
2002年第60卷第2期,221~227
化学学报
ACTA CHI M ICA SINICA
Vol.60,2002
No.2,221~227
MMP -2和Hydroxamate 类抑制剂绝对自由能的计算
侯廷军 章 威 徐筱杰
X
(北京大学化学与分子工程学院 北京100871)
摘要 采用基于线性响应近似的自由能计算方法计算了一类hydroxamate 抑制剂和MMP -2的绝对结合自由能.计算中,催化锌离子和MMP -2以及配体之间采用了非键模型.分子动力学模拟结果显示,采用非键模型时,催化Zn 离子采用五配位的形式,但配位键的形式和初始结构比较有很大的差别.通过拟合,分别得到了单参数、双参数以及三参数的自由能预测模型,其中,含有常数校正项的三参数模型具有最佳的预测能力,预测自由能和实际自由能之间平均绝对误差仅为2.38kJ/mol.
关键词 线性响应近似,结合自由能,hydroxamate 抑制剂,MMP -2,分子动力学
Binding Free Energy Calculations for MMP2-Hydroxamate Complexes
HOU,Ting -Jun ZHANG,Wei XU,Xiao -Jie
X
(College o f Chemistry an d Molecular Engineerin g ,Peking University ,Beijing 100871)
Abstract The absolute binding affinities of a series of hydroxa mate inhibitors with MMP -2were evaluated by molecular dyna mics (MD)simulations with a linear response approach.During MD simulations,a nonbonded
model for the catalytic zinc center was used to represent the interac tions between zinc center and enzyme/inhibitor.The trajectories from MD simulation show that using the nonbonded model the catalytic zinc ion adopts five coordination number,but the c oordination form e xists large difference with that of the initial model.After fittings,the models with one parameter,two parameters and three parameters were obtained.The calculated results indicate that the three -parameter model with a constant term bears the best predicting ability.The best model yields an average error of 2.38kJ/mol for the eight binding affinities of hydroxamtes.Keywords linear response approximation,binding free energy,hydroxamate inhibitors,MMP -2,molecular dynamics
受体和配体之间结合自由能评价是基于结构的计算机辅助药物设计的核心问题,精确的自由能预测方法能够大大提高药物设计的效率.在过去的20年中,随着受体-配体相互作用的理论研究以及计算机辅助药物分子设计方法的快速发展,自由能预测方法的研究受到了越来越多的关注.
一些分子对接的程序采用了多种药物分子和受体之间结合的评价方法,比如表面匹配,能量匹配(非键相互作用能)以及化学环境匹配等等[1~4]
.虽然这些评价标准能部分反映底物和蛋白质间的结合
程度,但由于它们的评价能力非常有限,应用范围受到了较大的局限.因此在目前粗筛方法日臻完善的
情况下,以自由能为代表的精确评价方法显得日益重要起来.在自由能的计算方法中,自由能微扰(free energy perturbation,FEP)和热力学积分(thermodynamic integration,TI)方法是最为经典的方法,为大家所广泛接受.但这类方法需要长时间的数据采集,而且只能适合较为简单的情况,在药物设计中并没有得到广泛的应用[5,6]
.基于经验方程的自由能计算方法也是很重要的一类方法[7~11].这类方法把结合自由
X E -mail:xiaoj xu@che https://www.doczj.com/doc/1316373737.html,
Received June 11,2001;revi sed and accepted August 8,2001.国家自然科学基金(Nos.29873003,29992590-2)资助项目.
能分解为不同的相互作用能量项,通过一组训练集并利用统计方法来得到自由能计算的经验公式.这类方法取样简单,计算量小.但这类方法得到的经验公式很依赖于训练集的选择,对不同的体系也具有不同的预测能力.而且这类方法不能很好地考虑体系的柔性以及溶剂效应.1994,!qvist 等[12]提出了一种基于线性响应近似(linear response approximation,LR)的自由能计算方法.这种方法理论基础来源于非平衡态统计物理学中的线性响应理论,把自由能分解为极性和非极性的贡献.虽然这种方法也需要进行分子动力学的采样,但和FEP 比较,它仅仅需要对体系的始态和终态做动力学采样,因此计算量比FEP 大大减少.和半经验方法比较,它的优势很明显.首先它用分子动力学方法考虑了体系的柔性,在正确预测复合物结构的基础上对体系的构象空间进行采样;其次它通过采用在溶剂环境以及蛋白环境两次分子动力学采样,考察了溶剂效应对结合自
由能的影响.
本文采用基于线性响应的自由能计算方法预测了一组MMP -2(gelatinase -A)抑制剂的绝对结合自由能.MMPs (gelatinase)是一大类含锌的金属蛋白酶,因为参与结蒂组织的分解反应而成为许多药物抑制剂的作用点[13].在MMPs 家族中MMP -2被认为是一个非常重要的药物设计靶点[14]
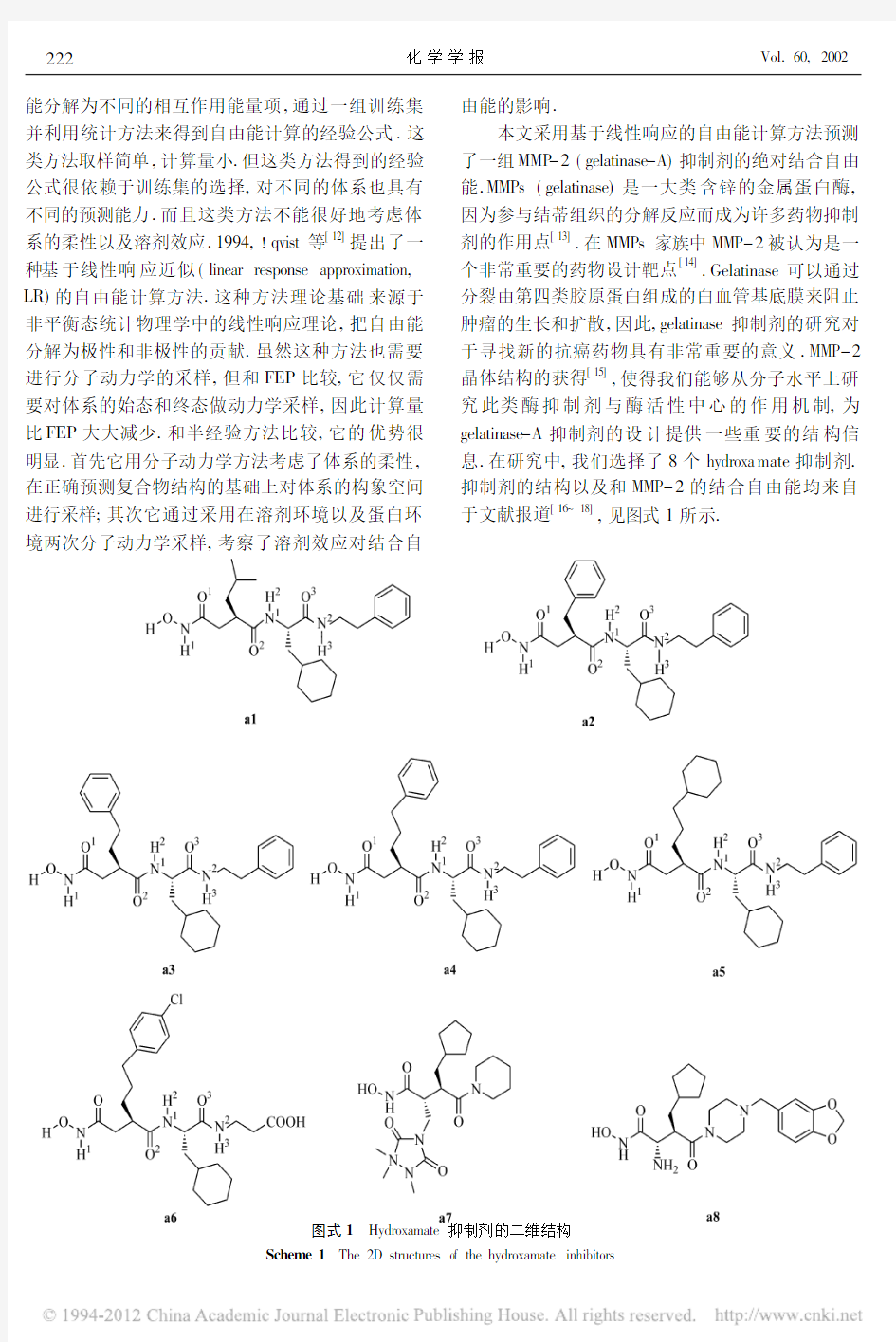
.Gelatinase 可以通过分裂由第四类胶原蛋白组成的白血管基底膜来阻止肿瘤的生长和扩散,因此,gelatinase 抑制剂的研究对于寻找新的抗癌药物具有非常重要的意义.MMP -2晶体结构的获得[15],使得我们能够从分子水平上研究此类酶抑制剂与酶活性中心的作用机制,为gelatinase -A 抑制剂的设计提供一些重要的结构信息.在研究中,我们选择了8个hydroxa mate 抑制剂.抑制剂的结构以及和MMP -2的结合自由能均来自于文献报道[16~
18]
,见图式1所示
.
图式1 Hydroxamate 抑制剂的二维结构Scheme 1 The 2D structures of the hydroxamate inhibitors
222
化学学报Vol.60,2002
1计算方法
1.1基于线性响应近似的自由能计算方法
根据线性响应近似的原理,结合自由能可以由下式计算得到[12]:
$G bi nd=1
2
其中$G bind是结合自由能,V el i是底物和溶剂分子之间的静电相互作用能,V vdw i是底物和溶剂分子之间的范德华相互作用能,V el s和V vdw s则分别是在复合物体系中底物和环境之间的静电相互作用能和范德华相互作用能,A是非极性相互作用对结合自由能的响应系数.上式中,静电相互作用和自由能之间为1/2的关系,可以根据线性响应近似的原理直接推导得出.
对于研究的体系,需要进行两次分子动力学采样,一次在溶剂环境下,一次在蛋白质环境下.从收集得到的轨迹中,可以得到$V el s和$V vdw s,通过线性拟合就可以得到A参数.用得到的模型就可以进行新化合物的结合自由能预测.
1.2模型的搭建以及电荷的计算
MMP-2的晶体结构直接取自蛋白质结构数据库(PDB),PDB编号为1QIB.和其它MMPs相似,MMP-2的催化区结构也包括5个筒状的B折叠,其中4个平行,1个为反平行,还有3个很长的A螺旋,MMP-2的催化区结构没有因为柔性loop区(FLD)的插入而发生大的变化.MMP-2和其抑制剂相互作用的复合物结构目前还没有文献报道,因此我们首先应该搭建出分子动力学模拟的复合物的初始模型.MMPs家族的成员在结构上非常保守,而且受体和底物之间的作用模式基本相似.可以通过MMPs家族其它成员复合物的晶体结构作为模板来搭建模拟的初始结构.我们选取了MMP-3和hydroxa mate抑制剂复合物的晶体结构作为参考结构(PDB编号:1BIW)[19].整个搭建过程分为三个步骤:首先,把1QIB和1BI W进行结构比对(structural alignment);把hydroxamate抑制剂从1BIW中取出,放入1QIB中得到复合物结构;把搭建得到的复合物模型进行人工调整以避免明显的原子碰撞,仅调整活性位点附近部分残基的两面角.
对于体系中的锌原子,计算则采用了非键模型,部分电荷即为形式电荷(+2|e|).抑制剂先采用半经验量化方法AM1优化[20],然后用Gaussian98中的Hartree-Fock方法来得到模型周围静电势分布,计算采用6-31*基组[21].最后采用约束静电势收敛方法(restrained electrostatic potential,RESP)来得到模型的部分电荷分布[22].
1.3分子动力学模拟
所有分子动力学模拟均采用AMBER6.0软件包[23].计算得到的部分电荷加入AMBER的数据库文件中.模拟温度为300K,溶剂采用TIP3P水模型.对于两个模拟体系,均在抑制剂周围加上2nm的水分子层.在分子动力学模拟之前,对模型进行一系列的约束分子力学优化,对模型中的重原子分别采用41.8, 4.18和0.42kJ/(mol#nm2)的谐振子回复力.然后,在没有约束条件下进行分子力学优化,最大优化步数为20000步,收敛条件为能量梯度小于0.21 kJ/(mol#nm).
分子动力学模拟分为两步.首先,进行100ps的分子动力学模拟使得体系达到平衡.接着,进行200 ps的分子动力学模拟进行数据的收集.在数据收集时,每隔200fs记录一次模拟体系的构象.在分子动力学模拟时,抑制剂、水分子以及抑制剂周围1.6nm 之内的残基没有任何约束,而其它的残基的重原子则外加一个2090kJ/(mol#nm2)的谐振子回复力.模拟中,采用SHAKE方法来约束体系中与氢原子有关的键伸缩.分子动力学模拟的步长为2.0fs,非键相互作用的截断值(cutoff)设为1.2nm.分子动力学模拟在22个Pó733组成的并行机群上进行.
1.4自由能计算模型的搭建
从分子动力学轨迹可以得到抑制剂和溶剂以及抑制剂和环境(包括蛋白质和溶剂)之间的平均非键相互作用能.通过线性拟合得到结合自由能和平均非键相互作用能之间的线性模型.拟合过程采用了遗传算法(genetic algorithm,GA)[24,25],线性回归系数定义为得分函数来评价每个自由能模型.模型的预测能力用Leave-one-out的交互验证回归系数来评价.
2结果与讨论
2.1催化锌离子的结构
在复合物初始模型的搭建过程中,选取了MMP-3和hydroxamate抑制剂复合物的晶体结构1BI W作为模板.在IBIW中,催化锌离子和周围的三个组氨酸以及配体上的两个氧原子形成了明显的五配位形式,因此搭建出来的MMP-2和抑制剂的初始结构中,催化锌离子也成五配位的形式.图1a显示了MMP-2和a1形成的复合物起始构象中催化锌离子的配位形式.本文中,催化锌离子采用了非键模型.但在前面的工作中,催化锌离子我们采用了键合的
223
No.2侯廷军等:MMP-2和Hyd roxamate类抑制剂绝对自由能的计算
模型.非键模型和键合模型比较各有其优缺点.键合模型能量项较为复杂,模型比较严格.但键合模型的势场参数难以获得,而且键合模型通过共价键的形式约束了金属离子配位数的变化.非键模型比较简单,金属离子和周围原子之间通过非键相互作用来描述,对金属约束较少,允许金属离子配位数有一定的变化.但非键模型非常依赖于范德华参数的精确度.在实际模拟中,非键模型应用得更为广泛一些.本文采用非键模型主要基于两个考虑:首先是基于线性响应近似的自由能计算方法在推导过程中仅仅只有考虑了非键相互作用项,采用非键模型就不会引入配键的能量项.其次是我们前面的一些模拟工作对催化锌离子采用了键合模型,采用非键模型能和前面的工作进行一些比较[26]
.
分子动力学模拟的轨迹表明,当体系达到平衡以后,催化锌离子的配位形式发生了明显的变化.图1b 显示经过200ps 分子动力学模拟以后,催化锌离子和周围残基以及抑制剂a1的三维结构.图1b 中的结构表明,Zn
原子基本上还是处于五配位的形
图1 催化锌离子和周围残基以及抑制剂a1之间的配位形式
(a)初始构象;(b)动力学200ps 时的构象
Figure 1 The coordination form of the catalytic zinc atom with MMP -2and a1
(a)the i nitial conformation;(b )the conformation after 200ps MD simulations
式,但配位结构和图1a 中的结构比较有了明显的变化.组氨酸201和205上的N 原子还是能够和Zn 离子形成稳定的配键,这两个残基上的N 原子与Zn 原子的距离分别为0.202和0.201nm.但His211上的N 原子和Zn 离子间的距离为0.585nm,已经不可能形成配键.图1b 中,Glu202上两个氧原子和Zn 离子的距离分别为0.175和0.185nm,显然能和Zn 离子形成稳定的配键.在我们前面的计算中,由于采用键合的Zn 离子模型,Zn 离子的配位形式和初始结构比较没有明显变化[26]
.但本文采用非键模型时,Zn 离子基本还是保持了五配位的形式,但周围配键的电子给体发生了一些变化,当然这种配位形式还需要实验进一步的证明.
2.2 能量项的波动
对于抑制剂和靶酶的体系,经过了100ps 的平衡以后,体系已经达到了比较稳定的状态.图2显示了抑制剂a1和MMP -2之间范德华和静电相互作用随时间变化的曲线,可以很清楚地看到,能量项基本上在较小的范围内波动.表1列出了范德华和静电相互作用在不同时间段下的统计平均值,0至50ps 之间以及0至200ps 之间统计的结果没有明显的差别.因此,200ps 的采样可以得到稳定的能量项数值.
表1 不同时间段下a1和MMP -2间的非键相互作用Table 1 The averaged nonbonded interactions between a 1and MMP -2using different time interval
t /ps E ele /(kJ #mol -1)E vdw /(kJ #mol -1)0~50-225.51(18.04)-404.91(37.05)0~100-226.62(21.00)-406.60(38.21)0~150-227.10(20.43)-406.99(37.52)0~200
-226.67(19.81)
-404.91(37.11)
2.3 自由能模型
表2列出了从分子动力学轨迹得到的抑制剂处于自由状态和结合状态下和环境之间的平均静电相互作用以及范德华相互作用之间的差值.
表2 由分子动力学轨迹得到的平均相互作用能Table 2 The averaged nonbonded interactions from MD trajectories
No./(kJ #mol -1)
/(kJ #mol -1)
a1-52.38-161.10a2-46.52-54.93a3
-89.49-91.50a4-100.32-103.29a5-58.23-115.28a6-121.30-69.10a7-44.56-83.68a8
-98.19
91.17
224
化学学报Vol.60,2002
图2分子动力学采样过程中非键相互作用能的波动Figure2Fluctuation of the nonbonded interactions during the MD sampling
!qvist最初提出的方程中,仅仅需要两个参数.而且,在前面的一些文献中,静电相互作用对自由能的响应系数也采用了0.5的固定值,对不同的体系仅仅需要变化B的数值.我们首先采用了!qvist最初提出的模型,A固定为0.5,拟合得到的B为0.39,用这个模型预测得到的抑制剂的结合自由能见表3.这个模型从统计结果上看很不理想.模型的线性回归系数仅为0.01,而模型的交互验证回归系数则为-26.16.这个双参数模型不具有任何预测能力.得到这样的结果是可以理解的.因为从前面的理论研究中可以发现,A=0.5只能对少数简单离子体系的水合过程比较合适,但对于一些比较复杂的体系,尤其是溶质分子会造成溶剂氢键结构发生明显重组时,A的取值会发生很大的偏离[27].对于抑制剂和受体的相互作用过程则更为复杂,简单地把A保持恒定在理论上存在较大的缺陷.
因此我们在拟合过程中,也尝试对A采用其它不同的取值.用遗传算法同时对A和B进行优化得到了模型2,见表3.这个模型和model1相比,回归系数和交互验证回归系数都有了很大的提高.模型的回归系数为0.88,交互验证回归系数为0.57,结合自由能的实验值以及预测值残差的绝对平均值为5.4kJ/mol.
表3结合自由能的实验值以及不同的拟合模型的预测值Table3Experimental and calculated$G b using different fitted models
No.
$G
b
/(kJ#mol-1)
Expt.
Model1
A=0.5,B=0.39
Model2
A=0.13,B=0.49
Model3
A=0.09,B=0.32,C=-17.18
a1-54.0510.32-46.77-48.24 a2-32.60-7.44-29.97-36.99 a3-49.32-1.38-55.80-54.09 a4-58.19-2.55-62.62-58.60 a5-46.52 4.35-43.60-46.11 a6-62.74-10.99-68.38-62.37 a7-38.79 1.17-32.77-38.92 a8-43.850.71-36.03-40.76 r20.010.880.88 q2-26.160.570.83 F032.8017.95
在方程中我们加入了一个常数项进行拟合,见方程2:
$G bi nd=A<$V el>+B<$V vdw>+D(2)新的自由能模型和前两个模型比较,其交互验证回归系数有了很大的提高(q2=0.83).该模型的三参数分别为A=0.09,B=0.32和D=- 4.11.8个抑制剂自由能的实验值和预测值之间的平均绝对误差为2.38kJ/mol.前面一些文献也报道了用该方法对几种体系的自由能预测结果.在!qvist的工作中,用双参数模型对四个endothiopepsin和三个HIV蛋白酶抑制剂进行了自由能预测,其预测值和实验值之间的绝对误差分别为1.67kcal/mol(6.99kJ/mol)和7.52 kcal/mol(31.5kJ/mol).在Jones-Hertzog的工作中,作者用三参数模型对7个sulfonamide抑制剂进行了活性预测,其预测值和实验值之间的绝对误差为3.22 kcal/mol(13.48kJ/mol).在Hansson等对一组18个抑制剂进行的自由能计算中,用双参数模型得到的预测值和实验值之间的绝对误差为3.01kcal/mol
225
No.2侯廷军等:MMP-2和Hyd roxamate类抑制剂绝对自由能的计算
(12.60kJ/mol),而采用三参数模型时,预测值和实验值之间的绝对误差则为2.26kcal/mol(9.46kJ/ mol).从计算结果上看,含一个常数项的三参数能够很好地预测结合自由能.
Model3中拟合得到常数项D为-17.18.除了静电和范德华对结合自由能的贡献外,其它因素对结合自由能的贡献为-17.18kJ/mol.这个常数项可能主要包括两个方面的因素.首先,熵效应对结合自由能的影响,包括体系平动熵和转动熵的变化以及去溶剂化过程中水分子的重排所引起的熵变.在我们研究的体系中,抑制剂上疏水的P1c基团能够和MMP-2上疏水的S1c口袋形成较大的表面接触.当抑制剂和受体相互作用时,P1c基团和S1c口袋周围的水分子会发生重排,分子的重排会导致熵值的降低.当然,体系构型熵对自由能也有非常大的影响.总的来讲,分子对接过程中的熵变是非常复杂的,现在还缺乏有效的方法来定量地考察.虽然范德华相互作用和疏水效应有密切的关系,疏水效应有相当一部分是隐含在范德华相互作用能量项中,但还没有证据表明范德华相互作用能量项和疏水效应有线性的关系,因此常数项中有应该包含熵效应的部分贡献.其次,在模型中的$V ele和$V v d w两项中,仅仅包括了自由状态下抑制剂和溶剂以及结合状态下抑制剂和环境之间能量的变化.其它的相互作用的变化,包括蛋白质和溶剂能量的变化都不能包括在A和B参数中,它们对自由能影响可能也体现在常数项之中.
从原理上讲,对于不同的受体-配体体系,熵效应对配体结合的影响是不同的.这就是说,model3中的D常数项会随着体系的不同而改变.我们研究的抑制剂基本上具有相同的结构,它们在分子对接过程中的熵效应可能比较相似,因此,引入一个常数项可以大大改善自由能模型.在前面的工作中,我们采用基于线性响应近似的自由能计算方法计算了15个MMP-2抑制剂的绝对结合自由能.催化锌离子和周围的配键采用了键合模型,得到的三参数模型和本文得到的模型具有相似的预测能力,但模型中的三个参数和本文模型中三个参数有较大的差别.这并不难理解,采用键合模型时,抑制剂和靶酶之间的非键相互作用有很大一部分都包含在配键相互作用中,因此也导致了模型的差别.当然,对催化锌离子采用不同的模型时,Zn离子周围的部分残基在动力学采样中构象空间会有一些差别,但这种影响对所有抑制剂应该是相似的,这也会导致模型中常数项的变化.References
1Shoichet,K.;Kuntz,I. D.J.Mol.Biol.1991,221, 327.
2Luty, B. A.;Wasserman,Z.R.;Stoutern,P. F.W.;
Hodge, C.N.;McCammon,J. https://www.doczj.com/doc/1316373737.html,put.Chem.
1995,16,454.
3Jiang, F.;Kim,S.H.J.Mol.Biol.1991,219,79.
4Hou,T.J.;Wang,J.M.;Chen,L.R.;Xu,X.J.
Protein Eng.1999,12,639.
5Jorgensen,W.L.Acc.Chem.Res.1989,22,184.
6Koll man,P.Chem.Re v.1993,93,2395.
7Horton,N.;Lewis,M.Protein Sci.1992,1,169.
8Bohacek,R.S.;McMartin, C.J.Med.Che m.1992, 35,1671.
9Krystek,S.;Stouch,T.;Novotny,J.J.Mol.Biol.1993, 234,661.
10Bêh m,https://www.doczj.com/doc/1316373737.html, p ut-A ided Mol.Design1994,8,243. 11Matthew, D. E.;Christopher,W.M.;Timoghy,R. A.;
Gaia,V.P.;Roger,https://www.doczj.com/doc/1316373737.html, put.-Aide d Mol.
Design1997,11,425.
12!qvist,J.;Medina, C.;Samuelsson,J. E.Pr otein Eng.
1994,7,385.
13Woessner,J. F.FASBE J.1991,5,2145.
14Li otta,L. A.;Steeg,P.S.;Stetler-Stevenson,W.G.Cell 1991,64,327.
15Morgunova, E.;Tuuttila, A.;Bergmann,U.;Isupov,M.;
Lindqvist,Y.;Schneider,G.;Tryggvason,K.Science 1999,284,1667.
16Porter,J.R.;Beeley,N.R. A.;Boyce, B. A.;Mason,
B.;M illican, A.;Millar,K.;Leonard,J.;Morphy,J.
R.;O.Connell,J.P.Bioorg.Med.Che m.Lett.1994, 4,2741.
17Broadhurst,M.J.;Brown,P. A.;Lawton,G.;
Ballantyne,N.;Borkakoti,N.;Bottomley,K.M.K.;
Cooper,M.I.;Eatherton, A.J.;Kilford,I.R.;M alsher, P.J.;Nixon,J.S.;Lewis, E.J.;Su tton, B.M.;
Johnson,W.H.Bioorg.Me d.Chem.Lett.1997,7, 2299.
18Alpegiani,M.;Bissolino,P.;Absate, F.;Perrone, E.;
Corigli,R.;Jabes, D.Chem.A bstr.1999,130,139360. 19Natchus,M.G.;Cheng,M.Y.;Wahl, C.T.Bioorg.
Med.Chem.Lett.1998,8,2077.
20MOPAC7.0User Guide,Quantu m Chemis try Program Exchange(QCPE),Indiana Universi ty,1993.
21Gaussian98User Guide,Gaussian,Inc.,Pittsburgh,1998. 22Cleplak,P.;Cornell,W. D.;Bayly, C.;Koll man,P. A.
https://www.doczj.com/doc/1316373737.html, put.Chem.1994,16,1357.
23Case, D. A.;Pearlman, D. A.;Caldwell,J.W.;
226化学学报Vol.60,2002
Cheatham III,T. E.;Ross,W.S.;Simmerling, C.L.;
Darden,T. A.;Merz,K.M.;Stanton,R.V.;Cheng, A.
L.;Vincent,J.J.;Crowley,M.;Tsui,V.;Radmer,R.
J.;Duan,Y.;Pitera,J.;Massova,I.;Seibel,G.L.;
Sin gh,U. C.;Weiner,P.K.;Kollman,P. A.AMBER6, University of California,San Francisco,1999.
24Hou,T.J.;Wang,J.M.;Xu,X.J.Chemometr.Inte ll.
Lab.1999,45,303.
25Hou,T.J.;Wang,J.M.;Liao,N.;Xu,X.J.J.
Chem.In https://www.doczj.com/doc/1316373737.html,p.Sci.2000,39,775.
26Hou,T.J.;Zhang,W.;Xu,X.J.J.Ph ys.Chem.
2001,105,4304.
27!qvist,J.;Hansson,T.J.Phys.Chem.1996,100, 9512.
(A0106011PAN, B. F.)
227
No.2侯廷军等:MMP-2和Hyd roxamate类抑制剂绝对自由能的计算
Study on Molecular Reaction Dynamics for PuCO System
LI,Quan;WANG,Hong -Yan;JIANG,Gang;ZHU Zheng -He
Acta Chimica Sinica 2002,60(2),
215
The results for the collision process indicate that the main channel is the complexing reaction wi th no threshold energy for the collision Pu (7F g )+C O(0,0)and the exchange reacti on O(3P g )+PuC(0,0)
Pu(7F g )+
C O(X 12+)wi th nothreshold energy for the collision O (3P g )+PuC (0,
0).The reactive cross section will decrease with increase of relative kinetic energy.
Binding Free Energy Calculations for MMP 2-Hydroxamate Complexes
HOU,Ting -Jun;ZHANG,Wei;XU,Xiao -Jie Acta Chimica Sinica 2002,60(2),
221The absolute binding affi nities of a series of hydroxamate inhibitors wi th MMP-2were evaluated by molecular dynamics (MD)simulations with a linear response approach.During MD simulations,a nonbonded model for the catalytic zinc cen ter
was used to represent the interactions between zinc center and enzyme/inhibitor.The best model yields an average error of 2.38kJ/mol for the eight bindi ng affinities of hydrox amtes.
The Conformation of N -(phenylmethylene)-2-Naphthaleneamine -Like Species and the P -Driving Force for Distorting Geometry
GUO,Yan -Shen;YU,Zhong -Heng;JIN,Xiang -Lin
Acta Chimica Sinica 2002,60(2),228The P system in the geometry with H =0b is mos t des tabilized no matter whether it is delocalized or not,and the P system
always prefers a
twi sted geometry.
Quantum Chem istry Calculation Research on Hypomycin
B .
s
Intramolecular
Proton
Transfer on Ground State
C HE N,De -Zhan;KONG,De -Xin;ZHANG,
Hong -Yu
Acta Chimica Sinica 2002,60(2),234
Quantu m chemistry calculation on hypromycin B .s intramolecular proton transfer,a novel perylenequinonoid pigment with only one in tramolecular hydrogen bond.
ò
Graphical Abstract Vol.60,No.2.
亥姆霍兹自由能(Helmholtz free energy): F=U-TS, U 是系统的内能,T 是温度,S 是熵。(注意与吉布斯自由能的区别) 吉布斯自由能(Gibbs free energy): G=H-TS , H为焓,S为熵,T为当前温度 由于吉布斯自由能G 可以表示为G = F + pV,另有G = μN,所以F = μN –pV;亥姆霍兹自由能的微分形式是:dF = - SdT - PdV + μdN 其中P 是压强,V 是体积,μ是化学势 在统计物理学中,亥姆霍兹自由能是一个最常用的自由能,因为它和配分函数Z直接关联:F = -kTlnZ 吉布斯自由能的微分形式是: dG = ? SdT + Vdp + μdN, 其中μ是化学势,也就是说每个粒子的平均吉布斯自由能等于化学势; ΔG叫做吉布斯自由能变(吉布斯自由能判据) 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 吉布斯自由能改变量。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行方向和方式判据。 (功函判据) 亥姆霍兹函数是一个重要的热力学参数,等于内能减去绝对温度和熵的乘积:两个状态差值的负数等于一个可逆等温等容过程的最大功输出。 亥姆霍兹自由能是等温下做所有功的能力,亦称功函 吉布斯自由能是等温等压下除体积功以外的功的能力 玻尔兹曼常数(Boltzmann constant)(k 或kB)是有关于温度及能量的一个物理常数: 记为“K”,数值为:K=1.3806488(13)×10^-23J/K 理想气体常数等于玻尔兹曼常数与阿伏伽德罗常数的乘积: R=kN; 熵函数 熵可以定义为玻尔兹曼常数乘以系统分子的状态数的对数值: S=k㏑Ω; 焓变熵变 焓 焓是物体的一个热力学能状态函数,即热函:一个系统中的热力作用,等于该系统内能加上其体积与外界作用于该系统的压力的乘积的总和(Enthalpy is a combination of internal energy and flow work.)。 焓是一个状态函数,也就是说,系统的状态一定,焓的值就定了。 焓的定义式(物理意义)是这样的:H=U+pV [焓=流动内能+推动功] 其中U表示热力学能,也称为内能(Internal Energy),即系统内部的所有能量; p是系统的压力(Pressure),V是系统的体积(V olume) 。 焓变 焓变(Enthalpy changes)即物体焓的变化量。 焓变是生成物与反应物的焓值差。作为一个描述系统状态的状态函数,焓变没有明确的物理意义。
@ 吉布斯自由能 定义 ΔG=ΔH-TΔS (Kj/mol) 吉布斯自由能相关书籍封面(1) G叫做吉布斯自由能。因为H、T、S均为状态函数,所以G为状态函数。 特点 ΔG叫做吉布斯自由能变化 、 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 吉布斯自由能改变量。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行方向和方式判据。 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 范特霍夫等温公式 吉布斯自由能随温度和压强变化很大。为了求出非标准状况下的吉布斯自由能,可以使用范特霍夫等温公式: ΔG = ΔG0 + RT \ln J 其中,ΔG0是同一温度、标准压强下的吉布斯自由能,R是气体常数,J是反应熵。 温度的变化在ΔG0的使用上表现出来,不同的温度使用不同的ΔG0。非标准状况的ΔG0需要通过定义式(即吉布斯等温公式)计算。压强或浓度的变化在J的表达上表现出来。 】 研究对象 >W非反应以不可逆方式自发进行 =W非反应以可逆方式进行 <0 反应以不可逆方式自发进行 =0 反应以可逆方式进行 >0 不能进行 * 等温等压下体系的吉布斯自由能减小的方向是不做非体积功的化学反应进行的方向。 任何等温等压下不做非体积功的自发过程的吉布斯自由能都将减少。 标准自由能 在温度T时,当反应物和生成物都处于标准态,发生反应进度 标准自由能推理过程 为1 mol的化学反应Gibbs自由能的变化值,称为标准摩尔反应吉布斯自由能变化值,用表示标准吉布斯自由能与一般反应的吉布斯自由能的关系: # 标准自由能变化 标准自由能变化(△GO):相应于在一系列标准条件(温度298K,压力1atm(=),所有溶质的浓度都是不是mol/L)下发生的反应自由能变化。△GO′表示条件下的标准自由能变化。 平衡常数 在等温等压反应中,如果吉布斯自由能为负,则正反应为自发,反之则逆反应自发。如果为0,则反应处于平衡状态。此时,根据范特霍夫等温公式,ΔG = ΔG0 + RT \ln J,J变成平衡常数,于是有: ΔG0 = -RT ln K 要注意,使用范特霍夫等温公式时,ΔG和ΔG0的温度一定要相等。 这样,我们可以推出以下结论: ΔG0>0时,K<1; ¥ ΔG0=0时,K=1; ΔG0<0时,K>1。 自由能做功 有人可能会问:为什么单单用等温等压过程系统向环境作最大有用功的能力而不用包括气体膨 吉布斯系列 学号:120103709014 摘要:在物理化学当中,吉布斯自由能是物理化学中的一个重要的热力学函数,虽然他只是定义的一个函数,是若干热力学函数的数学组合。但吉布斯自由能概念几乎贯穿在整个物理化学的学习过程中,加深对吉布斯自由能定义、性质和判据的掌握,正确理解体系的吉布斯自由能变化的计算公式及其使用范围和条件,是掌握事物内在本质和学好物理化学的基础。 关键字:吉布斯函数、范特霍夫等温方程、吉布斯自由能与熵和焓、吉布斯自由能与平衡常数、吉布斯自由能与化学势 一、吉布斯函数 吉布斯函数(Gibbs function),系统的热力学函数之一。又称热力势、自由焓、吉布斯自由能等。符号G,定义为:G=H-TS 式中H、T、S分别为系统的焓、热力学温度(开尔文温度K)和熵。吉布斯函数是系统的广延性质,具有能量的量纲。由于H,S,T都是状态函数,因而G也必然是一个状态函数。 当体系发生变化时,G也随之变化。其改变值△G,称为体系的吉布斯自由能变,只取决于变化的始态与终态,而与变化的途径无关:△G=G终一G始 按照吉布斯自由能的定义,可以推出当体系从状态1变化到状态2时,体系的吉布斯自由能变为:△G=G2-G1=△H -△(TS) 对于等温条件下的反应而言,有T2=T1=T 则△G=△H-T △S 上式称为吉布斯-亥姆霍兹公式(亦称吉布斯等温方程)。由此可以看出,△G包含了△H和△S的因素,若用△G 作为自发反应方向的判据时,实质包含了△H和△S两方面的影响,即同时考虑到推动化学反应的两个主要因素。因而用△G作判据更为全面可靠。而且只要是在等温、等压条件下发生的反应,都可用△G作为反应方向性的判据,而大部分化学反应都可归入到这一范畴中,因而用△G作为判别化学反应方向性的判据是很方便可行的。 如果一个封闭系统经历一个等温定压过程,则有: ΔG≤W′(2)式中ΔG为此过程系统的吉布斯函数的变化值,W′为该过程中的非体积功,不等号表示该过程为不可逆过程,等号表示该过程为可逆过程。式(2)表明,在等温定压过程中,一个封闭系统吉布斯函数的减少值等于该系统在此过程中所能做的最大非体积功。 如果一个封闭系统经历一个等温定压且无非体积功的过程,则根据式(2)可得: ΔG≤0(3)式(3)表明,在封闭系统中,等温定压且不作非体积功的过程总是自动地向着系统的吉布斯函数减小的 吉布斯自由能又叫做吉布斯函数,是热力学中一个重要的参量,常用G表示,它的定义是:G = U ? TS + pV = H ? TS, 其中U是系统的内能,T是温度,S是熵,p是压强,V是体积,H是焓。 吉布斯自由能的微分形式是: dG = ? SdT + Vdp + μdN, 其中μ是化学势,也就是说每个粒子的平均吉布斯自由能等于化学势。 定义:ΔG=ΔH-TΔS (kJ/mol) G叫做吉布斯自由能。因为H、T、S均为状态函数,所以G为状态函数。 ?G叫做吉布斯自由能变,可作为恒温、恒压过程自发与平衡的判据。 热力学第一定律表达式:Q=?U+W U是热力学能(亦称为内能),H是焓,Q为热量,W为功量 定义焓:H=U+pV,相应的比焓:h=u+pv 范特霍夫等温公式 吉布斯自由能随温度和压强变化很大。为了求出非标准状况下的吉布斯自由能,可以使用范特霍夫等温公式: ΔG = ΔG0 + RT·ln J 其中,ΔG0是同一温度、标准压强下的吉布斯自由能,R是气体常数,J是反应商。 温度的变化在ΔG0的使用上表现出来,不同的温度使用不同的ΔG0。非标准状况的ΔG0需要通过定义式(即吉布斯等温公式)计算。压强或浓度的变化在J的表达上表现出来。 反应进行的方向: 定义吉布斯自由能G=H-TS。因为H、T、S均为状态函数所以G为状态函数。 吉布斯自由能改变量-ΔG=-(G2-G1)>=W非。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量-ΔG是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行的方向和方式可以由ΔG进行判断: -ΔG>W非反应以不可逆方式自发进行 -ΔG=W非反应以可逆方式进行 -ΔG 化学反应的标准右布斯自由能变化 r G m 0是反应产物与反应物都处于标准态 下化学势之差。化学反应的吉布斯自由能变化r G m 是反应产物和反应物皆处于 任意状态下化学势之差。 r G m ?与 r G m 是两个含义不同的物理量。在等温等压条 件下,任何物质的i 0都有定值,所以任何反应的 r G m ?都有是常数;但由化学反 应的等温式可知 r G m 不但与r G m ?有关,即与Q a 有关,所以在等温等压条件下 r G m 不是常数。换言之, r G m ? >0的化学反应未必不能正向自发进行,可以通 过Q a 值的调整使反应的r G m <0,反应即能正向自发进行。例如氨合成反应,在 673K 时,如果N 2、H 2和NH 3的分压都是101325Pa ,此时r G m ? = kJ mol -1,这个 数值大于零,在该条件下r G m >0,反应不能正向自发进行。若改变N 2、H 2和NH 3 的分压,则可使r G m <0,反应便能正向自发进行。式业合成氨就是这种情况下实 现的。 r G m ? 虽然不能用来指示反应的方向,但可以用来估计反应的可能性。等温 式告诉我们,如果 r G m ? 的绝对值很大,则 r G m 的正负号在一般情况下可能与 r G m ? 一致。譬如, r G m ? 有很大的负值,若要使改变符号,Q a 必很大,这在实 际上有时是难以实现的。例如反应: 在298K 时,该反应的 r G m ? =-mol -1.根据r G m ? =-RTlnK ? ,K P P O 001 2 12 (/) , 解得氧气的平衡分压P O 2=×10-107 Pa.要使 r G m >0,即Zn 不被氧化,氧的分压要 小于×10-107 Pa ,因此Zn 在空气中能自动地被氧化,而且反应很彻底。 作品编号:DG13485201600078972981 创作者:玫霸* 亥姆霍兹自由能(Helmholtz free energy): F=U-TS, U 是系统的内能,T 是温度,S 是熵。(注意与吉布斯自由能的区别) 吉布斯自由能(Gibbs free energy): G=H-TS , H为焓,S为熵,T为当前温度 由于吉布斯自由能G 可以表示为G = F + pV,另有G = μN,所以F = μN –pV; 亥姆霍兹自由能的微分形式是:dF = - SdT - PdV + μdN 其中P 是压强,V 是体积,μ是化学势 在统计物理学中,亥姆霍兹自由能是一个最常用的自由能,因为它和配分函数Z直接关联:F = -kTlnZ 吉布斯自由能的微分形式是: dG = ? SdT + Vdp + μdN, 其中μ是化学势,也就是说每个粒子的平均吉布斯自由能等于化学势; ΔG叫做吉布斯自由能变(吉布斯自由能判据) 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 吉布斯自由能改变量。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行方向和方式判据。 (功函判据) 亥姆霍兹函数是一个重要的热力学参数,等于内能减去绝对温度和熵的乘积:两个状态差值的负数等于一个可逆等温等容过程的最大功输出。 亥姆霍兹自由能是等温下做所有功的能力,亦称功函 吉布斯自由能是等温等压下除体积功以外的功的能力 玻尔兹曼常数(Boltzmann constant)(k 或kB)是有关于温度及能量的一个物理常数:记为“K”,数值为:K=1.3806488(13)×10^-23J/K 理想气体常数等于玻尔兹曼常数与阿伏伽德罗常数的乘积: R=kN; 熵函数 熵可以定义为玻尔兹曼常数乘以系统分子的状态数的对数值: S=k㏑Ω; 焓变熵变 焓 焓是物体的一个热力学能状态函数,即热函:一个系统中的热力作用,等于该系统内能加上其体积与外界作用于该系统的压力的乘积的总和(Enthalpy is a combination of internal energy and flow work.)。 焓是一个状态函数,也就是说,系统的状态一定,焓的值就定了。 焓的定义式(物理意义)是这样的:H=U+pV [焓=流动内能+推动功] 其中U表示热力学能,也称为内能(Internal Energy),即系统内部的所有能量; p是系统的压力(Pressure),V是系统的体积(V olume) 。 定义 G叫做吉布斯自由能。因为H、T、S均为状态函数,所以G为状态函数。 特点 ΔG叫做吉布斯自由能变化 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 吉布斯自由能改变量。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行方向和方式判据。 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 范特霍夫等温公式 吉布斯自由能随温度和压强变化很大。为了求出非标准状况下的吉布斯自由能,可以使用范特霍夫等温公式: ΔG = ΔG0 + RT \ln J 其中,ΔG0是同一温度、标准压强下的吉布斯自由能,R是气体常数,J是反应熵。 温度的变化在ΔG0的使用上表现出来,不同的温度使用不同的ΔG0。非标准状况的ΔG0需要通过定义式(即吉布斯等温公式)计算。压强或浓度的变化在J的表达上表现出来。 研究对象 >W非反应以不可逆方式自发进行 =W非反应以可逆方式进行 标准自由能变化(△GO):相应于在一系列标准条件(温度298K,压力1atm(=),所有溶质的浓度都是不是mol/L)下发生的反应自由能变化。△GO′表示条件下的标准自由能变化。 平衡常数 在等温等压反应中,如果吉布斯自由能为负,则正反应为自发,反之则逆反应自发。如果为0,则反应处于平衡状态。此时,根据范特霍夫等温公式,ΔG = ΔG0 + RT \ln J,J变成平衡常数,于是有: ΔG0 = -RT ln K 要注意,使用范特霍夫等温公式时,ΔG和ΔG0的温度一定要相等。 这样,我们可以推出以下结论: ΔG0>0时,K<1; ΔG0=0时,K=1; ΔG0<0时,K>1。 自由能做功 有人可能会问:为什么单单用等温等压过程系统向环境作最大有用功的能力而不用包括气体膨胀功在内的总功来度量系统发生自发过程的可能性呢?原因在于,系统发生自发过程,膨胀功是可正可负的。可见单单考虑系统作有用功,排除了膨胀功,问题才更纯,更明确。 总之,在等温等压条件下系统自发过程的判断是: △G< 0 即:△G<0,过程自发;△G>0,过程不自发(逆过程自发);△G=0,达到平衡态。一个自发过程,随着过程的发展,△G的绝对值渐渐减小,过程的自发性渐渐减弱,直到最后,△G=0,达到平衡。 化学反应中的自由能 对于一个化学反应,可以像给出它的标准摩尔反应焓△rHmΘ一样给出它的标准摩尔反应自由能变化△rGmΘ(为简洁起见,常简称反应自由能)。 跟热力学能变△U、焓变△H随温度与压力的改变不会发生大的改变完全不同,反应自由能△rGm随温度与压力的改变将发生很大的改变。因此,从热力学数据表中直接查出或计算出来的,标态下的△rGmΘ()的数据,不能用于其它温度与压力条件下,必须进行修正。 用热力学理论可以推导出,求取T温度下的气体压力对△rGmΘ的影响的修正公式为: J=∏(pi/pΘ)vi 其中∏是算符,表示连乘积(例如,a1×a2×a3=╥ai;i=1,2,3),pΘ为标态压力=100kPa,pi为各种气体(与△rGm(T)对应)的非标态压力,vi是化学方程式中各气态物质的计量系数,故J是以计量系数为幂的非标态下各气体的分压与标准压力之比的连乘积。 若系统中还有溶液,上式应改为: J=∏(pi/pΘ)Vi?∏(ci/cΘ)Vi 若系统中只有溶液,则上式又应改为: J=∏(ci/cΘ)Vi 对大多数化学反应而言,温度对反应自由能的影响要大大超过反应物的分压(以及浓度)对反应自由能的影响。通过实验或热力学理论计算,可以得出各种反应的自由能受温度的影响情形。若 吉布斯自由能 定义 G叫做吉布斯自由能。因为H、T、S均为状态函数,所以G为状态函数。 特点 ΔG叫做吉布斯自由能变化 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 吉布斯自由能改变量。表明状态函数G是体系所具有的在等温等压下做非体积功的能力。反应过程中G的减少量是体系做非体积功的最大限度。这个最大限度在可逆途径得到实现。反应进行方向和方式判据。 吉布斯自由能的变化可作为恒温、恒压过程自发与平衡的判据。 范特霍夫等温公式 吉布斯自由能随温度和压强变化很大。为了求出非标准状况下的吉布斯自由能,可以使用范特霍夫等温公式: ΔG = ΔG0 + RT \ln J 其中,ΔG0是同一温度、标准压强下的吉布斯自由能,R是气体常数,J是反应熵。 温度的变化在ΔG0的使用上表现出来,不同的温度使用不同的ΔG0。非标准状况的ΔG0需要通过定义式(即吉布斯等温公式)计算。压强或浓度的变化在J的表达上表现出来。 研究对象 >W非反应以不可逆方式自发进行 =W非反应以可逆方式进行 任何等温等压下不做非体积功的自发过程的吉布斯自由能都将减少。 标准自由能 平衡常数 在等温等压反应中,如果吉布斯自由能为负,则正反应为自发,反之则逆反应自发。如果为0,则反应处于平衡状态。此时,根据范特霍夫等温公式,ΔG = ΔG0 + RT \ln J,J变成平衡常数,于是有: ΔG0 = -RT ln K 要注意,使用范特霍夫等温公式时,ΔG和ΔG0的温度一定要相等。 这样,我们可以推出以下结论: ΔG0>0时,K<1; ΔG0=0时,K=1; ΔG0<0时,K>1。 自由能做功 有人可能会问:为什么单单用等温等压过程系统向环境作最大有用功的能力而不用包括气体膨胀功在内的总功来度量系统发生自发过程的可能性呢?原因在于,系统发生自发过程,膨胀功是可正可负的。可见单单考虑系统作有用功,排除了膨胀功,问题才更纯,更明确。 总之,在等温等压条件下系统自发过程的判断是: △G< 0 即:△G<0,过程自发;△G>0,过程不自发(逆过程自发);△G=0,达到平衡态。一个自发过程,随着过程的发展,△G的绝对值渐渐减小,过程的自发性渐渐减弱,直到最后,△G=0,达到平衡。 吉布斯自由能 (2009-04-09 10:35:19) 转载 标签: 分类:科技探索 自由能 标准状况 化学势 等压 吉布斯 宇宙 杂谈 吉布斯自由能又叫吉布斯函数,是热力学中一个重要的参量,常用 G 表示,它的定义是: 其中 U 是系统的内能,T 是温度,S 是熵,p 是压强,V 是体积,H 是焓。吉布斯自由能的微分形式是: 其中是化学势。一个重要的推论是。也就是说每个粒子的平均吉布斯自由能等于化学势。 目录 [隐藏] ? 1 物理意义 ? 2 生成吉布斯自由能 ? 3 范特霍夫等温公式 ? 4 吉布斯自由能与熵和焓 o 4.1 推导 o 4.2 相变 ? 5 吉布斯自由能与平衡常数 ? 6 吉布斯自由能与电化学 7 参阅 [编辑] 物理意义 吉布斯自由能的物理含义是在等温等压过程中,除体积变化所做的功以外,从系统所能获得的最大功。换句话说,在等温等压过程中,除体积变化所做的功以外,系统对外界所做的功只能等于或者小于吉布斯自由能的减小。数学表示是: 如果没有体积变化所做的功,即 W=0,上式化为: 也就是说,在等温等压过程前后,吉布斯自由能不可能增加。如果发生的是不可逆过程,反应总是朝着吉布斯自由能减少的方向进行。 特别地,吉布斯自由能是一个广延量,单位摩尔物质的吉布斯自由能就是化学势μ,也就是说。 具体推导如下:假设,代入热力学第一定律的微分形式: 现在假想保证原来物体属性的情况下,切掉体系的一小部分dN。这时,dT,dp,dg 这些强度量的变化为零。所以必然有。 [编辑] 生成吉布斯自由能 由于自由能的绝对值很难求出,同时它又是一个状态函数,所以实际应用时,就采用各种物质与稳定单质的相对值,即生成吉布斯自由能,全称标准摩尔生成吉布斯自由能变。它是由处于标准状况下的稳定单质生成一摩尔标准状况下的化合物的吉布斯自由能变,用符号表示。 通过生成吉布斯自由能,我们能算出标准反应自由能。标准反应自由能是指在标准状况下,标况下的反应物生成标况下的生成物所需要的能量变化,即用生成物的生成吉布斯自由能与各自的化学计量系数相乘后减去的反应物的生成吉布斯自由能与各自的化学计量系数相乘后的乘积。常用符号表示。 对于一般反应:aA + bB → cC + dD 它的标准反应自由能 = [c(C)+ d(D)] - [a(A)+ b(B)] 一般化学教科书和化学手册中都列出常见物质的生成吉布斯自由能(同时还会列出生成焓和标准熵),用时直接查表便可。 [编辑] 范特霍夫等温公式 吉布斯自由能[编辑] 维基百科,自由的百科全书 (重定向自生成吉布斯自由能) 跳转至:导航、搜索 本条目需要补充更多来源。(2014年10月31日) 请协助添加多方面可靠来源以改善这篇条目,无法查证的内容可能会被提出异议而移除。 吉布斯自由能(英语:Gibbs free energy),或称吉布斯函数(Gibbs function)、自由焓(Free Enthalpy)是热力学中描述等温、等压过程的一个重要参量,常用表示,它的定义是: , 其中是系统的内能,是温度,是熵,是压强,是体积,是焓。 吉布斯自由能的微分形式是: , 其中是化学势。一个重要的推论是。也就是说每个粒子的平均吉布斯自由能等于化学势。 目录 [隐藏] ? 1 物理意义 ? 2 生成吉布斯自由能 o 2.1 部分物质的生成自由能[1] ? 3 范特霍夫等温公式 ? 4 吉布斯自由能与熵和焓 o 4.1 推导 o 4.2 相变 ? 5 吉布斯自由能与平衡常数 ? 6 吉布斯自由能与电化学 ?7 参阅 ?8 参考文献 物理意义[编辑] 在标准状况下,存在一个一般规律: “ 任何一个封闭系统都尽量使自由能最小 ” 因此,根据这个自然界的基本趋势,如果对于一个潜在反应,距离这个最小值进行定量测量,当热力学的计算显示吉布斯自由能ΔG的变化是负值的时候。本质上,这表明了那样一个反应更容易发生并且将释放能量。释放的能量等于这个化学反应所能够做的最大的功。相反,如果ΔG为正值,能量必须通过做功的方式进入反应系统使得此反应能够进行。 吉布斯自由能的物理含义是在等温等压过程中,除体积变化所做的功以外,从系统所能获得的最大功。换句话说,在等温等压过程中,除体积变化所做的功以外,系统对外界所做的功只能等于或者小于吉布斯自由能的减小。数学表示是: 如果没有体积变化所做的功,即,上式化为: 也就是说,在等温等压过程前后,吉布斯自由能不可能增加。如果发生的是不可逆过程,反应总是朝着吉布斯自由能减少的方向进行。 特别地,吉布斯自由能是一个广延量,单位摩尔物质的吉布斯自由能就是化学势,也就是说。 , 具体推导如下:假设,代入热力学第一定律的微分形式: , 现在假想保证原来物体属性的情况下,切掉体系的一小部分。这时, ,,这些强度量的变化为零。所以必然有, 生成吉布斯自由能[编辑] 附表18 一些物质的标准生成焓、标准吉布斯函数和25℃、100kPa 时的绝对熵 附表18 一些物质的标准生成焓、标准吉布斯函数和25℃、100kPa 时的绝对熵 0f H Δ 0f G Δ 0m S 物质 分子式 相对分子质量 M r J/mol J/mol J/(mol K)? 水(g) 水(l) 过氧化氢(g) 臭氧(g) 碳(石墨)(s) 一氧化碳(g) 二氧化碳(g) 甲烷(g) 乙炔(g) 乙烯(g) 乙烷(g) 丙烯(g) 丙烷(g) 丁烷(g) 戊烷(g) 苯(g) 己烷(g) 庚烷(g) 辛烷(g) 辛烷(l) 甲醇(g) 乙醇(g) 氨(g) 柴油(l) 硫(s) 二氧化硫(g) 三氧化硫(g) 氧化氮(g) 硝基甲烷(l) H 2O H 2O H 2O 2O 3C CO CO 2CH 4C 2H 2 C 2H 4C 2H 6C 3H 6 C 3H 8C 4H 10C 5H 12 C 6H 6C 6H 14C 7H 16 C 8H 18C 8H 18CH 3OH C 2H 5OH NH 3C 14.4H 24.9 S SO 2SO 3N 2O CH 3NO 2 18.015 18.015 34.015 47.998 12.011 28.011 44.010 16.043 26.038 28.054 30.070 42.081 44.094 58.124 72.151 78.114 86.178 100.20511 4.232 114.232 32.042 46.069 17.031 198.06 32.06 64.059 80.058 44.013 61.04 -241826-285830-136106+142674 0 -110527 -393522-74873 +226731 +52467 -84740 +20430 -103900-126200 -146500 +82980 -167300-187900 -208600-250105-201300 -235000-45720 -174000 0 -296842-395765+82050 -113100 -228582 -237141 -105445 +163184 0 -137163 -394389 -50768 +209200 +68421 -32885 +62825 -23393 -15970 -8208 +129765 +28 +8227 +16660 +6741 -162551 -168319 -16128 +178919 0 -300125 -371016 +104179 -14439 188.834 69.950 232.991 238.932 5.740 197.653 213.795 186.251 200.958 219.330 229.597 267.066 269.917 306.647 348.945 269.562 387.979 427.805 466.514 360.575 239.709 282.444 192.572 525.90 32.056 248.212 256.769 219.957 171.80 本表引自:C Borgnakke ,R E Sonntag. Thermodynamic and Transport Properties. New York :John Wiley & Sons Inc ,1997 37 天津大学 无机化学教学团队 第二章 高等教育出版社 化学反应的方向、 速率和限度 第一节 化学反应的方向和吉布斯自由能变 第二章 化学反应的方向、速率和限度 摩尔吉布斯自由能变量(简称自由能变),以?r G m 表示,单位:J·mol -1。p 吉布斯公式 在等温、等压下,不作非体积功的前提下,化学反应摩尔吉布斯自由能变(?r G m )与摩尔反应焓变(?r H m )、摩尔反应熵变(?r S m )、温度(T )之间有如下关系: ?r G m = ?r H m – T ?r S m 2.1.2影响化学反应方向的因素化学反应的吉布斯自由能变──热化学反应方向的判据 化学反应的吉布斯自由能变──热化学反应方向的判据 p在等温、等压的封闭体系内, 不作非体积功,?r G m可作为热化学反应自发过程的判据。 即:?r G m < 0 自发过程, 化学反应自发正向进行 ?r G m = 0 平衡状态 ?r G m > 0 非自发过程,化学反应逆向进行 化学反应的吉布斯自由能变──热化学反应方向的判据 p等温、等压的封闭体系内,不作非体积功的前提下,任何自发过程总是朝着吉布斯自由能(G)减小的方向进行。 r G m= 0 时, 体系的G降低到最小值, 反应达平衡。此即为著名的最小自由能原理。 = (–) – (+)?r G m =?r H m -T ?r S m 各种 情况符 号反应情况 ?r H m ?r S m ?r G m 1-+-任何温度下均为 自发反应2+-+ 任何温度下均为 非自发反应 3++常温(+) 高温(-)常温下为非自发反应 高温下为自发反应4 --常温(-) 高温(+) 常温下为自发反应 高温下为非自发反应 “高温”是指当T > 时 Δr H m Δr S m (–) = (–) – (+)吉布斯函数
吉布斯自由能
化学反应的吉布斯自由能变化
亥姆霍兹自由能和吉布斯自由能的区别
吉布斯自由能
吉布斯自由能
吉布斯函数
吉布斯自由能
一些物质的标准生成焓、标准吉布斯函数和 25℃、100kPa 时的绝对熵
2.1.2吉布斯自由能变
相关主题
文本预览