Hydrogen-Bonded Supramolecular Conjugated Polymer Nanoparticles for White Light-Emitting Devices
- 格式:pdf
- 大小:772.79 KB
- 文档页数:6


《两种制备方法对水飞蓟宾纳米混悬剂体内外行为的影响》篇一一、引言水飞蓟宾是一种具有重要药用价值的化合物,广泛应用于临床治疗多种疾病。
然而,由于水飞蓟宾的溶解性差,其生物利用度常常受到限制。
为了解决这一问题,研究者们尝试了多种方法制备水飞蓟宾纳米混悬剂,以提高其溶解度和生物利用度。
本文将探讨两种制备方法对水飞蓟宾纳米混悬剂体内外行为的影响。
二、制备方法一第一种制备方法主要采用乳化溶剂挥发法(Emulsification-Evaporization Method),简称E-E法。
这种方法主要包括将水飞蓟宾溶解在有机溶剂中,再通过高速搅拌或乳化使药物与辅料混合形成均匀的油滴状。
然后通过降低温度或使溶剂挥发的方式使油滴固化,最终形成纳米混悬剂。
三、制备方法二第二种制备方法为纳米沉淀法(Nanoprecipitation Method)。
该方法首先将水飞蓟宾与稳定剂混合,然后将其溶于有机溶剂中。
接着在搅拌条件下,将该混合物与另一溶液迅速混合,产生高浓度过饱和的体系。
过饱和体系中由于大量的沉淀过程导致形成稳定而小尺寸的颗粒。
再经一定的处理方法获得最终的产品纳米混悬剂。
四、两种制备方法对体内外行为的影响1. 体外释放研究对于两种制备方法所得的纳米混悬剂进行体外释放研究,可以观察到通过纳米沉淀法制备的混悬剂药物释放速率相对较快,因为其能够产生较高的过饱和度并迅速产生大量的药物沉淀颗粒。
而E-E法由于在固化过程中可能存在药物与辅料之间的相互作用,导致药物释放速率相对较慢。
2. 体内吸收研究在体内吸收方面,由于纳米混悬剂具有较小的颗粒尺寸和较大的表面积,能够显著提高药物的溶解度和生物利用度。
通过比较两种制备方法得到的纳米混悬剂在体内的吸收情况,可以观察到采用纳米沉淀法制备的混悬剂能够更好地促进药物在体内的吸收,提高生物利用度。
而E-E法虽然过程相对复杂,但其所得到的纳米混悬剂也能在体内产生一定的药效。
3. 体内外相关性研究在体内外相关性研究中,通过对两种制备方法得到的纳米混悬剂进行体内外释放及吸收的研究,发现其具有较好的相关性。

月桂醇基海藻酸钠与层状双金属纳米颗粒稳定载药Pickering乳液及其缓释性能刘若林;李嘉诚;冯玉红;颜慧琼;黄俊浩;刘艳凤;程春风;林强【期刊名称】《高分子材料科学与工程》【年(卷),期】2015(0)4【摘要】为了提高海藻酸钠(SA)对疏水性农药的负载量和释药缓释作用,将其与月桂醇通过偶联酯化反应进行疏水改性,对改性后的海藻酸钠进行红外光谱、核磁共振表征分析,结果证明月桂醇侧链成功接枝到海藻酸钠分子骨架上。
将月桂醇改性海藻酸钠(DA)和十六烷基三甲基溴化铵(CTAB)与层状双金属氢氧化物(LDH)纳米颗粒进行复配,其Zeta电位分别为+44.9 m V和-33.2 m V,同时其粒径分别增大到93.3 nm和659.8 nm。
结果表明带负电的月桂醇改性海藻酸钠吸附在层状双金属氢氧化物颗粒表面可以阻碍颗粒间的相互聚集,在分散体系中表现出了良好的稳定性能。
高速剪切下制备稳定Pickering乳液,对疏水性农药氯氟氰菊酯进行了释药试验,表明改性后的海藻酸钠与LDH颗粒制备Pickering乳液对氯氟氰菊酯具有较好的药物缓释作用。
【总页数】6页(P102-106)【关键词】月桂醇改性海藻酸钠;层状双金属氢氧化物纳米颗粒;Pickering乳液;缓释载药;氯氟氰菊酯【作者】刘若林;李嘉诚;冯玉红;颜慧琼;黄俊浩;刘艳凤;程春风;林强【作者单位】海南大学材料与化工学院化学系;海南师范大学化学与化工学院【正文语种】中文【中图分类】O631.3;TQ460.1【相关文献】1.Pickering乳液法原位制备载药磁性SiO2空心球及缓释性能 [J], 王志琰;毋伟;张魁;陈建峰;张鹏远2.改性海藻酸钠活化SiO2纳米粒子稳定载药Pickering乳液及释药性 [J], 颜慧琼;李嘉诚;冯玉红;胡文涛;刘若林;林强3.玉米醇溶蛋白-多酚纳米颗粒对Pickering乳液稳定性的调控 [J], 唐瑜婉;王启明;杨雅轩;李富华;赵吉春;明建4.基于玉米醇溶蛋白纳米颗粒稳定的Pickering乳液研究进展 [J], 王文莉;柴向华;范宇婷;吴克刚;段雪娟;于泓鹏;刘晓丽5.纳米纤维素颗粒稳定的Pickering乳液的性能研究 [J], 牛付阁;韩备竞;寇梦璇;樊佳玫;潘伟春因版权原因,仅展示原文概要,查看原文内容请购买。
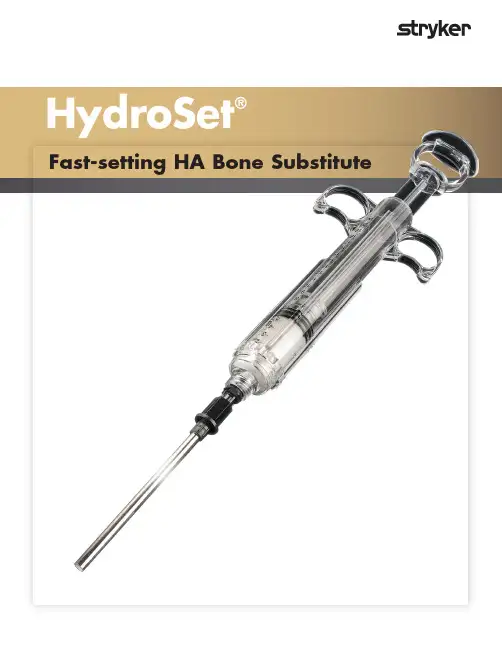
HydroSet®Fast-setting HA B one S ubstituteThe HydroSet material is formulated to harden in awet field environment.Features·Fast-setting1·Excellent wet-field properties2,3·Injectable or manual implantation 4·Osteoconductive 1·Isothermic1HydroSet®Fast-setting HA bone substitueIndicationsHydroSet is a self-setting, calcium phosphate cement intended for use in the repair of neurosurgical burr holes, contiguous craniotomy cuts and other cranial defects as well as in the augmentation or restorationof bony contour in the craniofacial skeleton.Features and benefitsFast-SettingHydroSet has been specifically designed to setquickly once implanted under normal physiological conditions, potentially minimi s ing procedure time.1 Excellent Wet Field CharacteristicsHydroSet is formulated to harden in the presence of water, blood, and CSF, which allows for irrigation/wash out of the surgery site during setting.2,3Injectable or Manual ImplantationHydroSet can be applied manually by hand/spatulaor injected through a syringe, which enablesusers to better meet unique closure needs. 4 OsteoconductiveHydroSet is a calcium phosphate cement thathardens to form hydroxyapatite and remodels tonatural bone through osteoclastic resorptionand new bone formation.1IsothermicHydroSet avoids thermal injury as it does notgive off any potentially damaging heat as ithardens.1RadiopaqueHydroSet is visible on postoperative X-rays.1Proven bone substitute technology for over a decadeHydroset is part of Stryker’s market leading bone cement portfolio and is an excellent substitute solution for a wide variety of clinical applications in multiple surgical specialties.2Scanning Electron Microscope image of HydroSet materialcrystalline microstructure at 15000x magnification2Part number DescriptionQty 79-43903HydroSet Bone Cement 3cc 79-43905HydroSet Bone Cement 5cc 79-43910HydroSet Bone Cement 10cc 79-43915HydroSet Bone Cement15ccOrdering informationImplantation instructionsAdd liquid to powder. Each kit contains one liquid filled glass syringe and one bowl of powder. Peel back the lid on the bowl; empty the liquid contents of the syringe into the bowl with powder.Mix liquid and powder. Mix the liquid and powderquickly (3-4 revolutions per second) in a circular motion for 45 seconds, ensuring that all the solution has been distributed throughout the powder. Compress the material against the sides of the bowl until a homogeneous, consistent paste is achieved.Note: The cement paste may look uniformly mixedafter 10-15 seconds of mixing; however, continue to mix for 45 seconds to ensure powder is thoroughly mixed intosolution. Care should be taken when handling and mixing the powder in the bowl. Losing powder could cause a wet cement mixture that may exhibit undesirable handling and setting characteristics.Transfer cement to delivery syringe. Place the cement delivery syringe barrel at an angled position using the fixture aid in the blister tray to hold the syringe securely. Note: Approach the fixture aid holding the syringe at a 45º angle and then push the syringe onto the fixture aid to achieve a stable footing.Transfer the cement from the mixing bowl to the cement delivery syringe using the supplied spatula. The funnel comes pre-attached to the syringe barrel. Once cement transfer is complete, remove the funnel from the end of the cement delivery syringe barrel (counter clockwise direction). Attach the supplied cannula to the end of the cement delivery syringe barrel (clockwise direction).Attach the plunger rod into the piston at the syringe barrel entrance by screwing into place while keeping the syringe system vertical with the cannula pointing up (clockwise direction). Now position the delivery syringe in a vertical position with the cannula pointing up and fully load the plunger rod into the syringe barrel to remove trapped air within the syringe assembly and to accumulate the cement to the base of the syringe ready for implantation.Note: Removing trapped air is necessary, as trapped air will compromise injectability.14532The loading process should be finished by 2 minutes 30 seconds from the start of mixing. Implantation and sculpting the cement; Prior to implantation, it is important to check for the desired cement consistency for times greater than or equal to 2 minutes 30 seconds after initial mixing. Cementshould ideally be fully injected by 4 minutes 30seconds from the start of mixing. After implantation,cement sculpting past 4 minutes 30 seconds from the start of mixing may disrupt the settingproperties of the cement.Note: Minimi s e contact and heat transfer between palm of hands and syringe barrel w ith cement within, as excessive heat will reduce theinjectability time window. In defects with exposed surface areas larger than 4cm 2, place supportive metal implants (titanium mesh) prior to applying the material.Note: Prior to injection, control active bleeding at the implant site. Suction, cautery, bone wax, and gel foam may be used.Warning: Remove gel foam and bone wax prior to implantation.Allow the material to set completely. Set time will occur between 4 minutes 30 seconds to 8 minutes 30 seconds from the start of mixing (potentially longer if the temperature at the defect site is below 32ºC). Leave the material undisturbed during the setting time. Close the surgical site. In defects with a surface area greater than or equal to 4cm 2, apply a closed suction drain to prevent excessive wound fluid accumulation.Note: HydroSet is a temperature sensitive product with ideal prod-uct and operating room temperatures being in the range of 18°-22° C (64.4° – 71.6° F). Product use below this temperature range will result in a runnier paste consistency and during injection could cause liquid to powder separation. Product use above this temperature range will result in a stiffer paste with reduced workingand injectability time.Note: Scultping and material manipulation must cease after 4 minutes and 30 secondsReferences:1. Larsson, S.: Injectable Phosphate Cements – A Review. 2006.2.Hannink, G., Wolke, J.G.C., Schreurs, B.W., Buma, P.: In Vivo Behavior of a Novel Injectable Calcium Phosphate Cement Compared with Two Other Commercially Available Calcium Phosphate Cements. 2007.3.HydroSet IFU4.Clarkin, O.M., Boyd, D., Madigan, S., and Towler, M.R.: Comparison of an Experimental Bone Cement with a Commercial Control,HydroSet, 2009.CMF-BR-148_Rev. None_17430_SSPCopyright © 2018 Stryker Printed in USACraniomaxillofacialThis document is intended solely for the use of healthcare professionals. A surgeon must always rely on his or her own professional clinical judgment when deciding whether to use a particular product when treating a particular patient. Stryker does not dispense medical advice and recommends that surgeons be trained in the use of any particular product before using it in surgery.The information presented is intended to demonstrate a Stryker product. A surgeon must always refer to the package insert, product label and/or instructions for use, including the instructions for cleaning and sterili s ation (if applicable), before using any Stryker product. Products may not be available in all markets because product availability is subject to the regulatory and/or medical practices in individual markets.Please contact your Stryker representative if you have questions about the availability of Stryker products in your area.Stryker Corporation or its divisions or other corporate affiliated entities own, use or have applied for the following trademarks or service marks: HydroSet, Stryker. All other trademarks are trademarks of their respective owners or holders.Distributed by:Stryker Australia 8 Herbert Street,St Leonards NSW 2065T: +61 2 9467 1000F: +61 2 9467 1010Stryker New Zealand Limited PO Box 17-136Greenlane, Auckland 1546 T: 64 9 573 1894F: 64 9 5731895。
![海藻酸钠-羟基磷灰石杂化纳米粒子及其制备方法[发明专利]](https://uimg.taocdn.com/b388876f84868762cbaed5dc.webp)
专利名称:海藻酸钠-羟基磷灰石杂化纳米粒子及其制备方法专利类型:发明专利
发明人:李旭东,任肖湘,王彦明,苏文
申请号:CN201510299338.6
申请日:20150603
公开号:CN104958766A
公开日:
20151007
专利内容由知识产权出版社提供
摘要:一种海藻酸钠-羟基磷灰石杂化纳米粒子,由质量分数为65%~99.5%的羟基磷灰石和质量分数为0.5%~35%的海藻酸钠组成,其形貌呈针状,针的长度为10~70nm,针的宽度为3~
20nm。
制备方法:将海藻酸类多糖溶于去离子水中得到质量浓度为0.2%~0.8%的海藻酸类多糖溶液,在搅拌下向所得海藻酸类多糖溶液中加入钙盐水溶液和磷酸盐水溶液,所述钙盐水溶液和磷酸盐水溶液的加入量以反应体系中Ca/P的摩尔比为(1~2.0):1为限,且钙盐水溶液与海藻酸类多糖溶液的体积比为1:(20~40),继后调节反应体系的pH值至10~12并停止搅拌,静置陈化至少5天,静置陈化结束后收集反应产物并将反应产物干燥。
上述海藻酸钠-羟基磷灰石杂化纳米粒子特别适合作为抗癌药物载体。
申请人:四川大学
地址:610065 四川省成都市武侯区一环路南一段24号
国籍:CN
代理机构:成都科海专利事务有限责任公司
更多信息请下载全文后查看。
![水基液体[发明专利]](https://uimg.taocdn.com/45a5c6e97cd184254a35359c.webp)
专利名称:水基液体
专利类型:发明专利
发明人:阿拉恩·露易斯·彼里,弗南德·杰罗米·凯克申请号:CN86104443
申请日:19860627
公开号:CN86104443A
公开日:
19870311
专利内容由知识产权出版社提供
摘要:水溶性羟基二羧酸或水溶性羟基三羧酸,一般是与链烷醇胺一起用于油/水液,特别是金属加工液或液压液,所得到的液体有极好的硬水相容性,在软水中发泡趋向低,并且生物稳定性好,一般含有如乳化剂、铜钝化剂等其它添加剂。
申请人:埃克森化学专利公司
地址:美国新泽西州
国籍:US
代理机构:中国国际贸易促进委员会专利代理部
更多信息请下载全文后查看。

hydrocarbon碳氢化合物:Hydrocarbons are composed of hydrogen and carbon. The intramolecular bonds are covalent.macromolecules高分子:a huge molecule made up of thousands of atoms. Macromolecules高分子(polymer)Polymer was coined to mean many mers.A compound consisting of long-chain molecules, each molecule made up of repeating units connected together.Mer(单体单元,结构单元):Structural entities, originates from the Greek word meros, which means part;Monomer(单体):A stable molecule from which a polymer is synthesized, such as ethylene. Polymerization(聚合反应):The synthesis of the large molecular weight polymers is termed polymerizationThermoplastics(热塑性聚合物)Thermoplastics soften when heated (and eventually liquefy) and harden when cooled—processes that are totally reversible and may be repeated. (加热变软冷却变硬可回收的)Thermosetting (热固性聚合物)Thermosetting polymers become permanently hard when heat is applied and do not soften upon subsequent heating.塑料形成方式:The most common method for forming plastic polymers. Include compression(压塑), transfer(传递模塑), blow(吹塑模塑法), injection (注塑模塑), and extrusion molding (挤压模塑)。

化学交联水凝胶英语English:Chemical crosslinking hydrogels are formed through the process of chemical crosslinking, which involves the creation of covalent bonds between polymer chains in the presence of a crosslinking agent. This method allows for the customization of hydrogel properties, such as mechanical strength, swelling behavior, and degradation rate, by adjusting the crosslinking density and type of crosslinking agent used. Chemical crosslinking of hydrogels can be achieved through various crosslinking mechanisms, including radical polymerization, Michael addition, click chemistry, and Schiff base reaction. These hydrogels have been widely used in various biomedical applications, such as drug delivery systems, tissue engineering scaffolds, wound dressings, and biosensors, due to their biocompatibility, tunable properties, and ability to mimic the extracellular matrix. Additionally, chemical crosslinking hydrogels can be designed to respond to specific stimuli, such as pH, temperature, or enzymatic activity, making them attractive candidates for controlled drug release or tissue regeneration. Overall, chemical crosslinking hydrogels haveshown great potential in advancing both basic science research and clinical applications in the field of biomaterials.中文翻译:化学交联水凝胶是通过化学交联过程形成的,这涉及在交联剂存在的情况下在聚合物链之间形成共价键。

05. 高分子化学05.1 高分子物质brush polymer coiling type polymer05.2 聚合与高分子化学反应17 环状单体cyclic monomer18 共聚单体comonomer19 聚合[反应]polymerization20 均聚反应homopolymerization21低聚反应,齐聚反应(曾用名)oligomerization22 调聚反应telomerization23 自发聚合spontaneous polymerization24 预聚合prepolymerization25 后聚合post polymerization26 再聚合repolymerization27 铸塑聚合, 浇铸聚合cast polymerization28 链[式]聚合chain polymerization29 烯类聚合,乙烯基聚合vinyl polymerization30 双烯[类]聚合diene polymerization31 加[成]聚[合]addition polymerization32自由基聚合,游离基聚合(曾用名) free radical polymerization, radical polymerization33控制自由基聚合,可控自由基聚合controlled radical polymerization,CRP34 活性自由基聚合living radical polymerization35 原子转移自由基聚合atom transfer radical polymerization,ATRP36 反向原子转移自由基聚合reverse atom transfer radical polymerization,RATRP37可逆加成断裂链转移reversible addition fragmentation chaintransfer,RAFT38 氮氧[自由基]调控聚合nitroxide mediated polymerization39 稳定自由基聚合stable free radical polymerization,FRP40 自由基异构化聚合free radical isomerization polymerization41 自由基开环聚合radical ring opening polymerization42 氧化还原聚合redox polymerization43无活性端聚合,死端聚合(曾用名)dead end polymerization44 光[致]聚合photo polymerization45 光引发聚合light initiated polymerization46 光敏聚合photosensitized polymerization47 四中心聚合four center polymerization48 电荷转移聚合charge transfer polymerization49 辐射引发聚合radiation initiated polymerization50 热聚合thermal polymerization51 电解聚合electrolytic polymerization52 等离子体聚合plasma polymerization53 易位聚合metathesis polymerization54 开环易位聚合ring opening metathesis polymerization,ROMP55 精密聚合precision polymerization56 环化聚合cyclopolymerization57 拓扑化学聚合topochemical polymerization58 平衡聚合equilibrium polymerization59 离子[型]聚合ionic polymerization60 辐射离子聚合radiation ion polymerization61 离子对聚合ion pair polymerization62正离子聚合,阳离子聚合cationic polymerization63 碳正离子聚合carbenium ion polymerization,carbocationicpolymerization64 假正离子聚合pseudo cationic polymerization65 假正离子活[性]聚合pseudo cationic living polymerization66 活性正离子聚合living cationic polymerization67 负离子聚合,anionic polymerization阴离子聚合68 碳负离子聚合carbanionic polymerization69 活性负离子聚合living anionic polymerization70 负离子环化聚合anionic cyclopolymerization71 负离子电化学聚合anionic electrochemical polymerization72 负离子异构化聚合anionic isomerization polymerization73 烯丙基聚合allylic polymerization74 活[性]聚合living polymerization75 两性离子聚合zwitterion polymerization76 齐格勒-纳塔聚合Ziegler Natta polymerization77 配位聚合coordination polymerization78 配位离子聚合coordinated ionic polymerization79 配位负离子聚合coordinated anionic polymerization80 配位正离子聚合coordinated cationic polymerization81 插入聚合insertion polymerization82定向聚合,立构规整聚合stereoregular polymerization, stereospecific polymerization83 有规立构聚合tactic polymerization84 全同立构聚合isospecific polymerization85 不对称诱导聚合asymmetric induction polymerization86 不对称选择性聚合asymmetric selective polymerization87 不对称立体选择性聚合asymmetric stereoselective polymerization88 对映[体]不对称聚合enantioasymmetric polymerization89 对映[体]对称聚合enantiosymmetric polymerization90 异构化聚合isomerization polymerization91 氢转移聚合hydrogen transfer polymerization92 基团转移聚合group transfer polymerization,GTP93 消除聚合elimination polymerization94 模板聚合matrix polymerization,templatepolymerization95 插层聚合intercalation polymerization96 无催化聚合uncatalyzed polymerization97 开环聚合ring opening polymerization98 活性开环聚合living ring opening polymerization99 不死的聚合immortal polymerization100 酶聚合作用enzymatic polymerizationpolyaddition聚加成反应,101逐步加成聚合(曾用名)102 偶联聚合coupling polymerization103 序列聚合sequential polymerization104 闪发聚合,俗称暴聚flash polymerization105 氧化聚合oxidative polymerization106 氧化偶联聚合oxidative coupling polymerization107 逐步[增长]聚合step growth polymerization缩聚反应condensation polymerization,108polycondensation酯交换型聚合transesterification type polymerization, 109ester exchange polycondensation110 自催化缩聚autocatalytic polycondensation111 均相聚合homogeneous polymerization112 非均相聚合heterogeneous polymerization113 相转化聚合phase inversion polymerization114 本体聚合bulk polymerization, mass polymerization 115 固相聚合solid phase polymerization气相聚合gaseous polymerization,116gas phase polymerization117 吸附聚合adsorption polymerization118 溶液聚合solution polymerization119 沉淀聚合precipitation polymerization120 淤浆聚合slurry polymerization121 悬浮聚合suspension polymerization122 反相悬浮聚合reversed phase suspension polymerization 123 珠状聚合bead polymerization, pearl polymerization 124 分散聚合dispersion polymerization125 反相分散聚合inverse dispersion polymerization126 种子聚合seeding polymerization127 乳液聚合emulsion polymerization128 无乳化剂乳液聚合emulsifier free emulsion polymerization 129 反相乳液聚合inverse emulsion polymerization130 微乳液聚合micro emulsion polymerization131 连续聚合continuous polymerization132 半连续聚合semicontinuous polymerization133 分批聚合,间歇聚合batch polymerization134 原位聚合in situ polymerization135 均相缩聚homopolycondensation136 活化缩聚activated polycondensation137 熔融缩聚melt phase polycondensation138 固相缩聚solid phase polycondensation139 体型缩聚three dimensional polycondensation140 界面聚合interfacial polymerization141 界面缩聚interfacial polycondensation142 环加成聚合cycloaddition polymerization143 环烯聚合cycloalkene polymerization144 环硅氧烷聚合cyclosiloxane polymerization145 引发剂initiator146 引发剂活性activity of initiator147 聚合催化剂polymerization catalyst148 自由基引发剂radical initiator149 偶氮[类]引发剂azo type initiator150 2,2′偶氮二异丁腈2,2'- azobisisobutyronitrile, AIBN151 过氧化苯甲酰benzoyl peroxide, BPO152 过硫酸盐引发剂persulphate initiator153 复合引发体系complex initiation system154 氧化还原引发剂redox initiator电荷转移复合物,charge transfer complex, CTC155电荷转移络合物156 聚合加速剂,聚合促进剂polymerization accelerator157 光敏引发剂photoinitiator158 双官能引发剂bifunctional initiator,difunctional initiator 159 三官能引发剂trifunctional initiator160 大分子引发剂macroinitiator161 引发-转移剂initiator transfer agent, inifer162 引发-转移-终止剂initiator transfer agent terminator, iniferter 163 光引发转移终止剂photoiniferter164 热引发转移终止剂thermoiniferter165 正离子催化剂cationic catalyst166 正离子引发剂cationic initiator167 负离子引发剂ionioic initiator168 共引发剂coinitiator169 烷基锂引发剂alkyllithium initiator170 负离子自由基引发剂anion radical initiator171 烯醇钠引发剂alfin initiator172 齐格勒-纳塔催化剂Ziegler Natta catalyst173 过渡金属催化剂transition metal catalyst174 双组分催化剂bicomponent catalyst175 后过渡金属催化剂late transition metal catalyst176 金属络合物催化剂metal complex catalyst177 [二]茂金属催化剂metallocene catalyst178 甲基铝氧烷methylaluminoxane, MAO179μ氧桥双金属烷氧化物催化剂bimetallic μ-oxo alkoxides catalyst180 双金属催化剂bimetallic catalyst 181 桥基茂金属bridged metallocene182限定几何构型茂金属催化剂constrained geometry metallocene catalyst183 均相茂金属催化剂homogeneous metallocene catalyst 184 链引发chain initiation185 热引发thermal initiation186 染料敏化光引发dye sensitized phtoinitiation187 电荷转移引发charge transfer initiation188 诱导期induction period189 引发剂效率initiator efficiency190 诱导分解induced decomposition191 再引发reinitiation192 链增长chain growth, chain propagation193 增长链端propagating chain end194 活性种reactive species195 活性中心active center196 持续自由基persistent radical197 聚合最高温度ceilling temperature of polymerization 198 链终止chain termination199 双分子终止bimolecular termination200 初级自由基终止primary radical termination201 扩散控制终止diffusion controlled termination202 歧化终止disproportionation termination203 偶合终止coupling termination204 单分子终止unimolecular termination205 自发终止spontaneous termination206 终止剂terminator207 链终止剂chain terminating agent208 假终止pseudotermination209 自发终止self termination210 自由基捕获剂radical scavenger211 旋转光闸法rotating sector method212 自由基寿命free radical lifetime213 凝胶效应gel effect214 自动加速效应autoacceleration effect215 链转移chain transfer216 链转移剂chain transfer agent217 尾咬转移backbitting transfer218 退化链转移degradation (degradative) chain transfer 219 加成断裂链转移[反应]addition fragmentation chain transfer 220 链转移常数chain transfer constant①缓聚作用retardation221②延迟作用222 阻聚作用inhibition223 缓聚剂retarder224 缓聚剂,阻滞剂retarding agent225 阻聚剂inhibitor226 封端[反应]end capping227 端基terminal group228 聚合动力学polymerization kinetics229 聚合热力学polymerization thermodynamics230 聚合热heat of polymerization231 共聚合[反应]copolymerization232 二元共聚合binary copolymerization233 三元共聚合ternary copolymerization234 竞聚率reactivity ratio235 自由基共聚合radical copolymerization236 离子共聚合ionic copolymerization237 无规共聚合random copolymerization 238 理想共聚合ideal copolymerization239 交替共聚合alternating copolymerization 240 恒[组]分共聚合azeotropic copolymerization 241 接枝共聚合graft copolymerization242 嵌段共聚合block copolymerization243 开环共聚合ring opening copolymerization 244 共聚合方程copolymerization equation 245 共缩聚copolycondensation246 逐步共聚合step copolymerization247 同种增长homopropagation248 自增长self propagation249 交叉增长cross propagation250 前末端基效应penultimate effect251 交叉终止cross termination252 Q值Q value253 e值e value254 Q,e概念Q, e scheme255 序列长度分布sequence length distribution 256 侧基反应reaction of pendant group 257 扩链剂,链增长剂chain extender258 交联crosslinking259 化学交联chemical crosslinking260 自交联self crosslinking261 光交联photocrosslinking262 交联度degree of crosslinking263 硫化vulcanization264 固化curing265 硫[黄]硫化sulfur vulcanization266 促进硫化accelerated sulfur vulcanization 267 过氧化物交联peroxide crosslinking268 无规交联random crosslinking269 交联密度crosslinking density270 交联指数crosslinking index271 解聚depolymerization272 ①降解②退化degradation273 链断裂chain breaking274 解聚酶depolymerase275 细菌降解bacterial degradation276 生物降解biodegradation277 化学降解chemical degradation278 辐射降解radiation degradation279 断链降解chain scission degradation280 自由基链降解free radical chain degradation 281 无规降解random degradation282 水解降解hydrolytic degradation283 热降解thermal degradation284 热氧化降解thermal oxidative degradation 285 光降解photodegradation286 光氧化降解photo oxidative degradation 287 力化学降解mechanochemical degradation 288 接枝聚合graft polymerization289 活化接枝activation grafting290 接枝点grafting site291 链支化chain branching05.3 高分子物理化学与高分子物理6等规度, 全同立构[规整]isotacticity度syndiotacticity7间同度,间同立构[规整]度8无规度,无规立构度atacticity9嵌段block10规整嵌段regular block11非规整嵌段irregular block12立构嵌段stereoblock13有规立构嵌段isotactic block14无规立构嵌段atactic block15单体单元monomeric unit16二单元组diad17三单元组triad18四单元组tetrad19五单元组pentad20无规线团random coil21自由连接链freely-jointed chain22自由旋转链freely-rotating chain23蠕虫状链worm-like chain24柔性链flexible chain25链柔性chain flexibility26刚性链rigid chain27棒状链rodlike chain28链刚性chain rigidity29聚集aggregation30聚集体aggregate31凝聚、聚集coalescence32链缠结chain entanglement33凝聚缠结cohesional entanglement 34物理缠结physical entanglement35拓扑缠结topological entanglement36凝聚相condensed phase37凝聚态condensed state38凝聚过程condensing process39临界聚集浓度critical aggregation concentration40线团-球粒转换coil-globule transition41受限链confined chain42受限态confined state43物理交联physical crosslinking44统计线团statistical coil45等效链equivalent chain46统计链段statistical segment47链段chain segment48链构象chain conformation49无规线团模型random coil model50无规行走模型random walk model51自避随机行走模型self avoiding walk model52卷曲构象coiled conformation53高斯链Gaussian chain54无扰尺寸unperturbed dimension55扰动尺寸perturbed dimension56热力学等效球thermodynamically equivalent sphere 57近程分子内相互作用short-range intramolecular interaction 58远程分子内相互作用long-range intramolecular interaction 59链间相互作用interchain interaction60链间距interchain spacing61长程有序long range order62近程有序short range order63回转半径radius of gyration64末端间矢量end-to-end vector65链末端chain end66末端距end-to-end distance67无扰末端距unperturbed end-to-end distance68均方根末端距root-mean-square end-to-end distance69伸直长度contour length70相关长度persistence length71主链;链骨架chain backbone72支链branch chain73链支化chain branching74短支链short-chain branch75长支链long-chain branch76支化系数branching index77支化密度branching density78支化度degree of branching79交联度degree of crosslinking80网络network81网络密度network density82溶胀swelling83平衡溶胀equilibrium swelling84分子组装,分子组合molecular assembly85自组装self assembly86微凝胶microgel87凝胶点gel point88可逆[性]凝胶reversible gel89溶胶-凝胶转化sol-gel transformation90临界胶束浓度critical micelle concentration,CMC91组成非均一性constitutional heterogenity, compositionalheterogenity92摩尔质量平均molar mass average 又称“分子量平均”93数均分子量number-average molecular weight,number-average molar mass94重均分子量weight-average molecular weight,weight-average molar mass95Z均分子量Z(Zaverage)-average molecular weight,Z-molar mass96黏均分子量viscosity-average molecular weight,viscosity-average molar mass97表观摩尔质量apparent molar mass98表观分子量apparent molecular weight99聚合度degree of polymerization100动力学链长kinetic chain length101单分散性monodispersity102临界分子量critical molecular weight103分子量分布molecular weight distribution,MWD104多分散性指数polydispersity index,PID105平均聚合度average degree of polymerization106质量分布函数mass distribution function107数量分布函数number distribution function108重量分布函数weight distribution function109舒尔茨-齐姆分布Schulz-Zimm distribution110最概然分布most probable distribution 曾用名“最可几分布”111对数正态分布logarithmic normal distribution 又称“对数正则分布”112聚合物溶液polymer solution113聚合物-溶剂相互作用polymer-solvent interaction114溶剂热力学性质thermodynamic quality of solvent115均方末端距mean square end to end distance116均方旋转半径mean square radius of gyration117θ温度theta temperature118θ态theta state119θ溶剂theta solvent120良溶剂good solvent121不良溶剂poor solvent122位力系数Virial coefficient 曾用名“维里系数”123排除体积excluded volume124溶胀因子expansion factor125溶胀度degree of swelling126弗洛里-哈金斯理论Flory-Huggins theory127哈金斯公式Huggins equation128哈金斯系数Huggins coefficient129χ(相互作用)参数χ-parameter130溶度参数solubility parameter131摩擦系数frictional coefficient132流体力学等效球hydrodynamically equivalent sphere133流体力学体积hydrodynamic volume134珠-棒模型bead-rod model135球-簧链模型ball-spring [chain] model136流动双折射flow birefringence, streaming birefringence137动态光散射dynamic light scattering138小角激光光散射low angle laser light scattering139沉降平衡sedimentation equilibrium140沉降系数sedimentation coefficient141沉降速度法sedimentation velocity method142沉降平衡法sedimentation equilibrium method143相对黏度relative viscosity144相对黏度增量relative viscosity increment145黏度比viscosity ratio146黏数viscosity number147[乌氏]稀释黏度计[Ubbelohde] dilution viscometer148毛细管黏度计capillary viscometer149落球黏度计ball viscometer150落球黏度ball viscosity151本体黏度bulk viscosity152比浓黏度reduced viscosity153比浓对数黏度inherent viscosity, logarithmic viscositynumber154特性黏数intrinsic viscosity, limiting viscosity number 155黏度函数viscosity function156零切变速率黏度zero shear viscosity157端基分析analysis of end group158蒸气压渗透法vapor pressure osmometry, VPO159辐射的相干弹性散射coherent elastic scattering of radiation160折光指数增量refractive index increment161瑞利比Rayleigh ratio162超瑞利比excess Rayleigh ratio163粒子散射函数particle scattering function164粒子散射因子particle scattering factor165齐姆图Zimm plot166散射的非对称性dissymmetry of scattering167解偏振作用depolarization168分级fractionation169沉淀分级precipitation fractionation170萃取分级extraction fractionation171色谱分级chromatographic fractionation172柱分级column fractionation173洗脱分级,淋洗分级elution fractionation174热分级thermal fractionation175凝胶色谱法gel chromatography176摩尔质量排除极限molar mass exclusion limit177溶剂梯度洗脱色谱法solvent gradient [elution] chromatography 178分子量排除极限molecular weight exclusion limit179洗脱体积elution volume180普适标定universal calibration181加宽函数spreading function182链轴chain axis183等同周期identity period184链重复距离chain repeating distance185晶体折叠周期crystalline fold period186构象重复单元conformational repeating unit 187几何等效geometrical equivalence188螺旋链helix chain189构型无序configurational disorder190链取向无序chain orientational disorder191构象无序conformational disorder192锯齿链zigzag chain193双[股]螺旋double stranded helix194[分子]链大尺度取向global chain orientation195结晶聚合物crystalline polymer196半结晶聚合物semi-crystalline polymer197高分子晶体polymer crystal198高分子微晶polymer crystallite199结晶度degree of crystallinity, crystallinity 200高分子[异质]同晶现象macromolecular isomorphism 201聚合物形态学morphology of polymer202片晶lamella, lamellar crystal203轴晶axialite204树枝[状]晶体dendrite205纤维晶fibrous crystal206串晶结构shish-kebab structure207球晶spherulite208折叠链folded chain209链折叠chain folding210折叠表面fold surface211折叠面fold plane212折叠微区fold domain213相邻再入模型adjacent re-entry model 214接线板模型switchboard model215缨状微束模型fringed-micelle model216折叠链晶体folded-chain crystal217平行链晶体parallel-chain crystal218伸展链晶体extended-chain crystal219球状链晶体globular-chain crystal220长周期long period221近程结构short-range structure222远程结构long-range structure223成核作用nucleation224分子成核作用molecular nucleation225阿夫拉米方程Avrami equation226主结晶primary crystallization227后期结晶secondary crystallization 228外延结晶,附生结晶epitaxial crystallizationepitaxial growth229外延晶体生长,附生晶体生长230织构texture231液晶态liquid crystal state232溶致性液晶lyotopic liquid crystal233热致性液晶thermotropic liquid crystal 234热致性介晶thermotropic mesomorphism 235近晶相液晶smectic liquid crystal236近晶中介相smectic mesophase237近晶相smectic phase238条带织构banded texture239环带球晶ringed spherulite240向列相nematic phase241盘状相discotic phase242解取向disorientation243分聚segregation244非晶相amorphous phase 曾用名“无定形相”245非晶区amorphous region246非晶态amorphous state247非晶取向amorphous orientation248链段运动segmental motion249亚稳态metastable state250相分离phase separation251亚稳相分离spinodal decomposition252bimodal decomposition253微相microphase254界面相boundary phase255相容性compatibility256混容性miscibility257不相容性incompatibility258不混容性immiscibility259增容作用compatiibilizationlower critical solution temperature, LCST260最低临界共溶(溶解)温度upper critical solution temperature , UCST261最高临界共溶(溶解)温度262浓度猝灭concentration quenching263激基缔合物荧光excimer fluorescence264激基复合物荧光exciplex fluorescence265激光共聚焦荧光显微镜laser confocal fluorescence microscopy266单轴取向uniaxial orientation267双轴取向biaxial orientation, biorientation268取向度degree of orientation269橡胶态rubber state270玻璃态glassy state271高弹态elastomeric state272黏流态viscous flow state273伸长elongation274高弹形变high elastic deformation275回缩性,弹性复原nerviness276拉伸比draw ratio, extension ratio277泊松比Poisson's ratio278杨氏模量Young's modulus279本体模量bulk modulus280剪切模量shear modulus281法向应力normal stress282剪切应力shear stress283剪切应变shear strain284屈服yielding285颈缩现象necking 又称“细颈现象”286屈服应力yield stress287屈服应变yield strain288脆性断裂brittle fracture289脆性开裂brittle cracking290脆-韧转变brittle ductile transition291脆化温度brittleness(brittle) temperature292延性破裂ductile fracture293冲击强度impact strength294拉伸强度tensile strength 又称“断裂强度,breaking strength”295极限拉伸强度ultimate tensile strength296抗撕强度tearing strength 又称“抗扯强度”297弯曲强度flexural strength, bending strength298弯曲模量bending modulus299弯曲应变bending strain300弯曲应力bending stress301收缩开裂shrinkage crack302剪切强度shear strength303剥离强度peeling strength304疲劳强度fatigue strength, fatigue resistance305挠曲deflection306压缩强度compressive strength307压缩永久变形compression set308压缩变形compressive deformation309压痕硬度indentation hardness310洛氏硬度Rockwell hardness311布氏硬度Brinell hardness312抗刮性scrath resistance313断裂力学fracture mechanics314力学破坏mechanical failure315应力强度因子stress intensity factor316断裂伸长elongation at break317屈服强度yield strength318断裂韧性fracture toughness319弹性形变elastic deformation320弹性滞后elastic hysteresis321弹性elasticity322弹性模量modulus of elasticity323弹性回复elastic recovery324不可回复形变irrecoverable deformation325裂缝crack 俗称“龟裂”326银纹craze327形变;变形deformation328永久变形deformation set329剩余变形residual deformation330剩余伸长residual stretch331回弹,回弹性resilience332延迟形变retarded deformation333延迟弹性retarded elasticity334可逆形变reversible deformation335应力开裂stress cracking336应力-应变曲线stress strain curve337拉伸应变stretching strain338拉伸应力弛豫tensile stress relaxation339热历史thermal history340热收缩thermoshrinking341扭辫分析torsional braid analysis,TBA 342应力致白stress whitening343应变能strain energy344应变张量strain tensor345剩余应力residual stress346应变硬化strain hardening347应变软化strain softening348电流变液electrorheological fluid349假塑性pseudoplastic350拉胀性auxiticity351牛顿流体Newtonian fluid352非牛顿流体non-Newtonian fluid353宾汉姆流体Bingham fluid354冷流cold flow355牛顿剪切黏度Newtonian shear viscosity 356剪切黏度shear viscosity357表观剪切黏度apparent shear viscosity358剪切变稀shear thinning359触变性thixotropy360塑性形变plastic deformation361塑性流动plastic flow362体积弛豫volume relaxation363拉伸黏度extensional viscosity364黏弹性viscoelasticity365线性黏弹性linear viscoelasticity366非线性黏弹性non-linear viscoelasticity367蠕变creep368弛豫[作用] relaxation 又称“松弛”369弛豫模量relaxation modulus370蠕变柔量creep compliance371热畸变温度heat distortion temperature372弛豫谱relaxation spectrum373推迟[时间]谱retardation [time] spectrum374弛豫时间relaxation time375推迟时间retardation time376动态力学行为dynamic mechanical behavior377动态黏弹性dynamic viscoelasticity378热-机械曲线thermo-mechanical curve379动态转变dynamic transition380储能模量storage modulus381损耗模量loss modulus382复数模量complex modulus383复数柔量complex compliance384动态黏度dynamic viscosity385复数黏度complex viscosity386复数介电常数complex dielectric permittivity387介电损耗因子dielectric dissipation factor388介电损耗常数dielectric loss constant389介电弛豫时间dielectric relaxation time390玻璃化转变glass transition05.4 高分子加工技术和应用。

大孔交联聚甲基丙烯酸甲酯对酚类物质的氢键吸附研究Ξ徐满才 姚似锦 曾 盈α(湖南师范大学化学系,长沙,410081)史作清 许名成 何炳林(南开大学高分子化学研究所,天津,300071)摘 要 通过测定大孔交联聚甲基丙烯酸甲酯对苯酚的吸附等温线,利用热力学函数关系计算了吸附热,推测出吸附过程为氢键吸附.通过比较大孔交联聚甲基丙烯酸甲酯对苯酚、邻苯二酚及间苯二酚的吸附性能的差别,进一步证明了大孔交联聚甲基丙烯酸甲酯对酚类物质的氢键吸附机理.关键词 聚甲基丙烯酸甲酯;酚类;吸附机理;氢键分类号 /631.1Hydrogen -Bond i ng Adsorption of M acroporousCrossl i nked Poly m ethyl M ethacryla te for PhenolsX u M anca i Y ao S h ij ing Z eng Y ing(D epartm ent of Chem istry ,H unan N o r m al U niversity ,Changsha ,4100081)S h i Z uoqing X u M ing cheng H e B ing lin(Institute of Po lym er Chem istry ,N ankaiU niversity ,T ianjin ,300071)Abstract T he adso rp ti on iso ther m s of the title adso rben t fo r pheno l w ere m easu red and the adso rp ti on heat w as calcu lated from the iso ther m s by u sing the ther m odynam ic func 2ti on .T he resu lt indicated a hydrogen 2bonding adso rp ti on m echan is m .Fu rther m o re ,difference of the adso rp ti on p roperty of the title adso rben t fo r p heno l ,catecho l and reso rcino l w as in 2vestigated ,and the resu lt p roved the adso rp ti on m echan is m w as th rough the fo r m ati on of hy 2drogen bond .Key words po lym ethyl m ethacrylate ;p heno ls ;adso rp ti on m echan is m ;hydrogen bond 吸附分离功能高分子是依靠原子或分子之间的相互作用而实现其吸附剂分离功能的.依靠离子键、共价键、配位键和范德华力等作用而实现其吸附分离功能的高分子分别是离子交换树脂、聚合物键联试剂、螯合树脂和吸附树脂,它们已广泛应用于化工、环保、热能、电力、食品、冶金、制药等自然科学研究中[1].氢键是一种分子间(或分子内)较强的相互作用,50年代在研究聚酰胺对酚类物质的吸附时,曾提出过氢键吸附理论[2];近年来,又有一些依据溶质和吸附1998年6月 A cta Sci N at U niv N o rn H unan Jun .1998Ξ国家自然科学基金资助项目剂之间形成氢键的强度不同而进行吸附分离的报道[3].但是,到目前为止,利用氢键进行吸附分离的高分子的合成、结构和应用等方面尚无系统的研究报道.若能研制出含氢键供体的高分子吸附剂(即酸性氢键吸附剂)、含氢键受体的吸附剂(即碱性氢键吸附剂)和既含氢键供体又含氢键受体的吸附剂(即两性氢键吸附剂),用于对含有氢键受体或氢键供体的物质的提取分离,将会丰富和完善氢键吸附理论,在应用上将为许多物质的提取分离增添一种行之有效的方法.本项研究合成了大孔交联聚甲基丙烯酸甲酯这种含氢键受体的吸附剂,通过测定它对苯酚的吸附等温线计算出吸附热,并比较了它对苯酚、邻苯二酚和间苯二酚的吸附性能的差别,从理论上证明了大孔交联聚甲基丙烯酸甲酯对酚类物质的氢键吸附机理,为研究氢键吸附剂的应用提供了理论依据.1 实验部分1.1 大孔交联聚甲基丙烯酸甲酯的合成[4]在致孔剂存在下,将甲基丙烯酸甲酯、二乙烯苯进行悬浮共聚,然后用石油醚抽提除去致孔剂,制得大孔交联聚甲基丙烯酸甲酯珠体.1.2 大孔交联聚甲基丙烯酸甲酯对酚类物质吸附等温线的测定准确称取一定量的大孔交联聚甲基丙烯酸甲酯珠体置于锥形瓶中,加入一系列不同浓度的苯酚(或邻苯二酚、间苯二酚)水溶液,于TH Z 8821型台式多用恒温振荡器中在一定温度下振荡20h 以上,待吸附达到平衡后,在Κ217型紫外可见分光光度计上用分光光度法测定吸附后残液中酚的浓度,计算吸附量,测得大孔交联聚甲基丙烯酸甲酯对酚类物质的吸附等温线.1.3 吸附热的计算根据C lau siu s 2C lapeyron 方程: ln C =-∃H A ,m R ・1T +K .(1)C 为吸附平衡时溶质的浓度,T 为绝对温度,∃H A ,m 为平衡浓度C 时的微分吸附热,K 为常数,通过测定各种温度下大孔交联聚甲基丙烯酸甲酯对苯酚的吸附等温线,再由吸附等温线作出等吸附量q 时的ln C ~1T 图,由ln C ~1T图的斜率即可求出吸附量为q 时的微分吸附热∃H A ,m .2 结果与讨论2.1 大孔交联聚甲基丙烯酸甲酯对苯酚的氢键吸附机理图1是温度为296K ,303K ,308K ,313K 和318K 时聚甲基丙烯酸甲酯对水溶液中苯酚的吸附等温线.根据图1,分别作出吸附量q 为1.50,2.00,2.25,2.50和2.75m g g 时的ln C ~1T图,如图2所示.从图2可以看出,ln C 与1T呈线性关系,说明吸附过程服从C lau siu s 2C lapeyron 方程.由直线的斜率计算出各个吸附量q 下的微分吸附热∃H A ,m ,如附表所示.附表 不同吸附量下聚甲基丙烯酸甲酯对苯酚的吸附热吸附量q m g g1.502.002.252.502.75微分吸附热∃H A ,m kJ mo l 23.423.824.024.224.9从表中可以看出,当吸附量在1.50~2.75m g g 之间时,微分吸附热的大小在23~25kJ m o l34第2期 徐满才等:大孔交联聚甲基丙烯酸甲酯对酚类物质的氢键吸附研究 图2 ln C ~1T 图1 聚甲基丙烯酸甲酯对苯酚的吸附等温线1.q =2.75m g g 2.q =2.50m g g 3.q =2.25m g g4.q =2.00m g g5.q =1.50m g g 之间,这个数值比范德华力要大,而且在氢键的键能范围(8~50kJ m o l )之内[5],吸附过程中又不存在形成离子键、共价键和配位键的因素,因此,聚甲基丙烯酸甲酯是通过氢键作用吸附图3 聚甲基丙烯酸甲酯对苯酚的氢键作用苯酚的.吸附过程的氢键作用如图3所示.2.2 大孔交联聚甲基丙烯酸甲酯对苯酚、邻苯二酚和间苯二酚的吸附性能比较图4是在pH =2,温度为308K 的条件下大孔交联聚甲基丙烯酸甲酯对水溶液中苯酚、邻苯二酚和间苯二酚的吸附等温线.由图4可知,摩尔浓度相同时,聚甲基丙烯酸甲酯对邻苯二酚和间苯二酚的吸附量明显小于对苯酚的吸附量.这是图4 聚甲基丙烯酸甲酯对苯酚、邻苯二酚和间苯二酚的吸附等温线(35℃)1:苯酚;2:间苯二酚;3:邻苯二酚因为:邻苯二酚含有分子内氢键,从而降低了与聚甲基丙烯酸甲酯形成分子间氢键的能力,因而聚甲基丙烯甲酯对邻苯二酚的吸附量较小;间苯二酚虽然不能形成分子内氢键,但由于它的亲水性较强,酚羟基与水通过氢键结合,因而与聚甲基丙烯酸甲酯形成氢键的能力较低,聚甲基丙烯酸甲酯对间苯二酚的吸附量也较小.聚甲基丙烯酸甲酯对上述三种酚类物质吸附性能的差别,进一步证明了聚甲基丙烯酸甲酯对水溶液中酚类物质的氢键吸附机理.利用大孔交联聚甲基丙烯酸甲酯作为氢键受体,可将能提供氢键供体的物质与不能提供氢键供体的物质进行吸附分离,这对有机物特别是天然产物的提取分离有着重要意义.(下转第48页)图4 D 72树脂与咖啡因的氢键作用3 结论D 72树脂不但可以用作离子交换树脂,还可作为氢键供体用作氢键吸附剂,用来分离含有氢键受体和不含氢键受体的化合物,这对有机化合物特别是天然产物的分离提取有着重要意义.参考文献1 何炳林,黄文强.离子交换与吸附树脂.上海:上海科技教育出版社,19952 王庆文,杨玉桓,高鸿宾.有机化学中的氢键问题.天津:天津大学出版社,19933 Grasselli T G .A tlas of Spectral D ata and Physical Constants fo r O rganic Compounds ,2th ed .CRCP ress ,1975(上接第44页)参考文献1 何炳林,黄文强.离子交换与吸附树脂.上海:上海科技教育出版社,19952 徐任生,陈仲良.中草药有效成分提取与分离.上海:上海科学技术出版社,19833 Payne G F ,N inom iya Y .Selective adso rp ti on of so lutes based on hydrogen bonding .Separati on SciT ech ,1990,25:11174 钱庭宝.离子交换剂应用技术.天津:天津科学技术出版社,19845 王庆文,杨玉桓,高鸿宾.有机化学中的氢键问题.天津:天津大学出版社,1993。

一种松香基小分子水凝胶剂及其形成的超分子水凝胶A pine resin-based small molecular hydrogelator and the supramolecular hydrogel formed by itHydrogels are a class of three-dimensional networks thatcan absorb a large amount of water while maintaining their solid-like structure. They have attracted significant attention in various fields, such as biomedical engineering, drug delivery, and tissue engineering, due to their unique properties. In recent years, there has been growinginterest in developing hydrogelators that are derived from natural sources.One particular type of natural material that has shown promise as a hydrogelator is pine resin. A hydrogelator isa compound capable of forming hydrogels through self-assembly. Pine resin, also known as rosin or colophony, is obtained from the sap of various types of pine trees. It consists mainly of resin acids, which are long-chain carboxylic acids.Research has shown that certain small molecules derivedfrom pine resin can act as effective hydrogelators. These small molecules have amphiphilic properties, meaning they possess both hydrophilic and hydrophobic regions. This property enables them to self-assemble in an aqueous environment and form a stable network structure within the gel.The formation of supramolecular hydrogels by these pineresin-derived small molecules involves several steps. First, the small molecules dissolve in an organic solvent such as ethanol or methanol. Then, water is added to the solutionto induce gelation. As water molecules interact with the hydrophilic regions of the small molecules, they disruptthe non-covalent interactions holding the gelator molecules together, leading to gel formation.The resulting supramolecular hydrogels exhibit several advantageous properties for various applications. For example, they have excellent mechanical strength andstability due to the network structure formed byintermolecular interactions between the gelator molecules. The gels also display good biocompatibility and biodegradability, making them suitable for biomedical applications.In addition, the porosity of the hydrogel network can be easily controlled by adjusting the concentration of the gelator or the solvent composition. This tunability allows for the encapsulation and controlled release of bioactive molecules, making these hydrogels promising candidates for drug delivery systems.Furthermore, these pine resin-derived hydrogels have shown potential in tissue engineering. The three-dimensional structure of the hydrogel provides a suitable environment for cell growth and proliferation. It can also mimic the extracellular matrix, facilitating cell adhesion and differentiation.In conclusion, pine resin-based small molecular hydrogelators have demonstrated their ability to form supramolecular hydrogels with unique properties. Thesehydrogels have great potential in various fields, including biomedical engineering, drug delivery, and tissue engineering. Further research and development in this area may lead to exciting advances and applications in the future.三维网络结构的,能吸收大量水分而保持固态结构的凝胶被称为水凝胶,并由其独特的性质在生物医学工程、药物传递和组织工程等领域引起了重要关注。
New1H-Pyrazole-Containing Polyamine Receptors Able ToComplex L-Glutamate in Water at Physiological pH ValuesCarlos Miranda,†Francisco Escartı´,‡Laurent Lamarque,†Marı´a J.R.Yunta,§Pilar Navarro,*,†Enrique Garcı´a-Espan˜a,*,‡and M.Luisa Jimeno†Contribution from the Instituto de Quı´mica Me´dica,Centro de Quı´mica Orga´nica Manuel Lora Tamayo,CSIC,C/Juan de la Cier V a3,28006Madrid,Spain,Departamento de Quı´mica Inorga´nica,Facultad de Quı´mica,Uni V ersidad de Valencia,c/Doctor Moliner50, 46100Burjassot(Valencia),Spain,and Departamento de Quı´mica Orga´nica,Facultad deQuı´mica,Uni V ersidad Complutense de Madrid,A V plutense s/n,28040Madrid,SpainReceived April16,2003;E-mail:enrique.garcia-es@uv.esAbstract:The interaction of the pyrazole-containing macrocyclic receptors3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene1[L1],13,26-dibenzyl-3,6,9,12,13,16,-19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene2[L2],3,9,12,13,16,22,-25,26-octaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene3[L3],6,19-dibenzyl-3,6,9,12,13,-16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene4[L4],6,19-diphenethyl-3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetraene5[L5],and 6,19-dioctyl-3,6,9,12,13,16,19,22,25,26-decaazatricyclo-[22.2.1.111,14]-octacosa-1(27),11,14(28),24-tetra-ene6[L6]with L-glutamate in aqueous solution has been studied by potentiometric techniques.The synthesis of receptors3-6[L3-L6]is described for the first time.The potentiometric results show that4[L4]containing benzyl groups in the central nitrogens of the polyamine side chains is the receptor displaying the larger interaction at pH7.4(K eff)2.04×104).The presence of phenethyl5[L5]or octyl groups6[L6]instead of benzyl groups4[L4]in the central nitrogens of the chains produces a drastic decrease in the stability[K eff )3.51×102(5),K eff)3.64×102(6)].The studies show the relevance of the central polyaminic nitrogen in the interaction with glutamate.1[L1]and2[L2]with secondary nitrogens in this position present significantly larger interactions than3[L3],which lacks an amino group in the center of the chains.The NMR and modeling studies suggest the important contribution of hydrogen bonding andπ-cation interaction to adduct formation.IntroductionThe search for the L-glutamate receptor field has been andcontinues to be in a state of almost explosive development.1 L-Glutamate(Glu)is thought to be the predominant excitatory transmitter in the central nervous system(CNS)acting at a rangeof excitatory amino acid receptors.It is well-known that it playsa vital role mediating a great part of the synaptic transmission.2However,there is an increasing amount of experimentalevidence that metabolic defects and glutamatergic abnormalitiescan exacerbate or induce glutamate-mediated excitotoxic damageand consequently neurological disorders.3,4Overactivation ofionotropic(NMDA,AMPA,and Kainate)receptors(iGluRs)by Glu yields an excessive Ca2+influx that produces irreversible loss of neurons of specific areas of the brain.5There is much evidence that these processes induce,at least in part,neuro-degenerative illnesses such as Parkinson,Alzheimer,Huntington, AIDS,dementia,and amyotrophic lateral sclerosis(ALS).6In particular,ALS is one of the neurodegenerative disorders for which there is more evidence that excitotoxicity due to an increase in Glu concentration may contribute to the pathology of the disease.7Memantine,a drug able to antagonize the pathological effects of sustained,but relatively small,increases in extracellular glutamate concentration,has been recently received for the treatment of Alzheimer disease.8However,there is not an effective treatment for ALS.Therefore,the preparation of adequately functionalized synthetic receptors for L-glutamate seems to be an important target in finding new routes for controlling abnormal excitatory processes.However,effective recognition in water of aminocarboxylic acids is not an easy task due to its zwitterionic character at physiological pH values and to the strong competition that it finds in its own solvent.9†Centro de Quı´mica Orga´nica Manuel Lora Tamayo.‡Universidad de Valencia.§Universidad Complutense de Madrid.(1)Jane,D.E.In Medicinal Chemistry into the Millenium;Campbell,M.M.,Blagbrough,I.S.,Eds.;Royal Society of Chemistry:Cambridge,2001;pp67-84.(2)(a)Standaert,D.G.;Young,A.B.In The Pharmacological Basis ofTherapeutics;Hardman,J.G.,Goodman Gilman,A.,Limbird,L.E.,Eds.;McGraw-Hill:New York,1996;Chapter22,p503.(b)Fletcher,E.J.;Loge,D.In An Introduction to Neurotransmission in Health and Disease;Riederer,P.,Kopp,N.,Pearson,J.,Eds.;Oxford University Press:New York,1990;Chapter7,p79.(3)Michaelis,E.K.Prog.Neurobiol.1998,54,369-415.(4)Olney,J.W.Science1969,164,719-721.(5)Green,J.G.;Greenamyre,J.T.Prog.Neurobiol.1996,48,613-63.(6)Bra¨un-Osborne,H.;Egebjerg,J.;Nielsen,E.O.;Madsen,U.;Krogsgaard-Larsen,P.J.Med.Chem.2000,43,2609-2645and references therein.(7)(a)Shaw,P.J.;Ince,P.G.J.Neurol.1997,244(Suppl2),S3-S14.(b)Plaitakis,A.;Fesdjian,C.O.;Shashidharan,S Drugs1996,5,437-456.(8)Frantz,A.;Smith,A.Nat.Re V.Drug Dico V ery2003,2,9.Published on Web12/30/200310.1021/ja035671m CCC:$27.50©2004American Chemical Society J.AM.CHEM.SOC.2004,126,823-8339823There are many types of receptors able to interact with carboxylic acids and amino acids in organic solvents,10-13yielding selective complexation in some instances.However,the number of reported receptors of glutamate in aqueous solution is very scarce.In this sense,one of the few reports concerns an optical sensor based on a Zn(II)complex of a 2,2′:6′,2′′-terpyridine derivative in which L -aspartate and L -glutamate were efficiently bound as axial ligands (K s )104-105M -1)in 50/50water/methanol mixtures.14Among the receptors employed for carboxylic acid recogni-tion,the polyamine macrocycles I -IV in Chart 1are of particular relevance to this work.In a seminal paper,Lehn et al.15showed that saturated polyamines I and II could exert chain-length discrimination between different R ,ω-dicarboxylic acids as a function of the number of methylene groups between the two triamine units of the receptor.Such compounds were also able to interact with a glutamic acid derivative which has the ammonium group protected with an acyl moiety.15,16Compounds III and IV reported by Gotor and Lehn interact in their protonated forms in aqueous solution with protected N -acetyl-L -glutamate and N -acetyl-D -glutamate,showing a higher stability for the interaction with the D -isomer.17In both reports,the interaction with protected N -acetyl-L -glutamate at physiological pH yields constants of ca.3logarithmic units.Recently,we have shown that 1H -pyrazole-containing mac-rocycles present desirable properties for the binding of dopam-ine.18These polyaza macrocycles,apart from having a highpositive charge at neutral pH values,can form hydrogen bonds not only through the ammonium or amine groups but also through the pyrazole nitrogens that can behave as hydrogen bond donors or acceptors.In fact,Elguero et al.19have recently shown the ability of the pyrazole rings to form hydrogen bonds with carboxylic and carboxylate functions.These features can be used to recognize the functionalities of glutamic acid,the carboxylic and/or carboxylate functions and the ammonium group.Apart from this,the introduction of aromatic donor groups appropriately arranged within the macrocyclic framework or appended to it through arms of adequate length may contribute to the recognition event through π-cation interactions with the ammonium group of L -glutamate.π-Cation interactions are a key feature in many enzymatic centers,a classical example being acetylcholine esterase.20The role of such an interaction in abiotic systems was very well illustrated several years ago in a seminal work carried out by Dougherty and Stauffer.21Since then,many other examples have been reported both in biotic and in abiotic systems.22Taking into account all of these considerations,here we report on the ability of receptors 1[L 1]-6[L 6](Chart 2)to interact with L -glutamic acid.These receptors display structures which differ from one another in only one feature,which helps to obtain clear-cut relations between structure and interaction(9)Rebek,J.,Jr.;Askew,B.;Nemeth,D.;Parris,K.J.Am.Chem.Soc.1987,109,2432-2434.(10)Seel,C.;de Mendoza,J.In Comprehensi V e Supramolecular Chemistry ;Vogtle,F.,Ed.;Elsevier Science:New York,1996;Vol.2,p 519.(11)(a)Sessler,J.L.;Sanson,P.I.;Andrievesky,A.;Kral,V.In SupramolecularChemistry of Anions ;Bianchi,A.,Bowman-James,K.,Garcı´a-Espan ˜a,E.,Eds.;John Wiley &Sons:New York,1997;Chapter 10,pp 369-375.(b)Sessler,J.L.;Andrievsky,A.;Kra ´l,V.;Lynch,V.J.Am.Chem.Soc.1997,119,9385-9392.(12)Fitzmaurice,R.J.;Kyne,G.M.;Douheret,D.;Kilburn,J.D.J.Chem.Soc.,Perkin Trans.12002,7,841-864and references therein.(13)Rossi,S.;Kyne,G.M.;Turner,D.L.;Wells,N.J.;Kilburn,J.D.Angew.Chem.,Int.Ed.2002,41,4233-4236.(14)Aı¨t-Haddou,H.;Wiskur,S.L.;Lynch,V.M.;Anslyn,E.V.J.Am.Chem.Soc.2001,123,11296-11297.(15)Hosseini,M.W.;Lehn,J.-M.J.Am.Chem.Soc.1982,104,3525-3527.(16)(a)Hosseini,M.W.;Lehn,J.-M.Hel V .Chim.Acta 1986,69,587-603.(b)Heyer,D.;Lehn,J.-M.Tetrahedron Lett.1986,27,5869-5872.(17)(a)Alfonso,I.;Dietrich,B.;Rebolledo,F.;Gotor,V.;Lehn,J.-M.Hel V .Chim.Acta 2001,84,280-295.(b)Alfonso,I.;Rebolledo,F.;Gotor,V.Chem.-Eur.J.2000,6,3331-3338.(18)Lamarque,L.;Navarro,P.;Miranda,C.;Ara ´n,V.J.;Ochoa,C.;Escartı´,F.;Garcı´a-Espan ˜a,E.;Latorre,J.;Luis,S.V.;Miravet,J.F.J.Am.Chem.Soc .2001,123,10560-10570.(19)Foces-Foces,C.;Echevarria,A.;Jagerovic,N.;Alkorta,I.;Elguero,J.;Langer,U.;Klein,O.;Minguet-Bonvehı´,H.-H.J.Am.Chem.Soc.2001,123,7898-7906.(20)Sussman,J.L.;Harel,M.;Frolow,F.;Oefner,C.;Goldman,A.;Toker,L.;Silman,I.Science 1991,253,872-879.(21)Dougherty,D.A.;Stauffer,D.A.Science 1990,250,1558-1560.(22)(a)Sutcliffe,M.J.;Smeeton,A.H.;Wo,Z.G.;Oswald,R.E.FaradayDiscuss.1998,111,259-272.(b)Kearney,P.C.;Mizoue,L.S.;Kumpf,R.A.;Forman,J.E.;McCurdy,A.;Dougherty,D.A.J.Am.Chem.Soc.1993,115,9907-9919.(c)Bra ¨uner-Osborne,H.;Egebjerg,J.;Nielsen,E.;Madsen,U.;Krogsgaard-Larsen,P.J.Med.Chem.2000,43,2609-2645.(d)Zacharias,N.;Dougherty,D.A.Trends Pharmacol.Sci.2002,23,281-287.(e)Hu,J.;Barbour,L.J.;Gokel,G.W.J.Am.Chem.Soc.2002,124,10940-10941.Chart 1.Some Receptors Employed for Dicarboxylic Acid and N -AcetylglutamateRecognitionChart 2.New 1H -Pyrazole-Containing Polyamine Receptors Able To Complex L -Glutamate inWaterA R T I C L E SMiranda et al.824J.AM.CHEM.SOC.9VOL.126,NO.3,2004strengths.1[L1]and2[L2]differ in the N-benzylation of the pyrazole moiety,and1[L1]and3[L3]differ in the presence in the center of the polyamine side chains of an amino group or of a methylene group.The receptors4[L4]and5[L5]present the central nitrogens of the chain N-functionalized with benzyl or phenethyl groups,and6[L6]has large hydrophobic octyl groups.Results and DiscussionSynthesis of3-6.Macrocycles3-6have been obtained following the procedure previously reported for the preparation of1and2.23The method includes a first dipodal(2+2) condensation of the1H-pyrazol-3,5-dicarbaldehyde7with the corresponding R,ω-diamine,followed by hydrogenation of the resulting Schiff base imine bonds.In the case of receptor3,the Schiff base formed by condensation with1,5-pentanediamine is a stable solid(8,mp208-210°C)which precipitated in68% yield from the reaction mixture.Further reduction with NaBH4 in absolute ethanol gave the expected tetraazamacrocycle3, which after crystallization from toluene was isolated as a pure compound(mp184-186°C).In the cases of receptors4-6, the precursor R,ω-diamines(11a-11c)(Scheme1B)were obtained,by using a procedure previously described for11a.24 This procedure is based on the previous protection of the primary amino groups of1,5-diamino-3-azapentane by treatment with phthalic anhydride,followed by alkylation of the secondary amino group of1,5-diphthalimido-3-azapentane9with benzyl, phenethyl,or octyl bromide.Finally,the phthalimido groups of the N-alkyl substituted intermediates10a-10c were removed by treatment with hydrazine to afford the desired amines11a-11c,which were obtained in moderate yield(54-63%).In contrast with the behavior previously observed in the synthesis of3,in the(2+2)dipodal condensations of7with 3-benzyl-,3-phenethyl-,and3-octyl-substituted3-aza-1,5-pentanediamine11a,11b,and11c,respectively,there was not precipitation of the expected Schiff bases(Scheme1A). Consequently,the reaction mixtures were directly reduced in situ with NaBH4to obtain the desired hexaamines4-6,which after being carefully purified by chromatography afforded purecolorless oils in51%,63%,and31%yield,respectively.The structures of all of these new cyclic polyamines have been established from the analytical and spectroscopic data(MS(ES+), 1H and13C NMR)of both the free ligands3-6and their corresponding hydrochloride salts[3‚4HCl,4‚6HCl,5‚6HCl, and6‚6HCl],which were obtained as stable solids following the same procedure previously reported18for1‚6HCl and2‚6HCl.As usually occurs for3,5-disubstituted1H-pyrazole deriva-tives,either the free ligands3-6or their hydrochlorides show very simple1H and13C NMR spectra,in which signals indicate that,because of the prototropic equilibrium of the pyrazole ring, all of these compounds present average4-fold symmetry on the NMR scale.The quaternary C3and C5carbons appear together,and the pairs of methylene carbons C6,C7,and C8are magnetically equivalent(see Experimental Section).In the13C NMR spectra registered in CDCl3solution, significant differences can be observed between ligand3,without an amino group in the center of the side chain,and the N-substituted ligands4-6.In3,the C3,5signal appears as a broad singlet.However,in4-6,it almost disappears within the baseline of the spectra,and the methylene carbon atoms C6and C8experience a significant broadening.Additionally,a remark-able line-broadening is also observed in the C1′carbon signals belonging to the phenethyl and octyl groups of L5and L6, respectively.All of these data suggest that as the N-substituents located in the middle of the side chains of4-6are larger,the dynamic exchange rate of the pyrazole prototropic equilibrium is gradually lower,probably due to a relation between proto-tropic and conformational equilibria.Acid-Base Behavior.To follow the complexation of L-glutamate(hereafter abbreviated as Glu2-)and its protonated forms(HGlu-,H2Glu,and H3Glu+)by the receptors L1-L6, the acid-base behavior of L-glutamate has to be revisited under the experimental conditions of this work,298K and0.15mol dm-3.The protonation constants obtained,included in the first column of Table1,agree with the literature25and show that the zwitterionic HGlu-species is the only species present in aqueous solution at physiological pH values(Scheme2and Figure S1of Supporting Information).Therefore,receptors for(23)Ara´n,V.J.;Kumar,M.;Molina,J.;Lamarque,L.;Navarro,P.;Garcı´a-Espan˜a,E.;Ramı´rez,J.A.;Luis,S.V.;Escuder,.Chem.1999, 64,6137-6146.(24)(a)Yuen Ng,C.;Motekaitis,R.J.;Martell,A.E.Inorg.Chem.1979,18,2982-2986.(b)Anelli,P.L.;Lunazzi,L.;Montanari,F.;Quici,.Chem.1984,49,4197-4203.Scheme1.Synthesis of the Pyrazole-Containing MacrocyclicReceptorsNew1H-Pyrazole-Containing Polyamine Receptors A R T I C L E SJ.AM.CHEM.SOC.9VOL.126,NO.3,2004825glutamate recognition able to address both the negative charges of the carboxylate groups and the positive charge of ammonium are highly relevant.The protonation constants of L 3-L 6are included in Table 1,together with those we have previously reported for receptors L 1and L 2.23A comparison of the constants of L 4-L 6with those of the nonfunctionalized receptor L 1shows a reduced basicity of the receptors L 4-L 6with tertiary nitrogens at the middle of the polyamine bridges.Such a reduction in basicity prevented the potentiometric detection of the last protonation for these ligands in aqueous solution.A similar reduction in basicity was previously reported for the macrocycle with the N -benzylated pyrazole spacers (L 2).23These diminished basicities are related to the lower probability of the tertiary nitrogens for stabilizing the positive charges through hydrogen bond formation either with adjacent nonprotonated amino groups of the molecule or with water molecules.Also,the increase in the hydrophobicity of these molecules will contribute to their lower basicity.The stepwise basicity constants are relatively high for the first four protonation steps,which is attributable to the fact that these protons can bind to the nitrogen atoms adjacent to the pyrazole groups leaving the central nitrogen free,the electrostatic repulsions between them being therefore of little significance.The remaining protonation steps will occur in the central nitrogen atom,which will produce an important increase in the electrostatic repulsion in the molecule and therefore a reduction in basicity.As stated above,the tertiary nitrogen atoms present in L 4-L 6will also contribute to this diminished basicity.To analyze the interaction with glutamic acid,it is important to know the protonation degree of the ligands at physiological pH values.In Table 2,we have calculated the percentages ofthe different protonated species existing in solution at pH 7.4for receptors L 1-L 6.As can be seen,except for the receptor with the pentamethylenic chains L 3in which the tetraprotonated species prevails,all of the other systems show that the di-and triprotonated species prevail,although to different extents.Interaction with Glutamate.The stepwise constants for the interaction of the receptors L 1-L 6with glutamate are shown in Table 3,and selected distribution diagrams are plotted in Figure 1A -C.All of the studied receptors interact with glutamate forming adduct species with protonation degrees (j )which vary between 8and 0depending on the system (see Table 3).The stepwise constants have been derived from the overall association constants (L +Glu 2-+j H +)H j LGlu (j -2)+,log j )provided by the fitting of the pH-metric titration curves.This takes into account the basicities of the receptors and glutamate (vide supra)and the pH range in which a given species prevails in solution.In this respect,except below pH ca.4and above pH 9,HGlu -can be chosen as the protonated form of glutamate involved in the formation of the different adducts.Below pH 4,the participation of H 2Glu in the equilibria has also to be considered (entries 9and 10in Table 3).For instance,the formation of the H 6LGlu 4+species can proceed through the equilibria HGlu -+H 5L 5+)H 6LGlu 4+(entry 8,Table 3),and H 2Glu +H 4L 4+)H 6LGlu 4(entry 9Table 3),with percentages of participation that depend on pH.One of the effects of the interaction is to render somewhat more basic the receptor,and somewhat more acidic glutamic acid,facilitating the attraction between op-positely charged partners.A first inspection of Table 3and of the diagrams A,B,and C in Figure 1shows that the interaction strengths differ markedly from one system to another depending on the structural features of the receptors involved.L 4is the receptor that presents the highest capacity for interacting with glutamate throughout all of the pH range explored.It must also be remarked that there are not clear-cut trends in the values of the stepwise constants as a function of the protonation degree of the receptors.This suggests that charge -charge attractions do not play the most(25)(a)Martell,E.;Smith,R.M.Critical Stability Constants ;Plenum:NewYork,1975.(b)Motekaitis,R.J.NIST Critically Selected Stability Constants of Metal Complexes Database ;NIST Standard Reference Database,version 4,1997.Table 1.Protonation Constants of Glutamic Acid and Receptors L 1-L 6Determined in NaCl 0.15mol dm -3at 298.1KreactionGluL 1aL 2aL 3bL 4L 5L 6L +H )L H c 9.574(2)d 9.74(2)8.90(3)9.56(1)9.25(3)9.49(4)9.34(5)L H +H )L H 2 4.165(3)8.86(2)8.27(2)8.939(7)8.38(3)8.11(5)8.13(5)L H 2+H )L H 3 2.18(2)7.96(2) 6.62(3)8.02(1) 6.89(5)7.17(6)7.46(7)L H 3+H )L H 4 6.83(2) 5.85(4)7.63(1) 6.32(5) 6.35(6) 5.97(8)L H 4+H )L H 5 4.57(3) 3.37(4) 2.72(8) 2.84(9) 3.23(9)L H 5+H )L H 6 3.18(3) 2.27(6)∑log K H n L41.135.334.233.634.034.1aTaken from ref 23.b These data were previously cited in a short communication (ref 26).c Charges omitted for clarity.d Values in parentheses are the standard deviations in the last significant figure.Scheme 2.L -Glutamate Acid -BaseBehaviorTable 2.Percentages of the Different Protonated Species at pH 7.4H 1L aH 2LH 3LH 4LL 11186417L 21077130L 3083458L 4083458L 51154323L 6842482aCharges omitted for clarity.A R T I C L E SMiranda et al.826J.AM.CHEM.SOC.9VOL.126,NO.3,2004outstanding role and that other forces contribute very importantly to these processes.26However,in systems such as these,which present overlapping equilibria,it is convenient to use conditional constants because they provide a clearer picture of the selectivity trends.27These constants are defined as the quotient between the overall amounts of complexed species and those of free receptor and substrate at a given pH[eq1].In Figure2are presented the logarithms of the effective constants versus pH for all of the studied systems.Receptors L1and L2with a nonfunctionalized secondary amino group in the side chains display opposite trend from all other receptors. While the stability of the L1and L2adducts tends to increase with pH,the other ligands show a decreasing interaction. Additionally,L1and L2present a close interaction over the entire pH range under study.The tetraaminic macrocycle L3is a better(26)Escartı´,F.;Miranda,C.;Lamarque,L.;Latorre,J.;Garcı´a-Espan˜a,E.;Kumar,M.;Ara´n,V.J.;Navarro,mun.2002,9,936-937.(27)(a)Bianchi,A.;Garcı´a-Espan˜a,c.1999,12,1725-1732.(b)Aguilar,J.A.;Celda,B.;Garcı´a-Espan˜a,E.;Luis,S.V.;Martı´nez,M.;Ramı´rez,J.A.;Soriano,C.;Tejero,B.J.Chem.Soc.,Perkin Trans.22000, 7,1323-1328.Table3.Stability Constants for the Interaction of L1-L6with the Different Protonated Forms of Glutamate(Glu) entry reaction a L1L2L3L4L5L6 1Glu+L)Glu L 3.30(2)b 4.11(1)2HGlu+L)HGlu L 3.65(2) 4.11(1) 3.68(2) 3.38(4) 3Glu+H L)HGlu L 3.89(2) 4.48(1) 3.96(2) 3.57(4) 4HGlu+H L)H2Glu L 3.49(2) 3.89(1) 2.37(4) 3.71(2)5HGlu+H2L)H3Glu L 3.44(2) 3.73(1) 2.34(3) 4.14(2) 2.46(4) 2.61(7) 6HGlu+H3L)H4Glu L 3.33(2) 3.56(2) 2.66(3) 4.65(2) 2.74(3) 2.55(7) 7HGlu+H4L)H5Glu L 3.02(2) 3.26(2) 2.58(3) 4.77(2) 2.87(3) 2.91(5) 8HGlu+H5L)H6Glu L 3.11(3) 3.54(2) 6.76(3) 4.96(3) 4.47(3) 9H2Glu+H4L)H6Glu L 2.54(3) 3.05(2) 3.88(2) 5.35(3) 3.66(4) 3.56(3) 10H2Glu+H5L)H7Glu L 2.61(6) 2.73(4) 5.51(3) 3.57(4) 3.22(8) 11H3Glu+H4L)H7Glu L 4.82(2) 4.12(9)a Charges omitted for clarity.b Values in parentheses are standard deviations in the last significantfigure.Figure1.Distribution diagrams for the systems(A)L1-glutamic acid, (B)L4-glutamic acid,and(C)L5-glutamicacid.Figure2.Representation of the variation of K cond(M-1)for the interaction of glutamic acid with(A)L1and L3,(B)L2,L4,L5,and L6.Initial concentrations of glutamate and receptors are10-3mol dm-3.Kcond)∑[(H i L)‚(H j Glu)]/{∑[H i L]∑[H j Glu]}(1)New1H-Pyrazole-Containing Polyamine Receptors A R T I C L E SJ.AM.CHEM.SOC.9VOL.126,NO.3,2004827receptor at acidic pH,but its interaction markedly decreases on raising the pH.These results strongly suggest the implication of the central nitrogens of the lateral polyamine chains in the stabilization of the adducts.Among the N-functionalized receptors,L4presents the largest interaction with glutamate.Interestingly enough,L5,which differs from L4only in having a phenethyl group instead of a benzyl one,presents much lower stability of its adducts.Since the basicity and thereby the protonation states that L4and L5 present with pH are very close,the reason for the larger stability of the L4adducts could reside on a better spatial disposition for formingπ-cation interactions with the ammonium group of the amino acid.In addition,as already pointed out,L4presents the highest affinity for glutamic acid in a wide pH range,being overcome only by L1and L2at pH values over9.This observation again supports the contribution ofπ-cation inter-actions in the system L4-glutamic because at these pH values the ammonium functionality will start to deprotonate(see Scheme2and Figure1B).Table4gathers the percentages of the species existing in equilibria at pH7.4together with the values of the conditional constant at this pH.In correspondence with Figure1A,1C and Figure S2(Supporting Information),it can be seen that for L1, L2,L5,and L6the prevailing species are[H2L‚HGlu]+and[H3L‚HGlu]2+(protonation degrees3and4,respectively),while for L3the main species are[H3L‚HGlu]+and[H4L‚HGlu]2+ (protonation degrees4and5,respectively).The most effective receptor at this pH would be L4which joins hydrogen bonding, charge-charge,andπ-cation contributions for the stabilization of the adducts.To check the selectivity of this receptor,we have also studied its interaction with L-aspartate,which is a competitor of L-glutamate in the biologic receptors.The conditional constant at pH7.4has a value of3.1logarithmic units for the system Asp-L4.Therefore,the selectivity of L4 for glutamate over aspartate(K cond(L4-glu)/K cond(L4-asp))will be of ca.15.It is interesting to remark that the affinity of L4 for zwiterionic L-glutamate at pH7.4is even larger than that displayed by receptors III and IV(Chart1)with the protected dianion N-acetyl-L-glutamate lacking the zwitterionic charac-teristics.Applying eq1and the stability constants reported in ref17,conditional constants at pH7.4of 3.24and 2.96 logarithmic units can be derived for the systems III-L-Glu and IV-L-Glu,respectively.Molecular Modeling Studies.Molecular mechanics-based methods involving docking studies have been used to study the binding orientations and affinities for the complexation of glutamate by L1-L6receptors.The quality of a computer simulation depends on two factors:accuracy of the force field that describes intra-and intermolecular interactions,and an adequate sampling of the conformational and configuration space of the system.28The additive AMBER force field is appropriate for describing the complexation processes of our compounds,as it is one of the best methods29in reproducing H-bonding and stacking stabiliza-tion energies.The experimental data show that at pH7.4,L1-L6exist in different protonation states.So,a theoretical study of the protonation of these ligands was done,including all of the species shown in5%or more abundance in the potentiometric measurements(Table4).In each case,the more favored positions of protons were calculated for mono-,di-,tri-,and tetraprotonated species.Molecular dynamics studies were performed to find the minimum energy conformations with simulated solvent effects.Molecular modeling studies were carried out using the AMBER30method implemented in the Hyperchem6.0pack-age,31modified by the inclusion of appropriate parameters. Where available,the parameters came from analogous ones used in the literature.32All others were developed following Koll-man33and Hopfinger34procedures.The equilibrium bond length and angle values came from experimental values of reasonable reference compounds.All of the compounds were constructed using standard geometry and standard bond lengths.To develop suitable parameters for NH‚‚‚N hydrogen bonding,ab initio calculations at the STO-3G level35were used to calculate atomic charges compatible with the AMBER force field charges,as they gave excellent results,and,at the same time,this method allows the study of aryl-amine interactions.In all cases,full geometry optimizations with the Polak-Ribiere algorithm were carried out,with no restraints.Ions are separated far away and well solvated in water due to the fact that water has a high dielectric constant and hydrogen bond network.Consequently,there is no need to use counteri-ons36in the modelization studies.In the absence of explicit solvent molecules,a distance-dependent dielectric factor quali-tatively simulates the presence of water,as it takes into account the fact that the intermolecular electrostatic interactions should vanish more rapidly with distance than in the gas phase.The same results can be obtained using a constant dielectric factor greater than1.We have chosen to use a distance-dependent dielectric constant( )4R ij)as this was the method used by Weiner et al.37to develop the AMBER force field.Table8 shows the theoretical differences in protonation energy(∆E p) of mono-,bi-,and triprotonated hexaamine ligands,for the (28)Urban,J.J.;Cronin,C.W.;Roberts,R.R.;Famini,G.R.J.Am.Chem.Soc.1997,119,12292-12299.(29)Hobza,P.;Kabelac,M.;Sponer,J.;Mejzlik,P.;Vondrasek,put.Chem.1997,18,1136-1150.(30)Cornell,W.D.;Cieplak,P.;Bayly,C.I.;Gould,I.R.;Merz,K.M.,Jr.;Ferguson,D.M.;Spelmeyer,D.C.;Fox,T.;Caldwell,J.W.;Kollman,P.A.J.Am.Chem.Soc.1995,117,5179-5197.(31)Hyperchem6.0(Hypercube Inc.).(32)(a)Fox,T.;Scanlan,T.S.;Kollman,P.A.J.Am.Chem.Soc.1997,119,11571-11577.(b)Grootenhuis,P.D.;Kollman,P.A.J.Am.Chem.Soc.1989,111,2152-2158.(c)Moyna,G.;Hernandez,G.;Williams,H.J.;Nachman,R.J.;Scott,put.Sci.1997,37,951-956.(d)Boden,C.D.J.;Patenden,put.-Aided Mol.Des.1999, 13,153-166.(33)/amber.(34)Hopfinger,A.J.;Pearlstein,put.Chem.1984,5,486-499.(35)Glennon,T.M.;Zheng,Y.-J.;Le Grand,S.M.;Shutzberg,B.A.;Merz,K.M.,put.Chem.1994,15,1019-1040.(36)Wang,J.;Kollman,P.A.J.Am.Chem.Soc.1998,120,11106-11114.Table4.Percentages of the Different Protonated Adducts[HGlu‚H j L](j-1)+,Overall Percentages of Complexation,andConditional Constants(K Cond)at pH7.4for the Interaction ofGlutamate(HGlu-)with Receptors L1-L6at Physiological pH[H n L‚HGlu]an)1n)2n)3n)4∑{[H n L‚HGlu]}K cond(M-1)L13272353 2.44×103L2947763 4.12×103L31101324 3.99×102L423737581 2.04×104L51010222 3.51×102L6121224 3.64×102a Charges omitted for clarity.A R T I C L E S Miranda et al. 828J.AM.CHEM.SOC.9VOL.126,NO.3,2004。
Two novel quadruple hydrogen-bonding motifs:the formation of supramolecular polymers,vesicles,and organogelsPing Du,Gui-Tao Wang,Xin Zhao *,Guang-Yu Li,Xi-Kui Jiang,Zhan-Ting Li *State Key Laboratory of Bioorganic and Natural Products Chemistry,Shanghai Institute of Organic Chemistry,Chinese Academy of Sciences,345Lingling Lu,Shanghai 200032,Chinaa r t i c l e i n f o Article history:Received 8September 2009Revised 21October 2009Accepted 27October 2009Available online 30October 2009a b s t r a c tTwo new hydrazide-based quadruple hydrogen-bonding motifs are described.Dipodals based on these two motifs are revealed to form supramolecular polymers,which can further aggregate to form vesicles and/or organogels in hydrocarbons.The quadruple hydrogen-bonding motifs are characterized by the X-ray diffraction and (2D)1H NMR experiments,while the vesicles and organogels are evidenced by SEM,AFM,TEM,and fluorescent microscopy.Ó2009Elsevier Ltd.All rights reserved.Hydrogen bonding is the most reversible noncovalent forces for forming well-defined supramolecular entities.In the past decades,the quadruple hydrogen-bonding motifs have received consider-able attention due to their generally increased stability compared to the double or triple hydrogen-bonding motifs.1Moreover,this series of binding patterns has been widely utilized to construct dy-namic supramolecular polymers.2We previously reported the self-assembly of a series of stable quadruple hydrogen-bonding pattern from complementary aromatic hydrazide-derived monomers.1h More recently,3we also constructed a variety of foldamers from aromatic hydrazide oligomers by using intramolecular hydrogen bonding as the driving force.We herein report two novel quadru-ple hydrogen-bonding motifs,whose monomers dimerize by adopting a twisted or perpendicular conformation.Notably,hydro-gen-bonded supramolecular polymers constructed based on these motifs can self-assemble into both vesicles and organogels.4Different from their aryl amide analogues,which usually form single C @O ÁÁÁH–N hydrogen-bonding chains,1,2-dibenzoylhydra-zines generate stronger double C @O ÁÁÁH–N hydrogen-bonding chains.5,6We therefore synthesized compounds 1a ,1b ,2a ,2b ,and 3a–c .We envisioned that 1a and 1b would form a homodimer that is stabilized by four C @O ÁÁÁH–N hydrogen bonds,while the dipodals might form supramolecular polymers that are linked by this motif.The two outside N H units are locked by intramolecular six-membered O ÁÁÁH–N hydrogen bonds,7which should suppress the formation of the intermolecular hydrogen bonding and thus improve their assembling selectivity.The crystal structure of 1a exhibited a dimeric motif that is sta-bilized by two pairs of C @O ÁÁÁH–N hydrogen bonds (d =2.03and 2.11Å)(Fig.1a).Notably,only one of the two iso -butoxy O atoms was engaged in the intramolecular hydrogen bonding,even thoughN ONH H O HN O N H O OR 2HNO N ON N H H O OOR 2OO OR 1OR 1NO N H 2OO N H OORROOCH 2CH(CH 3)21a :R =CH 2CH(CH 3)21b :R =n-C 8H 17OON NH N N ORO ROOOHN O HNON N H H OO OROR 2a :R =CH 2CH(CH 3)22b :R =n-C 8H 17H 2H 3a :R 1=R 2=CH 2CH(CH 3)23b :R 1=CH 2CH(CH 3)2,R 2=n-C 8H 173c :R 1=R 2=n-C 8H 17H 1H 1H 1H 4H 5H 6H 3H 7H 80040-4039/$-see front matter Ó2009Elsevier Ltd.All rights reserved.doi:10.1016/j.tetlet.2009.10.115*Corresponding authors.Tel.:+862154925023;fax:+862164166128(X.Z.).E-mail addresses:xzhao@ (X.Zhao),ztli@ (Z.-T.Li).Tetrahedron Letters 51(2010)188–191Contents lists available at ScienceDirectTetrahedron Lettersj ou r na l h om e pa ge :w w w.e lse v ie r.c om /lo c at e /t et l et四重图形it is very stable and occurs even in aqueous media.7,8The two cen-tral benzene rings of the dimer stacked in a face-to-face manner,which should also stabilize the hydrogen-bonding motif (Fig.2a).The neighboring dimers were further connected with two strong C @O ÁÁÁH–N hydrogen bonds (d =2.05Å),while the peripheral ben-zene rings also stacked (not shown).The crystal structure of 1b revealed another quadruple hydro-gen-bonding motif (Fig.1b).The two monomers were arranged perpendicularly (Fig.2b).The N–H ÁÁÁO distances were different slightly due to the unsymmetry of the arrangement.The octoxy O atoms were all engaged in intramolecular hydrogen bonding,while the octyl chains stacked with their counterparts of neighbor-ing molecules (not shown).Different from that of 1a ,no aromatic stacking or other intermolecular hydrogen bonding was observed for 1b .It seems that the larger peripheral aliphatic groups played a key role in promoting the formation of this pounds 1a and 1b are similar in shape to the V-styled N 1,N 3-bis(6-acylami-nopyridin-2-yl)isophthalamides that can bind barbiturates via six hydrogen bonds.9,10However,they form homodimers due to their self-complementary feature.The crystal structure of 2a (Fig.3a)revealed the formation of a stair-styled supramolecular polymer that was mediated by a qua-druple hydrogen-bonding motif very similar to that of 1a .The cen-tral benzene rings stacked with each other,while the peripheral benzene rings stacked alternately with those of the neighboring polymeric chains.The crystal structure of 2b exhibited another kind of supramolecular polymer (Fig.1d)formed through a hydro-gen-binding motif similar to that of 1b (Fig.2b).No stacking or other intermolecular hydrogen bonds were observed in the pack-ing structure.All the octyl chains were arranged along the poly-meric framework and stabilized the packing structure through intermolecular van der Waals force.1H NMR experiments in CDCl 3supported that the quadruple hydrogen-bonding motif also exists in solution.Diluting the solu-tion of 1a and 1b in CDCl 3from 64or 100mM to 0.25mM caused the H-1signal (see the structures)to shift upfield by 0.83or1.05ppm,indicating that it was strongly involved in intermolecu-lar hydrogen bonding.In contrast,the H-2signal was shifted downfield by only 0.24or 0.26ppm.The results were consistent with the above-mentioned X-ray results.By fitting the plots of D d versus the concentration to a nonlinear equation of the 1:1binding model,association constants (K assoc )of 1a–1a and 1b–1b were derived to be 600and 480M À1.11The values are modest,pos-sibly due to the twisting of the framework.2D NOESY experiment for 2b in CDCl 3revealed NOEs between the central ethylene hydrogen (H-3)and the H-4,H-5,and H-6atoms (see the structure),but not the H-2,H-7,or H-8atom,indicating that the peripheral aromatic units were confined and did not rotate around the inner-located (Ar)C–C(@O)bond.Be-cause the distances between H-3,H-4,H-5,and H-6in thecrystalFigure 1.The crystal structures of (a)1a and (b)1b (only the amide hydrogen atoms are shown for clarity).Figure 3.The quadruply hydrogen-bonded supramolecular polymers of (a)2a and (b)2b in the crystal structures (only the amide hydrogen atoms are shown for clarity).The single crystal of 2a was grown by slow evaporation of its methanol solution and that of 2b was grown by diffusing acetonitrile to its DMF solution.P.Du et al./Tetrahedron Letters 51(2010)188–191189structure of2a are generally shorter than the corresponding ones in the crystal structure of2b,these results evidenced that in solu-tion2b thefirst dimeric motif should also form.Diffusion-ordered 2D NMR(DOSY)experiments were further preformed for1b,2b, and3c at two different concentrations,12giving the corresponding diffusion coefficients of2.6Â10À9(1b,10mM),2.5Â10À9(1b, 100mM), 5.0Â10À9(2b,10mM), 6.0Â10À10(2b,100mM), 5.1Â10À9(3c,10mM),and1.2Â10À9(3c,30mM)m2/s.The val-ues of1b are close,implying that it formed a simple dimer at both concentrations.In contrast,the values of2b and3c at higher con-centration were remarkably smaller than those at lower concentra-tion,clearly supporting that the dipodal compounds could form supramolecular polymers.At higher concentrations,they had a higher degree of polymerization,a larger size,and thus a smaller diffusion coefficient.The values of isomers2b and3c at10mM are comparable,indicating that their degree of polymerization was comparable as a result of formatting the identical hydrogen bonding motif.Since the new dimeric segments have a relatively large aromatic framework,we further investigated the potential of the supramo-lecular polymers to generate ordered structures in nonpolar hydro-carbons,including n-hexane,n-octane,n-dodecane,decalin, tetralin,benzene,and toluene,in which the hydrogen bonding and aromatic stacking should be enhanced.At room temperature, all the dipodals displayed low solubility in these solvents.Upon heating,the solubility of2a,2b,and3a in decalin was increased considerably(>1.0mM).SEM images showed that all these com-pounds formed spherical vesicles with average diameters of ca.0.6,1.0,and1.4m(Fig.4a and b),respectively.SEM images also indicated that2b formed vesicles in n-dodecane(1mM),but the vesicles were generally separate.In all the hydrocarbons,1a and 1b did not form vesicles.Adding1b to the decalin solution of2b reduced the capacity of2b of forming vesicles,and4equiv of1b destroyed the vesicles completely.Adding chloroform to the deca-line solution also suppressed the formation of the vesicles and when its content was40%,no spherical structures were observed from the SEM images.Adding tetralin to the above-mentioned solutions in decalin also weakened,and at25%,it prevented the formation of vesicles.These results indicated that the formation of the supramolecular polymers and their aggregation were the keys for the generation of the vesicles:The addition of1b weak-ened the supramolecular polymers,while the addition of chloro-form and tetralin,which might competitively stack with the aromatic units of the dipodals,reduced the aggregation of the supramolecular polymers.13AFM also supported that2a,2b,and3a self-assembled into ves-icles in decalin(see the Supplementary data).The diameter/height ratio of the vesicles was5–15,evidencing their hollowness.Upon being transferred onto the surface,the vesicles were dried due to the evaporation of the entrapped solvent,leaving them likeflat balls.Thefluorescence micrographs also supported the hollowness. As shown for2b(Fig.4c),the light spots all exhibit obvious lumi-nance differences between the outer rings and inner areas,3b sug-gesting that they were hollow andfilled with the solvent in solution.When they were transferred onto the surface,the en-trapped solvent was evaporated,leading to the difference in thick-ness and luminance.TEM also evidenced the hollowness of the vesicles.Figure4d shows the image of a vesicle of2b,which had an average wall thickness of ca.2.6nm.The crystal structures of2a and2b revealed that the width of their aromatic segments was ca.2.0nm.Due to their dynamic feature,the supramolecular polymers should exhibit a larger apparent thickness.Therefore,this result supports that the vesicles might have a monolayer structure that was generated by the aggregation of the polymeric frameworks’(Fig.5).The mole-cules should also be movable in the membranes.Thus we propose that the two hydrogen-bonding motifs shown in Figure2should co-exist in the vesicles.Compounds1a,1b,2a,and3a did not gelate any of the above-mentioned solvents.In contrast,2b gelated the mixtures of decalin and tetralin when the content of tetralin was20–40%,while3b and 3c gelated both of them and their mixtures(see the Supplementary data for details).The long octyl chains should contribute essen-tially by increasing the solubility and promoting the formation of the entangled networks.14Adding1b to the above-mentioned organogels could cause the latter to turn to solution,againsupport-Figure4.SEM images of the samples of(a)2a(1.0mM)and(b)2b(1.0mM),(c)thefluorescence micrograph of the sample of2b(20mM),and d)TEM image of the sample of 2b(0.4mM)in decalin.190P.Du et al./Tetrahedron Letters51(2010)188–191ing that the supramolecular polymers accounted for the gelation.SEM images showed that all the gels exhibited fibrous structures (see the Supplementary data ),while concentration gradient exper-iments revealed that the fibrils could be formed by fusion of the smaller vesicles.In conclusion,two novel quadruple hydrogen-bonding motifs have been developed from aromatic hydrazide-based monomers,which can be utilized to construct supramolecular polymers.Although the stability of the novel motifs is modest,the supramo-lecular polymers exhibit unique self-assembling property,that is,forming both vesicles and organogels in hydrocarbons.Because the binding segments consist of three benzene units,they may stack to stabilize the intermolecular hydrogen bonding in polar media,15which will be exploited in the future.AcknowledgmentsWe thank NSFC (Nos.20942004,20621062,20672137,20732007,20872167),NBRP (2007CB808001)and the Science and Technology Commission of Shanghai Municipality (09XD1405300)for financial support.Supplementary dataCrystallographic data (excluding structure factors)for struc-tures 1a,1b,2a,and 2b in this Letter have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication DC 746809,725875,746808,and 725874,respectively.Copies of the data can be obtained free of charge,on application to CCDC,12Union Road,Cambridge CB21EZ,UK (fax:t44(0)1223336033or e-mail:deposit@).Syn-thesis and characterizations,selected (2D)1H and 13C NMR spectra,additional AFM and SEM images,gelation results,typical procedure for the evaluation of association constants.Supplementary data associated with this article can be found,in the online version,at doi:10.1016/j.tetlet.2009.10.115.References and notes1.For representative examples,see:(a)Beijer,F.H.;Kooijman,H.;Spek,A.L.;Sijbesma,R.P.;Meijer,E.W.Angew.Chem.,Int.Ed.1998,37,75–78;(b)Beijer,F.H.;Sijbesma,R.P.;Kooijman,H.;Spek,A.L.;Meijer,E.W.J.Am.Chem.Soc.1998,120,6761–6769;(c)Corbin,P.S.;Zimmerman,S.C.J.Am.Chem.Soc.1998,120,9710–9711;(d)Corbin,P.S.;Zimmerman,S.C.J.Am.Chem.Soc.2000,122,3779–3780;(e)Lüning,U.;Kühl,C.Tetrahedron Lett.1998,39,5735–5738;(f)Taubitz,J.;Lüning,U.;Grotemeyer,mun.2004,2400–2401;(g)Gong,B.;Yan,Y.;Zeng,H.;Skrzypczak-Jankunn,E.;Kim,Y.W.;Zhu,J.;Ickes,H.J.Am.Chem.Soc.1999,121,5607–5608;(h)Zhao,X.;Wang,X.-Z.;Jiang,X.-K.;Chen,Y.-Q.;Li,Z.-T.;Chen,G.-J.J.Am.Chem.Soc.2003,125,15128–15139;(i)Sun,H.;Steeb,J.;Kaifer,A.E.J.Am.Chem.Soc.2006,128,2820–2821;(j)Yang,Y.;Yan,H.-J.;Chen,C.-F.;Wan,.Lett.2007,9,4991–4994.2.For representative examples,see:(a)Hirschberg,J.H.K.K.;Brunsveld,L.;Ramzi,A.;Vekemans,J.A.J.M.;Sijbesma,R.P.;Meijer,E.W.Nature 2000,407,167–170;(b)Scherman,O.A.;Ligthart,G.B.W.L.;Sijbesma,R.P.;Meijer,E.W.Angew.Chem.,Int.Ed.2006,45,2072–2076;(c)Park,T.;Zimmerman,S.C.;Nakashima,S.J.Am.Chem.Soc.2005,127,6520–6521;(d)Park,T.;Zimmerman,S.C.;Ong,H.C.;Todd,E.M.;Kuykendall,D.W.;Quansah,K.PMSE Prepr.2007,96,138–139;(e)Roland,J.T.;Guan,Z.J.Am.Chem.Soc.2004,126,14328–14329;(f)Kushner,A.M.;Vossler,J.D.;Williams,G.A.;Guan,Z.J.Am.Chem.Soc.2009,131,8766–8768.3.(a)Li,Z.-T.;Hou,J.-L.;Li,C.Acc.Chem.Res.2008,41,1343–1353;(b)Cai,W.;Wang,G.-T.;Xu,Y.-X.;Jiang,X.-K.;Li,Z.-T.J.Am.Chem.Soc.2008,130,6936–6937;(c)Cai,W.;Wang,G.-T.;Du,P.;Wang,R.-X.;Jiang,X.-K.;Li,Z.-T.J.Am.Chem.Soc.2008,130,13450–13459.4.(a)Yoshikawa,I.;Sawayama,J.;Araki,K.Angew.Chem.,Int.Ed.2008,47,1038–1041;(b)Kolomiets,E.;Buhler,E.;Candau,S.J.;Lehn,J.-M.Macromolecules 2006,39,1173–1181;(c)Yang,Y.;Chen,T.;Xiang,J.-F.;Yan,H.-J.;Chen,C.-F.;Wan,L.-J.Chem.Eur.J.2008,14,5742–5746;(d)Ge,Z.;Hu,J.;Huang,F.;Liu,S.Angew.Chem.,Int.Ed.2009,48,1798–1802.5.Raj,S.S.S.;Yamin,B.M.;Boshaala,A.M.A.;Tarafder,M.T.H.;Crouse,K.A.;Fun,H.-K.Acta Crystallogr.,Sect.C 2000,56,1011–1012.6.Zhu,Y.-Y.;Wu,J.;Li,C.;Zhu,J.;Hou,J.-L.;Li,C.-Z.;Jiang,X.-K.;Li,Z.-T.Cryst.Growth Des.2007,7,1490–1496.7.(a)Gong,B.Chem.Eur.J.2001,7,4336–4342;(b)Huc,.Chem.2004,17–29;(c)Li,Z.-T.;Hou,J.-L.;Li,C.;Yi,H.-P.Chem.-Asian J.2006,1,766–778.8.Yi,H.-P.;Wu,J.;Ding,K.-L.;Jiang,X.-K.;Li,.Chem.2007,72,870–877.9.Chang,S.K.;Van Engen,D.;Fan,E.;Hamilton,A.D.J.Am.Chem.Soc.1991,113,7640–7645.10.Berl,V.;Schmutz,M.;Krische,M.J.;Khoury,R.G.;Lehn,J.-M.Chem.Eur.J.2002,8,1227–1244.11.Conners,K. A.Binding Constants:The Measurement of Molecular Complex Stability ;Wiley:New York,1987.12.(a)Dobrawa,R.;Lysetska,M.;Ballester,P.;Grne,M.;Würthner, F.Macromolecules 2005,38,1315–1325;(b)Zhu,Y.-Y.;Li,C.;Li,G.-Y.;Jiang,X.-K.;Li,.Chem.2008,73,1745–1751.13.Nelson,J.C.;Saven,J.G.;Moore,J.S.;Wolynes,P.G.Science 1997,277,1793–1796.14.George,M.;Weiss,R.G.Acc.Chem.Res.2006,39,489–497.15.(a)Brunsveld,L.;Vekemans,J.A.J.M.;Hirschberg,J.H.K.K.;Sijbesma,R.P.;Meijer,E.W.Proc.Natl.Acad.Sci.U.S.A.2002,99,4977–4982;(b)Obert,E.;Bellot,M.;Bouteiller,L.;Andrioletti,F.;Lehen-Ferrenbach,C.;Boué,F.J.Am.Chem.Soc.2007,129,15601–15605.Figure 5.Tentative models for the formation of vesicles and organogels from the quadruply hydrogen-bonded supramolecular polymers in apolar media.P.Du et al./Tetrahedron Letters 51(2010)188–191191。
专利名称:具有海绵状多孔结构的高分子凝胶专利类型:发明专利
发明人:酒井崇匡,郑雄一
申请号:CN201980049310.3
申请日:20190729
公开号:CN112533986A
公开日:
20210319
专利内容由知识产权出版社提供
摘要:本发明的目的在于,提供由具有微米级多孔结构的亲溶剂性高分子构成的凝胶材料。
本发明提供亲溶剂性聚合物单元相互交联而成的高分子凝胶,其特征在于,包含溶剂,具有包括第一区域及第二区域的两个区域的三维网状结构,上述第一区域密集地存在上述聚合物单元,上述第二区域稀疏地存在上述聚合物单元,由上述第一区域构成的网目大小为1~500μm。
申请人:国立大学法人东京大学
地址:日本东京都
国籍:JP
代理机构:上海一平知识产权代理有限公司
更多信息请下载全文后查看。
Communication Macromolecular Rapid Communications 895DOI: 10.1002/marc .201300831light-emitting devices (WLEDs) by high-throughout solu-tion processing, such as roll-to-roll or inkjet printing tech-niques. [7,8] To achieve the fabrication of such devices, a core issue is to construct optical nano-inks that consist of high-quality and emission color-tunable semiconducting polymer nanoparticles (SPNs) via nano-engineering. [9]The nanoparticle chemistry of conjugated polymersaffords diverse preparation methods [10–13] and modifi ca-tion strategies, such as doping, [14]nanocomposites,[15]encapsulation, [16] or surface modifi cation, [17] to con-trol exciton behavior and energy transfer. [18,19]Gen-erally, white-emitting polymer nanoparticles can be achieved by manipulating fl uorescence resonance energy transfer (FRET) via controlling intraparticle or interpar-ticle doping. [18,20,21] However, with regard to the exten-sive application of biomedicines, [11,18,22–25]conjugatedpoly m er nanoparticles (CPNs) for organic devices, [20,26–29] especially WL EDs, are severely lagging since the pio-neering exploration of polymer light-emitting devices (PL EDs) and polymer solar cells (PSCs) by Scherf and co-workers. [26,27] Fréchet et al. demonstrated a site isolation approach to control the energy transfer of encapsulating nanoparticles for WL EDs. [21] For the blue-emitting prop-erty of polyfl uorenes (PFs) nanoparticles, [10,11] Tuncel et al. S upramolecular polyfl uorenol enable assembly into conjugated polymer nanoparticles (CPNs). Poly{9-[4-(octyloxy)phenyl]fl uoren-9-ol-2,7-diyl} (PPFOH)-based supramolecular nanoparti-cles are prepared via reprecipitation. PPFOH nanoparticles with diameters ranging from 40 to 200 nm are obtained by adding different amounts of water into DMF solution. Size-dependent luminescence is observed in PPFOH-based hydrogen-bonded nanoparticles that is different from that of poly(9,9-dioctylfl uorenes). Finally, white light-emitting devices using CPNs with a size of 80 nm exhibitwhite emission with the CIE coordinates (0.31, 0.34).Amphiphilic conjugated polymer nanoparticles are poten-tial organic nano-inks for the fabrication of organic devicesin printed electronics.H ydrogen-Bonded Supramolecular Conjugated Polymer Nanoparticles for White Light-Emitting DevicesJ in-Yi L in ,J enIt W ong ,L ing-Hai X ie ,*X iao-Chen D ong ,H ui Ying Y ang ,*W ei H uang *D r. J.-Y. Lin, Prof. L.-H. Xie, Prof. W. Huang C enter for M olecular Systems and Organic Devices (CM SOD), Key Laboratory for Organic Electronics & Information Displays (KLOEID) and Institute of Advanced M aterials (IAM ), N anjing University of Posts and Telecommunications, N anjing 210003,P .R.China E-mail:i amlhxie@;iamwhuang@ D r J. Wong, Prof. H. Y. YangP illar of Engineering Product Development, S ingapore University of Technology and Design, S ingapore E-mail:y anghuiying@.sg P rof. X.-C. Dong, Prof. W. Huang J iangsu-Singapore Joint Research Center for Organic/Bio- Electronics & Information Displays, Institute of Advanced M aterials,N anjing Tech University N anjing 211816,P .R.China1.I ntrod uction L ow-cost, large-area, and solid-state light sources have attracted much scientifi c and industrial interest in the background of the energy crisis. [1–6] One appealing and potential method is ecofriendly nanoparticle-based white Macromol. Rapid Commun. 2014, 35, 895−900© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, WeinheimJ.-Y. Lin et al.Macromolecular Rapid Communicationswww.mrc-journal.de896demonstrated CPN-based WL EDs with CIE coordinates of (0.28, 0.34) by controlling the photochemical cross-linkingreaction to tune low-band emission at 530–550 nm. [28]In this context, it is signifi cant to explore delicate methodsfor white-emitting conjugated polymer nanoparticles. Herein, we constructed hydrogen-bonded supramolecularconjugated polymer nanoparticles (SCPNs) with size-dependent luminescence for WLEDs. S ize-dependent luminescence is the typical feature in inorganic quantum dot and nanoparticles when the size is smaller than the critical exciton Bohr radius. However, organic or polymer nanoparticles with the feature of the size-dependent emissions have not been observed in the literature systemically. [27,30–33] For example, poly(9,9-dioctylfl uorene) (PFO) nanoparticles have been obtained via reprecipitation, and demonstrated optical properties similar to bulk fi lm. [30] One reason is that polymer semi-conductors have intrinsic split energy levels. Besides, theother reason would be the driving force for the formationof CPNs, namely weaker hydrophobic and π–πstackinginteractions. [10,11] An alternative is to explore size-dependent FRET processes. Supramolecular approaches allow for not only affording robust non-covalent driving forces to make high-quality nanoparticles, but also manipulating exciton and FRET behaviours by subtle arrangement of chain packing during assembled aggre-gation. [34] As a result, supramolecular conjugated poly-mers (SCPs) are an appealing molecular building block forself-assembling into nanoparticles with stimuli-respon-sive luminescence changes that have obvious advan-tages over general conjugated polymers. [35] Herein, we designed a poly{9-[4-(octyloxy)phenyl]fl uoren-9-ol-2,7-diyl} (PPFOH) bearing hydrogen-bonded supramolecular steric carbinols at the 9-position of polyfl uorene (PFs) asa state-of-the-art model of SCPs to demonstrate distin-guished supramolecular assembly and intriguing par-ticle-size dependent behavior. PPFOHcan assemble into high-quality nano-particles via reprecipitation. Further-more, its amphiphilic features makePPFOH aggregate emissions with thegreen–yellow color that are combinedwith blue light-emitting backbone ofPFs acting as host to compose a perfectFRET nanosystem. As a result, the size-dependent emissions of the hydrogen-bonded SCPNs were observed that offeran unprecedented alternative strategyfor WLEDs. Devices with the confi gura-tion of Au/ p -SiC/PPFOH nanoparticles/ITO exhibited stable and purifi ed white emission with the CIE coordinates of (0.31, 0.34). 2.R esults and Discussion2.1. S ynthesis and Characterizations of PPFOH and PPFO8P PFOH, together with controlled poly{9-(octyloxy)-9-[4-(octyloxy) phenyl]fl uoren -2,7-diyl} (PPFO8), were preparedaccording to our previous work (Scheme 1 a ).[ 35 ] Gel permea-tion chromatography (GPC) shows that PPFOH has number-average molecular weight (M n ) and polydispersity index(PDI) of about 5.4 × 104 and 1.46, respectively (Figure S1 in the Supporting Information). The control sample PPFO8 hadM n of 6.92 × 10 4 and PDI of 1.62. As showed in Figure S2, the peaks at 3550 and 3450 cm−1 in the Fourier-transform infrared (FT-IR) spectra of PPFOH powders, involvinghydroxyl groups, suggested both free hydroxyl groups andassociation of hydroxyl groups. The solubility of PPFO8 is similar to poly(9,9-dioctylfl uorene) (PFO) that exhibits excel-lent solubility in common organic solvents such as chloro-form (CHCl 3) and toluene, but extremely poor solubility in N ,N -dimethylformamide (DMF) due to the larger difference of solubility parameters between PPFO8 or PFO (9.5) andDMF (12.1).[ 35 ] In contrast, PPFOH exhibited good solubility in DMF that probably contributed to its slightly amphiph-ilic feature and the formation of hydrogen bonds betweenhydroxyl groups and DMF molecules. As a result, amphiphilicPPFOH–DMF complexes are favorable for assembling by the control of external conditions, such as the addition of poor solvent. [ 12,36,37 ] However, the controlled PPFO8 is insoluble in DMF with poor capability of supramolecular assembly.P reparation of PPFOH NanoparticlesN anoparticles of PPFOH were achieved by the reprecipita-tion method. Size-controllable nanoparticles were obtained Macromol. Rapid Commun. 2014, 35, 895−900©2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim S cheme 1. a ) Synthetic routes of PPFOH and PPFO8. b) PPFOH nanoparticles obtained via re-precipitation, together with the photographs of nanoparticle suspensions by expo-sure to UV 365 nm. PPFOH nanoparticles consist of random polymer chains (host) and hydrogen bonded aggregates (guest).Macromolecular Rapid CommunicationsHydrogen-Bonded Supramolecular Conjugated Polymer Nanoparticles . . .www.mrc-journal.de 897by adding H 2 O into DMF solution. Colloidal nanoparticle suspensions exhibited a milky white color owing to light scattering compared to pristine colorless DMF solution (Scheme 1 b ). The general PFO nanoparticles were prepared via a two-steps reprecipitation as follows: dilute tetrahy-drofuran (THF) solution was added into poor solvent (H 2O ) and the THF was removed via heating under ultrasonica-tion. The number density of CPNs in the suspension was too low and it could not be spin-coated to obtain high-quality nanoparticle fi lms. [ 10,11 ] In contrast, for PPFOH it was hard to obtain nanoparticles via two-step reprecipi-tation but rather by direct precipitation under the samepreparation conditions. The sizes of PPFOH nanoparticles can be tuned from 40 to 200 nm by controlling the ratio of DMF:H 2 O . The sizes were determined to be about 200, 120, 80, and 40 nm at ratios of 300:6, 300:10, 300:20, and 300:50, respectively (Figure 1 ). The diameters of these nanoparti-cles were determined by dynamic light scattering (DLS) to be about 600, 170, 100, and 70 nm (Figure S3). In considera-tion of the kinetically frozen processing of reprecipitation, it is reasonable to explain that the diam-eter of colloidal nanoparticles decreasedwith the increasing amount of addedH 2 O owing to the increased number ofnucleic cores. [ 36,38 ] The diameters in theDL S analysis larger than those obtainedfrom SEM images can be explained bythe swelling effect of nanoparticles indispersion. In constrast, the controlledPPFO8 could not assemble into nano-particles via this method. These resultssuggest that supramolecular function-alization by tethering hydroxyl groupson the polymer backbones affords aneffective approach to improve the capa-bility of supramolecular assembly of hairy-rod conjugated polymers into size-controllable nanoparticles via theformation of amphiphilic polymer–sol-vent complexes. In contrast to PPFOHpowders, for the PPFOH nanoparticles,only the lower wavenumber at 3400cm −1 involving hydroxyl groups wasfound in the FT-IR spectra, indicatedthat only association of hydroxyl groupsexisted in the nanoparticles (Figure S2).In this context, there existed strongerhydrogen-bonding interactions in thenanoparticles than in the powder state,which would result in stronger energytransfer. Meanwhile, SEM analysis wasexplored to investigate the stability of the nanoparticles in suspension. Weused nanopaticles with a diameter of80 nm as an example. In Figure S4, the diameter of pristine nanoparticles was about 80 nm. For the nanoparticles suspension with aging of 0.5 h, the diam-eter was about 80 nm, similar to pristine nanoparticles but exhibiting aggregation. With prolonged aging to 2 h, the diameter was maintained at 80 nm but further signifi -cant aggregation was demonstrated. Thus, there was not growth of the nanoparticles in the suspension with pro-longing aging time, but aggregation of nanoparticles was exhibited.2.2. S teady and Transient Optical Properties of PPFOH NanoparticlesT he optical properties of PPFOH nanoparticle suspensions were investigated by electronic absorption spectra; the steady-state and transient photoluminescence (PL) spectra are shown in Figure 2 a , S5, S6, S7 and Table S1. PPFOH in DMF solution (< 0.1 mg mL –1 ) has the maximum absorb-ance peak at about 390 nm that is probably attributable to Macromol. Rapid Commun. 2014, 35, 895−900©2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim F igure 1. S EM images of the PPFOH-based nanoparticles. PPFOH nanoparticle suspen-sions with the condition of DM F: H 2O =300:50 (a), 300:20 (b), 300:10 (c) and 300:6 (d). The scale bars represent 200 nm. e) Diameters vs. the amounts of H 2O . DMF solution: 1 mg mL –1 ,300 μL. F igure 2. P L spectra and CIE coordinates of PPFOH nanoparticles suspension with dif-ferent sizes: a) PL spectra, and, b) their CIE coordinates. All dataa were collected withexcitation at 380 nm.J.-Y. Lin et al.Macromolecular Rapid Communicationswww.mrc-journal.de 898the main chains of polyfl uorene (Figure S5). The PL spectraof PPFOH in dilute solution have three emission peaks at419, 443, and 472 nm that were assigned, respectively, tothe 0–0, 0–1, and 0–2 intrachain singlet transitions of thepolyfl uorene backbone. [ 39 ] Three emission peaks exhib-ited a single-exponential ( 1 A *) decay, and their lifetimes were about 400 ps corresponding to three peaks of the polyfl uorene fl uorescent spectrum (Table S1). [ 40 ] The emis-sion spectra of PPFOH nanoparticles exhibited two energy regimes: blue emission at 440–460 nm and green–yellow at 530–540 nm; this was dramatically different from thepristine PFO nanoparticles prepared via reprecipitation without green–yellow band emission,[ 41 ] but similar to the nanoparticles of amphiphilic oligofl uorene or polyfl u-orenes, suggested the existence of aggregates.[ 12,36,37 ]For nanoparticle suspensions obtained via reprecipitation, emission peaks at ca. 460 and 535 nm had lifetimes of ca. 112 ps and 2.92 ns (Figure S6 and Table S1), affording evi-dence for the aggregates rather than fl uorenone, according to previous investigations. [ 42–44 ] These assumptions are supported by the FT-IR spectra in which the feature peakof ketone at 1720 cm −1 was not observed (Figure S2). It canbe deduced that the aggregate species, together with thepristine polyfl uorene chains in nanoparticles, consist of aself-doping energy transfer host–guest nanosystem.S ize-dependent emissions of PPFOH nanoparticles have been observed in Figure 2 and S7. For nanoparticlesuspensions obtained via reprecipitation, the intensity of green–yellow band emission was increased with the decreased size of nanoparticles (Figure 2 a ). For reprecipi-tation with the frozen process, the intensity of green–yellow band emissions is associatedwith the amount of aggregates. Thiscan be explained by a proportionalrelation between the number of nucleiand the amount of aggregates withincreasing amount of H 2O . From Figure S7, the intensity of green–yellow emis-sion of PPFOH nanoparticle fi lms spin-coated from nanoparticle suspensionalso increased with decreasing size ofnanoparticles, and was stronger thanthe intensities of the correspondingnanoparticle suspension. PPFOH nano-particle suspension and fi lm with diam-eter of 200 nm exhibited blue emissionwith the CIE coordinates (0.17, 0.16) andnear-white emission with CIE coordi-nates of (0.24, 0.33), respectively. PPFOHnanoparticle suspensions with sizeof 80 nm exhibit white color with CIE coordinates (0.30, 0.42) but slight green color with CIE coordinates (0.29, 0.48). The particle thin fi lms exhibited similar size-dependent photoluminescence with high intensity of green emission with reduced particle size. The intensi-ties of green–yellow emission of nanoparticle fi lms spin-coated from suspension was stronger than those of the corresponding nanoparticle suspensions. Size-dependent photoluminescence offers an alternative flexible method to tune emission colors.2.3.E lectroluminescence Properties of PPFOH Nanoparticle-Based WLEDsH ybrid WL EDs with the confi guration Au/ p -SiC/PPFOH nanoparticles/ITO were fabricated in order to explore the potential applications of nanoparticles thin-fi lm devices (Figure 3 ). According to previous works, [ 45–48 ] the electrolu-minescent (EL) spectra in this kind of hybrid nanostructure-based LED exhibit ineffective energy transfer with respect to the photoluminescence. Therefore, PPFOH colloidal repre-cipitated nanoparticles with size of 80 nm (PL spectral withCIE coordinates (0.30, 0.42)) were selected to fabricate theWLEDs, in which nanoparticle thin fi lms were obtained via spin-coating from suspensions on the p -SiC (Figure S8). The current–voltage ( I –V ) curves in the hybrid WL EDs exhib-ited typical rectifying characteristics (Figure 3 b ). When the hybrid WLEDs was forward biased to about 15 V, the current reached up to 2.33 mA, while only a small reverse leakage current of 0.02 mA was observed at −15 V. The hybrid WLEDs has turn-on voltage (current) of ca. 15 V (2.33 mA).T he EL spectra of hybrid WL EDs based on the nanopar-ticles at various forward biased voltages are showed in Figure 3 c and no light emission was observed from the Macromol. Rapid Commun. 2014, 35, 895−900©2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim F igure 3. a ) The device confi guration of the hybrid WLEDs based on PPFOH nanoparticles. b) Current–voltage ( I –V ) curve of the hybrid WLEDs; the inset is the enlarged current–voltage curve at the reverse bias. c) EL spectra at different voltages. d) CIE coordinates of hybrid WLEDs biased at various voltages.MacromolecularRapid Communications Hydrogen-Bonded Supramolecular Conjugated Polymer Nanoparticles . . .www.mrc-journal.de 899hybrid WLEDs under reverse bias. The EL spectra of hybrid WL EDs can be fi tted by three Gaussian curves with peak wavelengths at around 433, 474, and 535 nm that wereconsistent with the PL profi les (Figure 3 c ). The EL spectra showed lower emission intensities at long wavelength andbroad emission as compared with the PL spectra. For devicesbased on PPFOH nanoparticles, EL spectra have the CIE chromaticity coordinates of (0.28, 0.32), (0.31, 0.34), (0.31, 0.32), and (0.35, 0.36) at 15, 20, 27.5, and 30 V, respectively. These values are well matched with the most optimized white light, with the CIE coordinates (0.33, 0.33) (Figure 3 d ). In addition, it should be noted that the EL spectra of PPFOH thin fi lm spin-coated from DMF solution for comparison exhibited only one emission peak at 560 nm under these experimental conditions. This supports the notion that nanoparticles are favorable for the white emission. 3.C onclusions W e have demonstrated state-of-the-art supramolecular conjugated polymer nanoparticles for application as WLEDs. In comparison with the controlled PPFO8 without hydroxyl group, hydrogen-bonded supramolecular conju-gated polymer PPFOH exhibited excellent supramolecular assembly behavior. PPFOH nanoparticles were obtained with size ranging from 40 to 200 nm via reprecipitation. PPFOH nanoparticles have fascinating size-dependent luminescent properties, affording a fl exible method to tune their emission color from blue–green, yellow to white. PPFOH nanoparticles with size of ca. 80 nm were observed to emit nearly white light with the CIE coordinates (0.30, 0.42). Prototype WL EDs using PPFOH nanoparticles with size of 80 nm exhibited white emission color with the CIE coordinates (0.31, 0.34) at 20 V. Supramolecular conjugated polymers are intriguing and versatile building blocks for self-assembly, and supramolecular polymer nanoparti-cles that will be promising organic nanoinks not only for WLEDs but also for organic nanodevices. S upporting Information S upporting Information is available from the Wiley Online Library or from the author. A cknowledgements: T he project was supported by the National Natural Science Funds for Excellent Young Scholar (21322402), the National Natural Science Foundation of China (21274064, 21144004, 61177029, 20974046), The Program for New Century Excellent Talents in University (NCET-11–0992), the Doctoral Fund of Ministry of Education of China (20133223110007), the Natural Science Foundation of Jiangsu Province (BK2011761, BK2008053, SJ209003), and the Program for Postgraduates Research Innovation in University of Jiangsu Province (CXLX11_0413, CXLX11_0423).[1]E .F.S chubert ,J .K.K im ,S cience 2005,308,1274.[2]D .I.S on ,B .W.K won ,D .H.P ark ,W .-S.S eo ,Y .Y i ,B .A ngadi ,C .-L .L ee ,W .K.C hoi ,N at. Nanotechnol. 2012,7,465.[3] Y . J. H ong ,C .-H. L ee ,A . Y oon ,M . K im ,H .-K. S eong ,H . J. C hung ,C .S one ,Y .J.P ark ,G .-C.Y i ,A dv. Mater. 2011,23,3284.[4]L .-H.X ie ,C .-R.Y in ,W .-Y.L ai ,Q .-L .F an ,W .H uang ,P rog. Polym. Sci. 2012,37,1192.[5]H .W u ,L .Y ing ,W .Y ang ,Y .C ao ,C he m. Soc. Re v. 2009,38,3391.[6]S .R eineke ,F .L indner ,G .S chwartz ,N .S eidler ,K .W alzer ,B .L uessem ,K .L eo ,N ature 2009,459,234.[7]E .F isslthaler ,A .B lumel ,K .L andfester ,U .S cherf ,E .J.W.L ist ,S oft Matter 2008,4,2448.[8]E .F isslthaler ,S .S ax ,U .S cherf ,G .M authner ,E .M oderegger ,K .L andfester ,E .J.W.L ist ,A ppl. Phys. Lett. 2008,92,183305.[9]L .-H.X ie ,S .-H.Y ang ,J .-Y.L in ,M .-D.Y i ,W .H uang ,P hilos.Trans. R. Soc. A 2013,371,20120337.[10]J .P echer ,S .M ecking ,C hem. Rev. 2010,110,6260.[11]D .T uncel ,H .V.D emir ,N anoscale 2010,2,484.[12]Y .K oizumi ,S .S eki ,S .T sukuda ,S .S akamoto ,S .T agawa ,J . Am.Chem. Soc. 2006,128,9036.[13]K .L andfester ,R .M ontenegro ,U .S cherf ,R .G untner ,U .A sawapirom ,S .P atil ,D .N eher ,T .K ietzke ,A dv. Mate r. 2002,14,651.[14]B .B alan ,C .V ijayakumar ,S .O gi ,M .T akeuchi ,J . Mater. Chem.2012,22,11224.[15]C .W u ,H .P eng ,Y .J iang ,J .M cNeill ,J . Phys. Chem. B 2006,110,14148.[16]C .F.W u ,C .S zymanski ,J .M cNeill ,L angmuir 2006,22,2956.[17] K . P etkau ,A . K aeser ,I . F ischer ,L . B runsveld ,A . P . H. J. S chenning , J . Am. Chem. Soc. 2011,133,17063 .[18]Z .T ian ,J .Y u ,C .W u ,C .S zymanski ,J .M cNeill ,N anoscale2010,2,1999.[19] R . A bbel ,R . v an der Weegen ,E . W. M eijer ,A . P. H. J. S chenning , C hem. Commun. 2009,1697 .[20]C .F.H uebner ,R .D.R oeder ,S .H.F oulger ,A dv. Funct. Mater. 2009,19,3604.[21]H .G ao ,D . A.P oulsen ,B .M a ,D . A.U nruh ,X .Z hao ,J .E.M illstone ,J .M.J.F réchet ,N ano Lett. 2010,10,1440.[22]C .W u ,C .S zymanski ,Z .C ain ,J .M cNeill ,J . Am. Che m. Soc.2007,129,12904.[23]C .W u ,B .B ull ,C .S zymanski ,K .C hristensen ,J .M cNeill ,A CSNano 2008,2,2415.[24]J .Y u ,C .W u ,S .P.S ahu ,L .P.F ernando ,C .S zymanski ,J .M cNeill ,J . Am. Chem. Soc. 2009,131,18410.[25]C .W u ,T .S chneider ,M .Z eigler ,J .Y u ,P .G.S chiro ,D .R.B urnham ,J .D.M cNeill ,D .T.C hiu ,J . Am. Che m. Soc.2010,132,15410.[26]T .K ietzke ,D .N eher ,K .L andfester ,R .M ontenegro ,R .G untner ,U .S cherf ,N at. Mater. 2003,2,408.[27]T .P iok ,S .G amerith ,C .G adermaier ,H .P lank ,F .P.W enzl ,S .P atil ,R .M ontenegro ,T .K ietzke ,D .N eher ,U .S cherf ,K .L andfester ,E .J.W.L ist ,A dv. Mater. 2003,15,800.[28]E .-J.P ark ,T .E rdem ,V .S.I brahimova ,S .N izamoglu ,H .V.D emir ,D .N.S.T uncel ,A CS Nano 2011,5,2483.[29]C .Z heng ,X .X u ,F .H e ,L .L i ,B .W u ,G .Y u ,Y .L iu ,L angmuir 2010,26,16730.[30]A .K aeser ,A .P.H.J.S chenning ,A dv. Mater. 2010,22,2985.R eceived:N ovember 6,2013;Revised:D ecember 16,2013; P ublished online: March 7, 2014; DOI: 10.1002/marc.201300831Keywords:s upramolecular conjugated polymers;n anoparticles;h ydrogen bonds;l uminescence;l ight-emitting diodesMacromol. Rapid Commun. 2014, 35, 895−900© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, WeinheimJ.-Y. Lin et al.Macromolecular Rapid Communicationswww.mrc-journal.de 900Macromol. Rapid Commun. 2014, 35, 895−900© 2014 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim [31]J .K.G rey ,D .Y.K im ,B .C.N orris ,W .L .M iller ,P .F.B arbara ,J . Phys. Chem. B 2006,110,25568.[32]N .K urokawa ,H .Y oshikawa ,N .H irota ,K .H yodo ,H .M asuhara ,C hemPhysChem 2004,5,1609.[33]Y .-Z.L ong ,M .-M.L i ,C .G u ,M .W an ,J .-L .D uvail ,Z .L iu ,Z .F an ,P rog. Polym. Sci. 2011,36,1415.[34] F . J. M. H oeben ,P . J onkheijm ,E . W. M eijer ,A . P. H. J. S chenning ,C hem. Rev. 2005,105,1491 .[35]J .L in ,Z .Y u ,W .Z hu ,G .X ing ,Z .L in ,S .Y ang ,L .X ie ,C .N iu ,W .H uang ,P olym. Chem. 2013,4,477.[36]L .Z hu ,J .Q in ,C .Y ang ,J . Phys. Chem. B 2010,114,14884.[37]L .Z hu ,C .Y ang ,J .Q in ,C hem. Commun. 2008,6303.[38]F .W ang ,M .-Y.H an ,K .Y.M ya ,Y .W ang ,Y .-H.L ai ,J . Am. Chem.Soc. 2005,127,10350.[39]Z .-Q.L in ,N .-E.S hi ,Y .-B.L i ,D .Q iu ,L .Z hang ,J .-Y.L in ,J .-F.Z hao ,C . W ang ,L .-H. X ie ,W . H uang ,J . Phys. Chem. C 2011,115,4418 .[40]J .T eetsov ,M .A nne Fox ,J . Mater. Chem. 1999,9,2117.[41]C .W u ,J .M cNeill ,L angmuir 2008,24,5855.[42]J .P ei ,X .-L .L iu ,Z .-K.C hen ,X .-H.Z hang ,Y .-H.L ai ,W .H uang , M acromolecules 2002,36,323.[43]X .-H.Z hou ,Y .Z hang ,Y .-Q.X ie ,Y .C ao ,J .P ei ,M acromolecules 2006,39,3830.[44]A .P.K ulkarni ,X .K ong ,S .A.J enekhe ,J . Phys. Chem. B 2004, 108,8689.[45]J .X iao ,H .Y ang ,Z .Y in ,J .G uo ,F .B oey ,H .Z hang ,Q .Z hang , J . Mater. Chem. 2011,21,1423.[46]J .X iao ,B .Y ang ,J .I.W ong ,Y .L iu ,F .W ei ,K .J.T an ,X .T eng ,Y .W u ,L .H uang ,C .K loc ,F .B oey ,J .M a ,H .Z hang ,H .Y.Y ang ,Q .Z hang ,O rg. Lett. 2011,13,3004.[47]B .Y ang ,J .X iao ,J .I.W ong ,J .G uo ,Y .W u ,L .O ng ,L .L .L ao ,F .B oey ,H .Z hang ,H .Y.Y ang ,Q .Z hang ,J . Phys. Chem. C 2011, 115,7924.[48]J .Z hao ,J .I.W ong ,C .W ang ,J .G ao ,V .Z.Y.N g ,H .Y.Y ang ,S .C.J.L oo ,Q .Z hang ,C hem. Asian J. 2013,8,665.。