic defects.FASEB J.9,1164–1172.
Patel,M.S.,and Korotchkina,L.G.(2001).Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation:complexity of multiple phos-phorylation sites and kinases.Exp.Mol.Med.33,191–197.
Qi,J.,Nakayama,K.,Gaitonde,S.,Goydos,J.S.,Krajewski,S.,Eroshkin,A., Bar-Sagi,D.,Bowtell,D.,and Ronai,Z.(2008).The ubiquitin ligase Siah2 regulates tumorigenesis and metastasis by HIF-dependent and-independent https://www.doczj.com/doc/2518821123.html,A105,16713–16718.Shah,M.,Stebbins,J.L.,Dewing,A.,Qi,J.,Pellecchia,M.,and Ronai,Z.A. (2009).Inhibition of Siah2ubiquitin ligase by vitamin K3(menadione)attenu-ates hypoxia and MAPK signaling and blocks melanoma tumorigenesis. Pigment Cell Melanoma Res22,799–808.
Son,J.,Lyssiotis,C.A.,Ying,H.,Wang,X.,Hua,S.,Ligorio,M.,Perera,R.M., Ferrone,C.R.,Mullarky,E.,Shyh-Chang,N.,et al.(2013).Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature496,101–105.
Vin?as-Castells,R.,Beltran,M.,Valls,G.,Go′mez,I.,Garc?′a,J.M.,Montserrat-Sent?′s,B.,Baulida,J.,Bonilla,F.,de Herreros,A.G.,and D?′az,V.M.(2010).The hypoxia-controlled FBXL14ubiquitin ligase targets SNAIL1for proteasome degradation.J.Biol.Chem.285,3794–3805.
Wise,D.R.,and Thompson,C.B.(2010).Glutamine addiction:a new therapeu-tic target in cancer.Trends Biochem.Sci.35,427–433.
Wise,D.R.,DeBerardinis,R.J.,Mancuso,A.,Sayed,N.,Zhang,X.Y.,Pfeiffer, H.K.,Nissim,I.,Daikhin,E.,Yudkoff,M.,McMahon,S.B.,and Thompson,C.B. (2008).Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine https://www.doczj.com/doc/2518821123.html,A 105,18782–18787.
Wise,D.R.,Ward,P.S.,Shay,J.E.,Cross,J.R.,Gruber,J.J.,Sachdeva,U.M., Platt,J.M.,DeMatteo,R.G.,Simon,M.C.,and Thompson, C.B.(2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of a-ketoglutarate to citrate to support cell growth and viability.Proc.Natl. https://www.doczj.com/doc/2518821123.html,A108,19611–19616.
Cell Metabolism
Hypoxia Downregulates a KGDH Activity
无论原发性肿瘤还就是继发性肿瘤,一旦生长直径超过1~2 mm,都会有血管生成。这就是由于肿瘤细胞自身可分泌多种生长因子,诱导血管生成。多数恶性肿瘤的血管生成密集且生长迅速。因此,血管生成在肿瘤的发展转移过程中起到重要作用,抑制这一过程将能明显阻止肿瘤组织的发展与扩散转移。于就是体外的血管生成实验就能很好的模拟肿瘤的血管发生过程,并且适合研究药物对这一过程的影响实验。 图一血管生成镜检图 一、实验材料与实验方法 1、实验材料 2、实验方法: 2、1实验流程介绍
图二实验流程图 (提前将Matrigel融化,铺于ibidi血管生成载玻片的下孔中,待胶凝后,将细胞悬液加入血管生成载玻片上孔中,成管后使用显微镜观察。) 2、2耗材结构介绍 图三血管生成载玻片纵截面示意图 (Matrigel铺在下孔,细胞铺在Matrigel上,上孔充满培养基) 2、3数据分析流程介绍
图四实验结果收集与分析流程图 (在特定的时间点采集图片,并且进行图像分析(Wimasis全自动分析)测量小管长度,成环数,细胞覆盖面积与结点。之后在对测量结果进行统计分析以说明实验结果。) 一、实验步骤 1、准备基质胶 1、实验前一天将Matrigel置于冰盒中,放入4。C冰箱,使胶能过夜缓慢融化。(注意:同样要准备一些4。C预冷的枪头用于吸取Matrigel) 2、开始实验前,将Matrigel始终保持放在冰盒中。 3、打开灭菌包装,取出ibidi血管生成载玻片。 4、每孔中加入10μl Matrigel。注意枪头要垂直于内孔的正上方加入Matrigel,防止有Matrigel流经上孔而留下残留胶。 由于Matrigel流动性不强,并且有可能移液枪不准确,有可能打入10μl的胶,却不能填满血管生成载玻片的下孔——这样,必然会影响到实验的成像结果。
肿瘤细胞诱导血管生成模型具体步骤及详细说明 肿瘤血管生成是指肿瘤微环境诱导的在原有血管基础上生成以毛细血管为主的血管系统,并在肿瘤组织内建立血液循环的过程。肿瘤血管生成与肿瘤微环境密切相关,受多种促血管生成因子和(或)血管生成抑制因子的调节。 1、鸡胚准备和接种组织:选择北京白鸡种蛋,洗净,用l:1000苯扎溴铵(新洁尔灭)液浸泡3分钟,置37.8土0.5℃培养箱中孵育。鸡胚发育第7天,蛋壳消毒后,标记制作2cmX3cm左右观察窗。接种组织视研究目的而定,一定要在接种当日取材,充分清洗除去血污和粘液,再剪成1——2mm 组织小块。 2、组织接种:在制备鸡胚观察窗的次日,即鸡胚发育第8天进行组织接种。每个鸡胚可接种5—7个组织小块于接种部位CAM表面,再用透明胶纸封好,继续孵育。 3、接种组织的收获与观察:组织接种后每12小时观察一次,到组织接种第11天(鸡胚发育第18天)收获接种组织,取出鸡胚,置接种组织连同周围的CAM于解剖显微镜下观察,并用10%甲醛固定保存,部分组织可作石蜡切片,用作血管生成研究的免疫组化染色等。 4、CAM的血管生成表现:组织接种24小时后,CAM血管开始向接种组织生长,随培养时间的延长,血管数目及直径均明显增加;第2天,新细小血管直接趋向组织;第3天,新生血管形成以接种组织为中心,10——20条放射状
血管网;尔后,CAM血管继续明显增加。非接种部位与接种部位比较,CAM 血管数目少,大血管与小血管呈脉样均匀分布;而接种部位的CAM血管在接种组织周围弥散样增加,以组织为中心向周围呈放射状分布,在首先与组织发生联系的区域CAM血管更多。第4天,可见新生细小CAM血管生长形成中、粗血管,并在中、粗血管继续分支出细小血管,形成新的血管网。CAM血管管腔结构清晰,可区别动、静脉。 注意事项 该类体外实验研究,有很多人为因素影响,不能代表体内生理反应,因为内皮细胞在生长因子存在的条件下培养了很长一段时间,已被激活。因此,有必要建立一类与体内生理反应有关的血管生成实验方法,尤其是与人类血管生成有关的血管生成实验。 CAM血管生成模型用于血管生成的研究,可取得两方面结果: ①检测评价接种物刺激CAM血管增生的血管生成作用,用于分析研究血管生成促进因子、抑制因子的活性和作用的检测,如肿瘤血管生成的研究等; ②对接种物,如肿瘤组织、自身的血管生成进行研究,用于分析不同组织的血管生成活性和作用,如肿瘤组织有引发新生血管生成的作用。
本实验技术来源于SciMall科学在线 血管生成(Angiogenesis)信号通路图 血管生成是通过人体中存在的诸多互补和复杂的信号途径调节的.血管内皮生长因子(VEGF)-血管内皮生长因子受体(VEGFR)、血管生成素(Ang)-Tie2轴和Dll4-Notch这3个复杂的、相辅相成的信号传导通路可在调节血管生成中发挥重要作用. VEGF与内皮细胞上的两种受体KDR和Flt-1高亲和力结合后,直接刺激血管内皮细胞增殖,并诱导其迁移和形成官腔样结构;同时还可增加微血管通透性,引起血浆蛋白(主要是纤维蛋白原)外渗,并通过诱导间质产生而促进体内新生血管生成。VEGF在血管发生和形成过程中起着中枢性的调控作用,是关键的血管形成刺激因子。碱性成纤维细胞生长因子(bFGF)。TNF-α是一类具有血管活性的细胞因子,可诱导异位子宫内膜炎性细胞因子MCP-1,IL-6和IL-8等的释放,促进异位内膜及基质细胞增殖及炎性细胞浸润,新生血管形成,组织粘连,从而形成异位病灶。 (来源:Scimall科学在线) 本信号转导涉及的信号分子主要包括: HIF1α,PHDs,HIF1β,PI3K,Akt,mTOR,S6K,4E-BP1,eIF4E1,elF4E1,Ras,MEK1,MEK2,Erk1,Erk2,MNK,CBP,P300,TCEB1,TCEB2,Rbx1,Cul2,VHL,MMP,Cox2,PAI-1,VEGF,PDGFR-β,VEGFR2,Tie2,FGFR,IGFR,TGFα-R,SLIT,ROBO,Src,FAK,p38,MAPK,Smad2,Smad3,PLCγ,NOS等。 点击图中信号分子,自动寻找相关试剂
肿瘤血管生成和抗血管生成治疗癌症的机制 主要研究者 Yihai Cao MTC 卡罗林斯卡学院 总目标: 我们研究项目的目标是研究肿瘤血管生成的复杂机制。通过了解病理性肿瘤血管生成的机制,我们希望能攻克血管来明确新的治疗靶点,优化当前治疗癌症的抗血管生成疗法,确定可靠的生物标记来指导这些新药的临床意义。因此,我们的研究目的本质上是翻译性质的并且与临床相关,如果成功,这个项目将造福数百万癌症患者。 具体目标: 1.研究在肿瘤生长与转移过程中血管和淋巴管生成的机制 2.研究抗血管生成药物的耐药机制和优化抗血管生成疗法 3.确定脱靶肿瘤为抗血管生成治疗的潜在有利部位 4.研究肿瘤血管和促进肿瘤生长转移的间质组织之间的作用 背景和理由 血管生成,就是新血管从现有的血管生长的过程,它对胚胎发育、女性生殖、伤口愈合、肿瘤生长和转移、慢性炎症、肥胖、糖尿病并发症和眼科疾病都至关重要[1]。1971年,Judah Folkman提出一个新概念,将抑制肿瘤血管生成作为治疗癌症的新策略[2]。经过40年该领域的研究后,临床前和临床数据提供了可靠的证据,证明抗血管生成疗法是治疗恶性和非恶性肿瘤有效合理的方法。如今,一些基于抗血管生成原理的靶向药物主要包括贝伐单抗,舒尼替尼,和索拉非尼,它们已结合传统疗法如化疗,成为人类肿瘤一线治疗手段的关键部分[3] 。此外,抗血管生成药物已被成功用于眼科疾病的治疗,比如老年性黄斑变性[ 4 ]。在癌症领域,尽管抗血管生成药物结合化疗的联合疗法能显著的提高各类癌症患者的生存率,但是抗血管生成疗法治疗大多数类型的癌症包括直肠癌、肺癌和乳腺癌的临床效果仍然不理想,只有少数癌症患者(大约30%)受益[5]。大量临床相
无论原发性肿瘤还是继发性肿瘤,一旦生长直径超过1~2 mm,都会有血管生成。这是由于肿瘤细胞自身可分泌多种生长因子,诱导血管生成。多数恶性肿瘤的血管生成密集且生长迅速。因此,血管生成在肿瘤的发展转移过程中起到重要作用,抑制这一过程将能明显阻止肿瘤组织的发展和扩散转移。于是体外的血管生成实验就能很好的模拟肿瘤的血管发生过程,并且适合研究药物对这一过程的影响实验。 图一血管生成镜检图
一.实验材料和实验方法 1.实验材料 2.实验方法: 2.1实验流程介绍 图二实验流程图 (提前将Matrigel融化,铺于ibidi血管生成载玻片的下孔中,待胶凝后,将细胞悬液加入血管生成载玻片上孔中,成管后使用显微镜观察。)
2.2耗材结构介绍 图三血管生成载玻片纵截面示意图 (Matrigel铺在下孔,细胞铺在Matrigel上,上孔充满培养基)2.3数据分析流程介绍 图四实验结果收集和分析流程图 (在特定的时间点采集图片,并且进行图像分析(Wimasis全自
动分析)测量小管长度,成环数,细胞覆盖面积和结点。之后在对测量结果进行统计分析以说明实验结果。) 一.实验步骤 1、准备基质胶 1.实验前一天将Matrigel置于冰盒中,放入4。C冰箱,使胶能过夜缓慢融化。(注意:同样要准备一些4。C预冷的枪头用于吸取Matrigel) 2.开始实验前,将Matrigel始终保持放在冰盒中。 3.打开灭菌包装,取出ibidi血管生成载玻片。 4.每孔中加入10μl Matrigel。注意枪头要垂直于内孔的正上方加入Matrigel,防止有Matrigel流经上孔而留下残留胶。 由于Matrigel流动性不强,并且有可能移液枪不准确,有可能打入10μl的胶,却不能填满血管生成载玻片的下孔——这样,必然会
血管生成实验模型研究进展 吴家明1 ,陆 茵 1,2 ,郜 明1,张伟伟 1 (1.南京中医药大学中医药研究院,江苏南京 210029;2.江苏省方剂研究重点实验室,江苏南京 210029) 收稿日期:2007-09-21,修回日期:2007-11-01 基金项目:国家自然科学基金资助项目(No 30371727,30772766);江 苏省自然科学基金资助项目(No BK2003113) 作者简介:吴家明(1980-),男,硕士生,研究方向:肿瘤血管生成与 抗肿瘤转移研究,E 2mail:nj w ujia m ing@https://www.doczj.com/doc/2518821123.html,; 陆 茵(1963-),女,教授,博士生导师,研究方向:肿瘤血管生成与抗肿瘤转移研究,通讯作者,Tel:0252 86798154,E 2mail:luyingreen@https://www.doczj.com/doc/2518821123.html, 中国图书分类号:R 205;R 332;R 3632332;R 36413 文献标识码:A 文章编号:1001-1978(2008)01-0011-04摘要:抗血管生成已经成为治疗肿瘤转移、糖尿病视网膜病变、风湿性关节炎等疾病的重要策略之一。血管生成模型作为一种研究工具在探讨血管形成机制、发现促进或抑制血管生成药物等研究中发挥十分积极的作用。如何寻找适合的血管生成模型是研究人员在研究中常遇到的问题。该文就主要常用模型做较全面的介绍,并对其优缺点进行评价。关键词:血管生成;模型 血管生成(angi ogenesis )是指在原有的毛细血管和(或)微静脉基础上通过血管内皮细胞的迁移和增殖,从已存在的血管处以芽生或非芽生(套迭)形式形成新的、以毛细血管为主的血管系统过程 [1] 。血管生成是许多促进或抑制血管 生成的分子参与调节的一个平衡过程[2]。血管生成过多与肿瘤、糖尿病性视网膜病变等疾病有关[3],抑制血管生成已经成为治疗这些疾病的重要策略。因此寻找血管生成抑制剂成为研究热点。血管生成研究需借助血管生成模型进行,血管形成的许多过程都可以在血管生成模型中模拟完成,包括内皮细胞增殖、迁移、毛细血管网状结构的形成等。本文就常用体内、体外及整体模型进行综述。 1 体外模型 体外模型主要分为细胞水平及组织学水平两类,常用的体外模型有以下4种。 1.1 内皮细胞增殖实验(cell proli fera ti on a ss ay) 内皮细 胞活化增殖是血管生成的起始阶段。目前主要有两种测定细胞增殖的方法即净细胞数测定和细胞周期分析。 1.1.1 净细胞数测定 M TT 法(四甲基偶氮唑比色法)是 测定活细胞数的化学定量方法,操作简便,价格低廉。但它不适合测定对细胞代谢有影响的药物,因为这类药物能影响 MTT 测定的活细胞数。另外通过胸腺嘧啶核苷参入法测定DNA 合成来测定细胞增殖能力。抑制细胞增殖可能是由于 药物的抑制作用,也可能是药物的毒性作用引起。因此,这种方法常需要结合细胞凋亡实验的数据才可以更好地评价药物对细胞增殖的影响。 1.1.2 细胞周期分析 近年来有报道通过细胞周期分析来 评价药物对细胞增殖的影响,细胞短时间暴露到溴脱氧尿苷 (B rd U )中可以促使B rdU 参入细胞DNA 中。碘化丙啶(P I ) 染色后测定细胞的总DNA 量,再用荧光激活细胞分析仪测定细胞中B rd U 和P I 的量可得出细胞周期信息[4]。但实验用的内皮细胞处于增殖状态,与体内静止状态的内皮细胞是不同的,实验结果与体内还是存在一定差异。 1.2 内皮细胞迁移实验(cell m i gra ti on a ss ay) 常用迁移 模型有两种:①细胞损伤模型:在培养血管内皮细胞的培养皿上用刀片划出#形区,经P BS 洗涤后再用含011%明胶的 ME M 培养20h,细胞用甲醇固定,Gie m sa 染色。在光镜下计 数从损伤边缘迁移出的细胞数,需要注意的是划出的损伤区域一定要精确。②Boyden 室模型,Boyden 室由两层组成,两层之间为胶原包被的多孔的多聚碳酸盐滤膜;血管内皮细胞放于上层,同时加入待测药物,共同培养6h 后,除去上层的细胞,下层细胞用甲醇固定,HE 染色,光镜下计算下层的血管内皮细胞数。龙淼云等[5]采用本模型研究证明了血管生成抑制因子arresten 对huvec 迁移有抑制作用。Boyden 室模型优点在于它对药物浓度梯度差异很敏感。但对实验技术要求较高且计数方法不同可能会造成统计结果误差较大。因此有必要建立一个严格的计数标准以减少因方法不同产生的误差。 1.3 小管形成实验(tube for ma ti on a ss ay) 小管形成实验 能模拟人体内毛细血管生成的过程,包括内皮细胞出芽增殖和毛细血管网结构形成等步骤,接近人体内血管生成的实际过程。内皮细胞在基质胶、纤维蛋白胶、胶原等基质上培养时能形成网状结构。用电子显微镜来分析小管间的紧密连接,从而定量血管生成情况[6]。需要指出的是似乎所有的内皮细胞都能在细胞外基质上形成管状结构,有些非内皮细胞也能在基质胶上形成管腔结构[7]。实验一般采用24孔培养板,但它底面积大,Matrigel 用量多,计数区域大只能随机选择几个典型区域且数据分析耗时长。Sanz 等[8]采用384孔和1536孔培养板培养可节约胶的用量,借助计算机可以计算整个孔的小管及小管之间的连接数及小管的长度和面积。改进方法后减少了原来分析困难及重现性差的缺点。 1.4 大鼠动脉环实验(ra t aorti c r i n g a ss ay) 1990年N ic 2osia 等首次将此模型应用于血管生成研究中。取下大鼠主 动脉后,剪成1mm 宽的血管环,再用纤维蛋白胶或胶原蛋白胶包埋,然后以无血清的MC DB131培养液培养。培养过程中每天计算主动脉环产生的新生微血管数并进行定量分析。用不同胶培养的大鼠主动脉环新生血管的生长曲线不同。胶原包埋的主动脉环,培养1wk 时血管数达到顶峰,第 2wk 开始萎缩。用纤维蛋白胶包埋时能使血管数顶峰期维 ? 11?中国药理学通报 Chinese Phar m acological B ulletin 2008Jan;24(1):11~4
血管生成(angiogenesis)是从已有血管发芽生成新血管的过程。这一过程与血管内皮细胞迁移和增殖相关。胚胎发育过程伴随着血管生成(angiogenesis)。但除了伤口愈合、女性经期及一些病理过程(如癌症)外,成人很少出现血管生成(angiogenesis)。Angiogenesis is the sprouting process from the existing blood vessels to new ones, which is associated with the migration and proliferation of endothelial cells, and always accompanied with embryonic development. However, in addition to the wound healing, the female menstrual period and some pathological processes (e.g., cancer), angiogenesis rarely appears in adults. 血管生成对恶性实体肿瘤的生长、转移乃至预后都有着极其重要的意义。目前国内在研究肿瘤中新生血管时,常用的血管内皮细胞标记物包括CD34 、CD31、CD105、CD146和vWF 等。义翘神州现已制备了一批高灵敏度、高特异性的血管生成相关抗体供科研人员选择:Angiogenesis has extremely important significance in the growth, metastasis and prognosis of malignant solid tumors. At present, in the study of new blood vessels in tumors, researchers commonly use vascular endothelial cell markers, including CD34, CD31, CD105 and CD146 and vWF, etc. Sino Biological Inc. has prepared a batch of antibodies process of high sensitivity and high specificity related to angiogenesis for researchers to choose:
人血管生成素2(ANG-2)酶联免疫分析 试剂盒使用说明书 本试剂盒仅供研究使用 产品编号:CSB-E04500h 检测范围:0.78 ng/ml - 50 ng/ml 最低检测限:0.195 ng/ml 特异性:本试剂盒可同时检测天然或重组的人ANG-2,且与其他相关蛋白无交叉反应。 有效期:6个月 预期应用:ELISA法定量测定人血清、血浆、心包积液、细胞培养上清或其它相关生物液体中ANG-2含量。 说明 1.试剂盒保存:-20℃(较长时间不用时);2-8℃(频繁使用时)。 2.浓洗涤液低温保存会有盐析出,稀释时可在水浴中加温助溶。 3.中、英文说明书可能会有不一致之处,请以英文说明书为准。 4.刚开启的酶联板孔中可能会含有少许水样物质,此为正常现象,不会对实验结果造成任何影响。 概述 血管生成素(angiopoietin,Ang)是近年来发现的与血管新生密切相关的一个新家族,包括Ang-1、Ang-2、Ang-3、Ang-4,参与胚胎血管的发生及成年机体生理、病理性血管形成,尤其Ang-2,在肿瘤组织中的表达明显高于周围正常组织。Ang-2是特异性血管生成刺激因子,也是恶性肿瘤早期的标志分子。Ang-2与Ang-1 60%同源,相互间易形成异二聚体结构,自身也常以同源二聚体及多聚体结构存在。Ang-2的生物学功能与Ang-1完全相反。Ang-1与血管内皮细胞的Tie-2受体结合,促进血管成熟及稳定,Ang-2与Tie-2结合,不使Tie-2磷酸化而激活Tie-2,反而拮抗Ang-1的生物活性,破坏血管完整性,影响内皮细胞之间及其与支持细胞之间的连接。在肿瘤发生的早期,Ang-2参与破坏瘤体周边原有的正常血管,而促进肿瘤新生血管的生成,在瘤体周边形成所谓的血管共择(co-option)区。当肿瘤形成以后,Ang-2与血管内皮生长因子(VEGF)有协同作用,共同促进肿瘤血管生成,并阻碍血管的完整性,使得肿瘤新生血管能在各种因子的刺激下不断增生。 实验原理 用纯化的抗体包被微孔板,制成固相载体,往包被抗ANG-2抗体的微孔中依次加入标本或标准品、生物素化的抗ANG-2抗体、HRP标记的亲和素,经过彻底洗涤后用底物TMB 显色。TMB在过氧化物酶的催化下转化成蓝色,并在酸的作用下转化成最终的黄色。颜色的深浅和样品中的ANG-2呈正相关。用酶标仪在450nm波长下测定吸光度(OD值),计算样品浓度。 试剂盒组成及试剂配制 1.酶联板(Assay plate ):一块(96孔)。 2.标准品(Standard):2瓶(冻干品)。 3.样品稀释液(Sample Diluent):1×20ml/瓶。 4.生物素标记抗体稀释液(Biotin-antibody Diluent):1×10ml/瓶。 5.辣根过氧化物酶标记亲和素稀释液(HRP-avidin Diluent):1×10ml/瓶。 6.生物素标记抗体(Biotin-antibody):1×120μl/瓶(1:100) 7.辣根过氧化物酶标记亲和素(HRP-avidin):1×120μl/瓶(1:100) 8.底物溶液(TMB Substrate):1×10ml/瓶。
内皮祖细胞与新生血管生成 发表时间:2016-05-12T16:44:02.303Z 来源:《系统医学》2016年第4期作者:李倩[导读] 包括EPC的动员、迁移、粘附和在新血管形成中的作用,以便更好的理解EPC在血管形成中的作用机制,为血管性疾病治疗提供新的治疗思路。 李倩 南宁95178部队医院 530050 【摘要】内皮祖细胞(endothelial progenitor cells,EPCs)是一类可以从骨髓动员入血,聚集至血管受损部位以参与血管形成的细胞群。由于缺乏独特的表面标记和分离方法,EPCs包含骨髓和内皮来源的一大类异质性细胞群。研究表明EPCs在出生后血管形成和血管内稳态中发挥关键作用,为血管性疾病提供新的治疗思路。然而,EPCs参与新血管形成的机制仍不完全清楚。我们回顾了EPCs参与新生血管形成的生物学过程,包括EPC的动员、迁移、粘附和在新血管形成中的作用,以便更好的理解EPC在血管形成中的作用机制,为血管性疾病治疗提供新的治疗思路。 【中图分类号】S891+.1【文献标识码】A【文章编号】2096-0867(2016)-04-356-02 1前言 血管损伤性疾病具有高发病率和死亡率的特点。受损内皮的有效修复和新生血管生成是这类疾病的治疗关键。当内皮完整性破坏,内皮细胞(endothelial cells,ECs)从邻近血管中增殖和迁移,促使受损血管内皮修复。以往认为这是新血管形成的唯一方式。然而,这种血管形成的传统观念目前正面临挑战。Asahara[1]等第一个描述了骨髓来源的细胞群体有助于新血管形成,并命名这一细胞群为内皮祖细胞(endothelial progenitor cells,EPCs)。相对于血管形成,来源于骨髓并增殖和分化成熟的EPCs形成的新血管被定义为血管发生。因此,EPCs被认为是治疗血管性疾病的新方法。然而,由于缺少表面标志,这些祖细胞代表了包含骨髓和内皮来源的异源性细胞群。最近针对EPCs的研究主要包含三种细胞类型,分别为克隆形成单元细胞(colony-forming unit-Hill,CFU-Hill)、循环的血管源细胞(circulating angiogenic cells,CACs)和内皮克隆形成细胞(endothelial colony-forming cells,ECFCs)。CFU-Hill细胞和CACs通常指早期成熟的EPCs。相对的,ECFCs被定义为晚期成熟EPCs。早期成熟的EPCs和晚期成熟的EPCs均有助于新血管形成,但这类骨髓源性EPC诱导血管生成的机制仍不清楚,需要进一步研究。 2内皮祖细胞的动员 有研究显示外周循环中的EPCs在生理状态下水平很低,大部分EPCs在骨髓微环境中由结合素粘结在骨髓基质细胞中。EPCs由骨髓动员至外周循环是其参与血管形成的关键步骤,但其动员的关键机制仍不完全清楚。有研究显示EPC在各种细胞因子的作用下可由干细胞转变为功能细胞。 血管内皮生长因子(vascular endothelial growth factor,VEGF)是一种功能复杂的细胞因子,在血管形成过程中具有重要调节作用。VEGF可通过动员骨髓中的EPCs而促进新生血管生成。在人类受试者中转染VEGF基因可上调循环EPCs水平。在烧伤部位,血浆中VEGF水平升高可动员VEGFR2+的EPCs进入循环。另外,VEGF具有上调粒细胞集落刺激因子(granulocyte colony-stimulating factor,G-CSF)的作用。G-CSF可诱导祖细胞从骨髓释放。VEGF和VEGFR相互作用可使骨髓NOS活化,从而产生一氧化氮(nitric oxide,NO),进而激活MMP-9。活化的MMP-9有利于释放可溶性Kit配体(sKitL),增强VEGFR2+的EPCs动员,刺激这些细胞从骨髓向外周血循环迁移。 炎症趋化因子CXC作为关键调节因子在动员骨髓干细胞或祖细胞进入外周循环中发挥作用。基质细胞趋化因子-1(Stromal cell-derived factor-1,SDF-1)是EPC动员最具特征的活性因子,也是EPC粘附和迁移的潜在细胞因子,它在血管生成中具有促进作用,而某些刺激如炎症和乏氧可促使SDF-1表达上调。局部缺血的环境促进SDF-1释放。在缺血发生的第一小时内,局部缺血的内皮细胞中SDF-1 mRNA生成增加。血浆中SDF-1刺激CXCR4+骨髓细胞动员,诱导定向造血干细胞和EPCs。CXCR4是SDF-1的一个受体,高表达于定向造血干细胞前体细胞和内皮祖细胞表面。SDF-1和CXCR4相互作用不仅促使EPCs从骨髓动员,而且刺激干细胞向缺血部位募集和固位。SDF-1可由血小板释放,诱导EPCs趋化。EPCs本身也以旁分泌的形式释放SDF-1。血管损伤的第一个反应是血小板向暴露的内皮下膜粘附,借此为干细胞向损伤部位动员和归巢提供靶向信号。在局部缺血的动物模型中,SDF-1α基因转录促进EPCs向外周血动员,增强新生血管生成。但在无损伤时SDF-1是否可动员EPCs仍不清楚。此外,VEGF或NOS信号障碍可阻断SDF-1诱导功能,提示VEGF/eNOS产生涉及新生血管生成中的SDF-1上调。研究显示由骨髓分化的干细胞或祖细胞动员需要骨髓NOS活化,活化的NOS可通过NO-MMP-9可溶性级联信号通路促进骨髓释放祖细胞。SDF-1也可以通过上调VEGF水平增强EPCs的动员,VEGF促进EPCs从骨髓释放入外周循环。白细胞介素-8(Interleukin-8,IL-8)是一种炎症因子,最初仅被认为是白细胞趋化因子。然而,最近研究表明IL-8是EPCs动员至外周血的调节因子,在动物模型中这个功能可由G-SCF协同。 一氧化氮(Nitric oxide,NO)被标记为是内皮趋化释放因子,是调节血管生理学特性的关键因子,包括血管舒张、血管通透性和抗血栓形成特性(35)。NO也可通过调节血小板和内皮的相互作用而保持血管完整性和血流。目前研究表明NO是EPC从骨髓动员进入循环,进而促进缺血四肢再灌注和创伤修复的关键决定因子。NO的产生依赖于一氧化氮合酶催化下的L-精氨酸向L-瓜氨酸转化。一氧化氮合酶具有四种亚型:神经元性一氧化氮合酶(nNOS)、可诱导的一氧化氮合酶(iNOS)、内皮一氧化氮合酶(eNOS)和线粒体一氧化氮合酶(mtNOS)。在这些亚型中,eNOS选择性表达于血管内皮细胞和周围间质干细胞中,在血管生物学中发挥关键作用。NO也可由各种EPC 亚型释放。eNOS在调节EPCs动员和功能上发挥关键作用。许多糖尿病患者受迟发性或不愈性肢端症状或糖尿病足困扰,可能与 EPCs在高血糖环境下功能受损相关。高血糖使eNOS功能受损,而eNOS功能损伤可致使EPC向周围循环动员受抑。磷酸化eNOS水平,而不是eNOS蛋白水平是阻碍EPCs动员的关键。出生后包括EPCs在内的干细胞存在于骨髓中,由结合素粘附于基质细胞,在细胞因子和其他血管源性因子的作用下释放至外周循环。EPC从骨髓动员的机制仍不完全清楚。NO-MMP-9-sKitL-ckit级联系统可能在这一过程中发挥关键作用。骨髓中eNOS可被这一过程中的多种细胞因子刺激从而产生NO。NO可刺激MMP-9,致使sKitL从基质细胞膜结合配体上释放。EPCs表达的c-Kit有助于维持骨髓内EPCs稳定。c-Kit也是sKitL的配体,结合sKitL后可从骨髓释放,导致c-Kit+的EPCs进入循环。当然,由这些因子诱导的EPCs动员在新血管形成中的机制仍需进一步研究。
血管生成素:抗血管生成药物的新靶点 生意社11月7日讯阿瓦斯丁是目前市场上抗血管生成生物药物的典范,该人源化单克隆抗体靶向作用于血管内皮生长因子。尽管美国食品药品管理局最近撤销了阿瓦斯汀治疗乳腺癌的适应证,但此药在世界各地仍广泛用于治疗大肠癌、脑癌、肺癌和肾细胞癌。而且,抗血管生成药物也可用于其他疾病的适应证,例如,雷珠单抗是 1
一种来自于贝伐单抗的单克隆抗体片段,已被批准用于治疗湿性年龄相关性黄斑变性。拜耳和Regeneron公司联合开发的湿性AMD药物Eylea,也是一种VEGF受体1和2的胞外结构域融合人IgG1的Fc部分组成的重组融合蛋白。 随着对抗血管生成药物研究的不断深入,科学家发现,血管生成素有望成为抗血管生成药物的新靶点。 血管生成素途径受到关注 开发更安全和更有效的抗血管生成药物一直是制 2
药行业努力的方向。血管生成素途径近年来受到越来越多的关注,有望改变VEGF通路已作为重要靶点的现状。对几种血管生成素家族成员的研究已经确定,血管生成素1和血管生成素2与其酪氨酸蛋白激酶受体TIE-2已成为研究热点。血管生成素-TIE通路被认为是一个特别有吸引力的治疗干预系统,因为其重要性不仅表现在对血管生成和血管内环境稳定上,同时也是血管生成和炎症通路的重要环节。 3
ANG-1和ANG-2是TIE-2受体酪氨酸激酶的功能性配体。ANG-1表达于许多类型的细胞,如周皮细胞、平滑肌细胞和成纤维细胞,作为TIE-2激动型配体。ANG-1介导的TIE-2激活可导致血管内皮细胞通透性和血管发育稳定性下降。另外,ANG-2由血管内皮细胞表达,可阻断ANG-1介导的TIE-2激活,作为TIE-2的拮抗剂发挥作用。ANG-2上调与不同类型的癌症转移和恶化相关。 而且,血管生成疾病都发现了ANG-2上调的现象。 4
1 血管生成相关机制 1.1 新血管的形成与结构 一个细胞需要生存,则必须围绕血管,即靠近血管约100~200μm。这个距离是氧气弥散距离的极限。如果没有血管供应,单个肿瘤的极限体积大小介于0.2~3mm之间,依肿瘤细胞来源不同而大小有所差异。处于这个极限大小下,肿瘤细胞的增生与死亡达到平衡。如果肿瘤想扩大自身体积,则必需求助于新生血管。 血管生成过程实际上就是沿着血管排列的血管内皮细胞增殖过程。血管内皮细胞是人体内寿命最长的细胞之一。正常情况下,它们每七年才分裂增殖一次。如果照这种速度产生新生血管,那么血管的更新过程则非常缓慢,所以该过程一定得加快。血管生成过程是受严密调控的过程,该过程处于血管生成激活物或促进因子和其它必需过程(促进血管内皮细胞增殖)与血管生成抑制因子(阻止血管生成过程)的共同调控之下。该调控也被称为―血管生成开关‖(angiogenic switch)(图1)。 通常情况下,抑制因子的作用都要强于促进因子,也就是说―开关‖常常处于关闭状态。如果出现了足够多的促进因子,则―开关‖被打开,开始形成新血管。 血管生成过程中最重要的一环就是血管内皮细胞的增殖和迁移。肿瘤细胞缺氧或受到其它一些信号调节时会合成、分泌血管生成促进因子。血管内皮细胞迁移与分裂增殖机制见图2。
肿瘤细胞分泌的内皮细胞生长因子等物质与血管内皮细胞上的受体分子结合,刺激其释放蛋白水解酶(proteolytic enzyme)。该蛋白水解酶可以降解血管周围的基质。这样,为血管内皮细胞的迁移和进一步分裂做好了准备。血管内皮细胞经过不断的分裂增殖以及向前迁移,逐渐形成管状结构,最终形成新生血管。 由于肿瘤组织中生成新生血管的过程没有受到严密调控,因而肿瘤组织中的新生血管与正常组织中的新生血管在结构上差异明显。肿瘤组织中的新生血管非常不规则,有很多分支和旁路。血管不完全由血管内皮细胞构成,有些地方的管壁竟然由肿瘤细胞覆盖而成。血管的通透性非常高,因为没有正常的基底膜围绕在血管周围,血管内皮细胞间的连接非常少。另外,血管周围也没有正常运作的控制血压的平滑肌细胞。 1.2血管生成因子(angiogenic factor) 有数十个不同的分子参与血管生成过程的调控。肿瘤细胞、血管内皮细胞、基质细胞、血细胞或细胞外基质都可以合成血管生成促进因子和抑制因子。在所有这些调控因子中,有两大家族是最重要的血管生成促进因子,那就是血管内皮细胞生长因子(Vascular Endothelial Growth Factor, VEGF,图3)和成纤维细胞生长因子(Fibroblast Growth Factor, FGF)。 VEGF家族拥有6名成员(图4),而FGF家族则包括酸性和碱性成纤维细胞生长因子。
促血管生成素(Ang) DOI:10.3760/cma.j.issn.1671-0282.2015.02.003 作者单位:100029 北京,卫计委中日友好医院急诊科 脓毒症(sepsis)是指由感染引起的全身炎症反应综合征,病情凶险,病死率高。目前,脓毒症的全球病死率高达30%~70%,脓毒症的高发病率与病死率逐渐成为威胁人民健康的全球性问题之一。最新美国国家卫生研究院资料显示,在美国ICU?澳甏笤加?70万至90万脓毒症患者入院,其中大约有20万患者死于脓毒症。目前脓毒症确切的发病机制尚未完全阐明,有大量的研究表明,细菌内毒素、失控的炎症反应、凝血功能紊乱、免疫功能紊乱、细胞凋亡、血管内皮细胞功能障碍、高代谢状态、基因多态性等因素与其发病机制均密切相关。促血管生成素(angiopoietin,Ang)是一个与新生血管生成密切相关的家族,近期的研究显示细菌内毒素可以调控Ang系统,影响脓毒症的血管内皮细胞功能,破坏血管内皮完整性,导致毛细血管通透性增加,最终引起多器官功能障碍和衰竭<sup>[1]</sup>。因此,Ang 系统在脓毒症的发生和发展过程中发挥了重要作用。 1Ang的编码基因、结构和表达 目前,已知Ang家族包括Ang-1 、Ang-2、Ang-3 和
Ang-4四个成员,主要在胚胎发育期表达,促进心血管系统的发育成熟,其中,Ang-1和Ang-2与血管生成的关系较为密切。成年以后,除在女性生殖系统(卵巢、子宫)表达水平较高外,在其他组织呈低水平表达。Ang各成员的蛋白结构基本相同,都由3部分组成:N-端疏水性分泌信号肽、α-螺旋的卷曲结构域及C-纤维蛋白原样结构域。卷曲结构域主要促进蛋白分子的多聚化;C-端纤维蛋白原样结构域是Ang中最具保守性的一部分,其中包含受体结合部分,决定某种Ang是否起激动作用。 1.1Ang-1,Ang-2的编码基因、结构和表达 人的Ang-1基因位于染色体8q22.3-23,包括9个外显子和8个内含子,其相对分子质量约为60 000~75 000。Ang-1是由498个氨基酸组成的同源六聚体,其卷曲结构域大约180个氨基酸,呈四面体结构,与肌球蛋白有弱的同源性;其C-纤维蛋白原样结构域大约有200个氨基酸,与纤维蛋白原、tenascin、hfrep、ficolin 及果蝇的SCABROUS有相似性,与受体的结合及磷酸化有关。胚胎心血管发育早期Ang-1主要在包绕心内膜的心肌上表达,后期则在血管周细胞上表达。其表达受缺氧、表皮生长因子(epidermal growth factor,EGF)、转化生长因子-β(transforming growth factor-β,TGF-β)等的调控。 Ang-2基因位于染色体8p21,编码496个氨基酸,与
肿瘤发生后生长过程大致可分为:第一时期为无血管期或称为血管前期,该期肿瘤细胞的营养供给及代谢产物的排泄主要靠简单的物理弥散作用,临床表现为原位癌或微转移灶,此期持续时间长,肿瘤生长受限,体积停滞在1~2mm3耐大小。第二时期为血管期也称血管浸润性生长期,当实体肿瘤直径达到或超过1一2mm时,肿瘤已不能单纯依靠弥散作用获取氧气和营养物质,新生血管的不断形成及其营养支持作用是肿瘤生长的必要条件,有实验证明肿瘤的生长依赖于血管生成:在鸡胚绒毛尿膜囊(CAM)上生长的肿瘤,体积大于1mm3后,如果3天内无血管长入,肿瘤将发生坏死和溶解;如果血管长入,肿瘤体积快速增长。鼠皮下移植瘤的实验亦证实:肿瘤在无血管时呈线性生长,血管生成时呈指数生长。第三时期为转移期,由于肿瘤诱发的新生血管不同于正常血管,其结构与功能异常,如扭曲、扩张、动静脉短路及分又,不能适当吻合,使血流积聚于盲端,易引起局部坏死;另一方面肿瘤组织内微血管基质不完善,如血管壁缺乏平滑肌支持,壁很薄,易通透,使瘤细胞产生的各种因子和蛋白酶类渗透到血管内,同时瘤细胞易进入血管顺血流转移到远隔部位,因此,新生的微血管是肿瘤浸润、转移的第一站,肿瘤微血管数量越多,肿瘤细胞进入血液循环的机会就越大,转移概率也越大。瘤细胞进入血循环后,自身形成同聚物或与白细胞、血小板形成异聚合物,通常这些聚合物被称之为癌栓,癌栓留驻在远端血流缓慢的毛细血管处,进而黏附血管内皮并诱导内皮崩解,癌细胞穿出微血管后,与基底膜接触通过特殊膜受体结合基质蛋白,并通过与最初侵袭原发组织同样的机制,完成在远端组织的转移。转移灶形成过程也会出现第一、二期的发展阶段,并在一定条件下发生再度转移。许多研究证实,实体瘤只有具备了血管生成表型后才能恶性生长、扩散及转移,新生血管通过灌注形式为肿瘤细胞提供所必需的营养,也是肿瘤细胞代谢产物排泄的有效渠道,同时新生血管为肿瘤细胞向远处转 主要包括脱离、转运和生长三个主要环节,基本过程大致可分为以下几个阶段:首先是原发瘤增殖、肿瘤新生血管生长;肿瘤细胞表面再生黏附分子降低,细胞之间黏附性减小,使肿瘤细胞从原发部位脱落,粘连侵袭基底膜并在周围间质中浸润生长;肿瘤细胞对周围组织、血管、淋巴管的压迫和浸润,与局部毛细血管或毛细淋巴管内皮细胞密切接触并穿透其管壁,或突入腔道;肿瘤细胞在血管淋巴管内继续存活并被转运,同时启动血小板聚集,形成小瘤栓,到达原隔靶组织并滞留于靶器官的微小血管中;肿瘤细胞与血管或淋巴管内皮细胞和基底膜粘连,穿透毛细血管或毛细淋巴管壁,并产生蛋白溶解酶,破坏组织结构;肿瘤细胞生长、繁殖及转移灶的形成;肿瘤间质内新血管形成及转移灶的快速生长。由此可见,肿瘤转移是一具有内在联系的复杂的。 , 包括出芽式血管生成(sprouting angiogenesis)和套入式血管生成。出芽血管生成一般包括五个步骤,即肿瘤诱导释放多种血管生成因子;血管内皮细胞因血管生成因子的作用而激活,血管扩张、渗透性增高,在血管周围形成富含纤维的临时基质;内皮细胞和肿瘤细胞释放蛋白水解酶降解血管基底膜和细胞外基质;内皮细胞增殖、迁移并形成血管芽;血管分化成型和新基质的再充填,形成新生血管网系统。特点是局部血管舒张、血管通透性升高和内皮细胞的增殖,在体内启动出芽式血管生成是个较为迟缓的过程。套入式血管生成:不是以大范围的内皮细胞增殖、基底膜降解以及侵袭周围组织为基础, 而是通过在已有的血管管腔内形成大量的跨血管组织微柱使毛细血管在自身基础上扩张, ,而通过肿瘤细胞自身变形和与细胞外基质作用, 模拟血管壁结构形成可输送血液的管道系统, 重塑肿瘤 微循环, 胞相间排列在肿瘤血管壁上, 共同围成肿瘤的血管腔;形成的机制大致有以下三种:①由于内皮细胞的脱落, 肿瘤细胞就会暴露于管腔; ②一些内皮细胞在肿瘤的演进过程中丧失了免疫
易必迪ibidi μ-Slide Angiogenesis 体外血管生成实验 产品特点: ibidi血管生成系列产品,包括μ-Slide Angiogenesis和μ-Plate Angiogenesis 96 well,是ibidi 公司根据血管生成实验的要求的改进型设计。 它较之传统的血管生成实验,有以下优点: 1、节省基质凝胶--Gel matrix(自行准备),比传统96孔板实验节省9/10(传统实验用量100ul,ibidi血管生成板—ibidi μ-Slide Angiogenesis 用量10ul); 2、特殊的双层孔洞设计,可形成厚度均匀(0.8mm)的平整Gel matrix表面,使细胞落于同一层物镜对焦平面,细胞形态更易观察; 3、紧密的上盖设计,有效的减缓液体蒸发; 4、适用多通道微量分注器; 注:配合ibidi 加热与孵育系统,可以进行清晰、长期的活细胞显微拍摄。 ibidi “Well-in-a-Well” 和普通标准孔的对比:
订购信息: 产品规格: μ-Plate Angiogenesis 96 well 1. Planar air-liquid interface: good phase contrast all over the observation area 2. Planar gel surface: all cells are in one optical plan V olume of Matrigel: 10?μl Standard well 1. Meniscus on air-liquid interface: poor phase contrast in most of the observation area 2. Mensicus on the gel surface: not possible to focus on all cells simultaneously V olume of Matrigel: 100?μl
[52]Ray MA,Trammell RA,Verhulst S,et al.Development of a mouse model for assessing fatigue during chemotherapy[J].Comp Med, 2011,61(2):119-130. [53]White PD.Chronic fatigue syndrome:Is it one discrete syndrome or many?Implications for the"one vs.many"functional somatic syndromes debate[J].J Psychosom Res,2010,68(5):455- 459. [54]Romito F,Cormio C,Giotta F,et al.Quality of life,fatigue and de-pression in Italian long-term breast cancer survivors[J].Support Care Cancer,2012,3:8. [55]Dewys WD,Begg C,Lavin PT,et al.Prognostic effect of weight loss prior to chemotherapy in cancer patients.Eastern Cooperative Oncology Group[J].Am J Med,1980,69(4):491-497.[56]Wang X,Pickrell AM,Zimmers TA,et al.Increase in muscle mito- chondrial biogenesis does not prevent muscle loss but increased tumor size in a mouse model of acute cancer-induced cachexia [J].PLoS One,2012,7(3):e33426. [57]Mercuriali F,Inghilleri G.Fatigue and cancer.European School of Oncology Scientific Updates,5.Treatment of anaemia in cancer patients:Transfusion of rHuEPO[M].Amsterdam:Elsevier,2001: 185-200. [58]Schubert C,Hong S,Natarajan L,et al.The association between fatigue and inflammatory marker levels in cancer patients:a quan- titative review[J].Brain Behav Immun,2007,21:413-427.[59]Pusztai L,Mendoza TR,Reuben JM,et al.Changes in plasma lev-els of inflammatory cytokine-s in response to paclitaxel chemo- therapy[J].Cytokine,2004,25:94-102. (编校:谈静) 肿瘤血管生成拟态研究新进展 刘见荣,侯风刚 Research advance of the mechanism of tumor vasculogenic mimicry Liu Jianrong,Hou Fenggang Department of Oncology,Traditional Chinese Medical Hospital of Shanghai Municipal Affilialed to Shanghai University of Tradtional Chi-nese Medicine,Shanghai200071,China. 【Abstract】Vasculogenic mimicry(VM)is a brand-new tumor blood essels mode which is independent of angiogen- esis.More and more research found that the formation of VM have a close relation with the plasticity of tumor cells themselves and tumor stem cells.In addition,the tumor microenvironment such as the extracellular matrix remodeling and hypoxia also play an important role in the forming process of vasculogenic mimicry.In recent years,the related re- search about mechanism to tumor vasculogenic mimicry has made a lot of progress.This provides new ideas and direc- tion for seeking the key role of VM inhibition targets,screening effective drug.This article gave an outline of VM and summarize the formation mechanism of vasculogenic mimicry and the research status about the oncotherapy. 【Key words】vasculogenic mimicry;tumor;forming mechanism;treatment Modern Oncology2013,21(04):0898-0902 【指示性摘要】血管生成拟态是独立于血管生成的一种全新肿瘤血管模式。越来越多的研究发现血管生成拟 态的形成机制和肿瘤细胞自身的可塑性及肿瘤干细胞关系密切,另外,肿瘤微环境如细胞外基质重塑、缺氧 等在血管生成拟态形成过程中也有很重要的作用。近年来,血管生成拟态形成机制的相关研究已经取得了 很大进展,为寻找抑制VM的关键作用靶点、筛选有效的治疗药物提供了新的思路和方向。本文就血管拟态 的发现、形成机制及其在肿瘤治疗中的研究现状等方面做一综述。 【关键词】血管生成拟态;肿瘤;形成机制;治疗 【中图分类号】R730【文献标识码】A DOI:10.3969/j.issn.1672-4992.2013.04.78 【文章编号】1672-4992-(2013)04-0898-05 【收稿日期】2012-12-06 【基金项目】国家自然基金资助项目(编号:81173221) 【作者单位】上海中医药大学附属上海市中医医院肿瘤科,上海200071 【作者简介】刘见荣(1986-),女,河南郑州人,硕士研究生,主要从事大肠癌中医临床与实验研究。E-mail:liujianrong-zz@163.com 【通讯作者】侯风刚(1972-),男,河北衡水人,副主任医师,硕士研究生导师,博士,主要从事中西医结合消化道肿瘤临床和实验研究。