分子生物学实验设计报告
绿色荧光蛋白的克隆表达
一、引言
荧光蛋白是海洋生物体内的一类发光蛋白,分为绿色荧光蛋白、蓝色荧光蛋白、黄色荧光蛋白和红色荧光蛋白。
绿色荧光蛋白是由日本学者下村修于1962年从多管水母中发现并分离得到的一种发光蛋白。它是由238个氨基酸构成的“β-桶”型三维立体结构,其中65至67位氨基酸(丝氨酸-酪氨酸-甘氨酸)形成发光团,为主要的发光位置。
通过生物化学的方法将基因做小小的改变,就可以改变GFP中的氨基酸,得到变异GFP。目前应用较多的GFP的突变体-增强型绿色荧光蛋白(简称EGFP)
传统的PCR产物克隆方法主要有两种:一种是PCR引物设计时引入载体上的酶切位点,PCR产物经双酶切后定向克隆到目的载体上;另一种是TA载体连接。这两种方法费时费力,过程繁冗。
而本实验采用的无缝克隆和组装技术是一种新的、快速、简洁的克隆方法,旨在克服上述缺陷,它可以在质粒的任何位点进行一个或多个目标DNA的片断的插入,而不需要任何限制性内切酶和连接酶。突破传统的双酶切再加上连接,只需要一步重组法,即可得到高效率克隆的重组载体,这个重组载体能够在一定的宿主细胞中进行扩增,形成大量的子代分子。
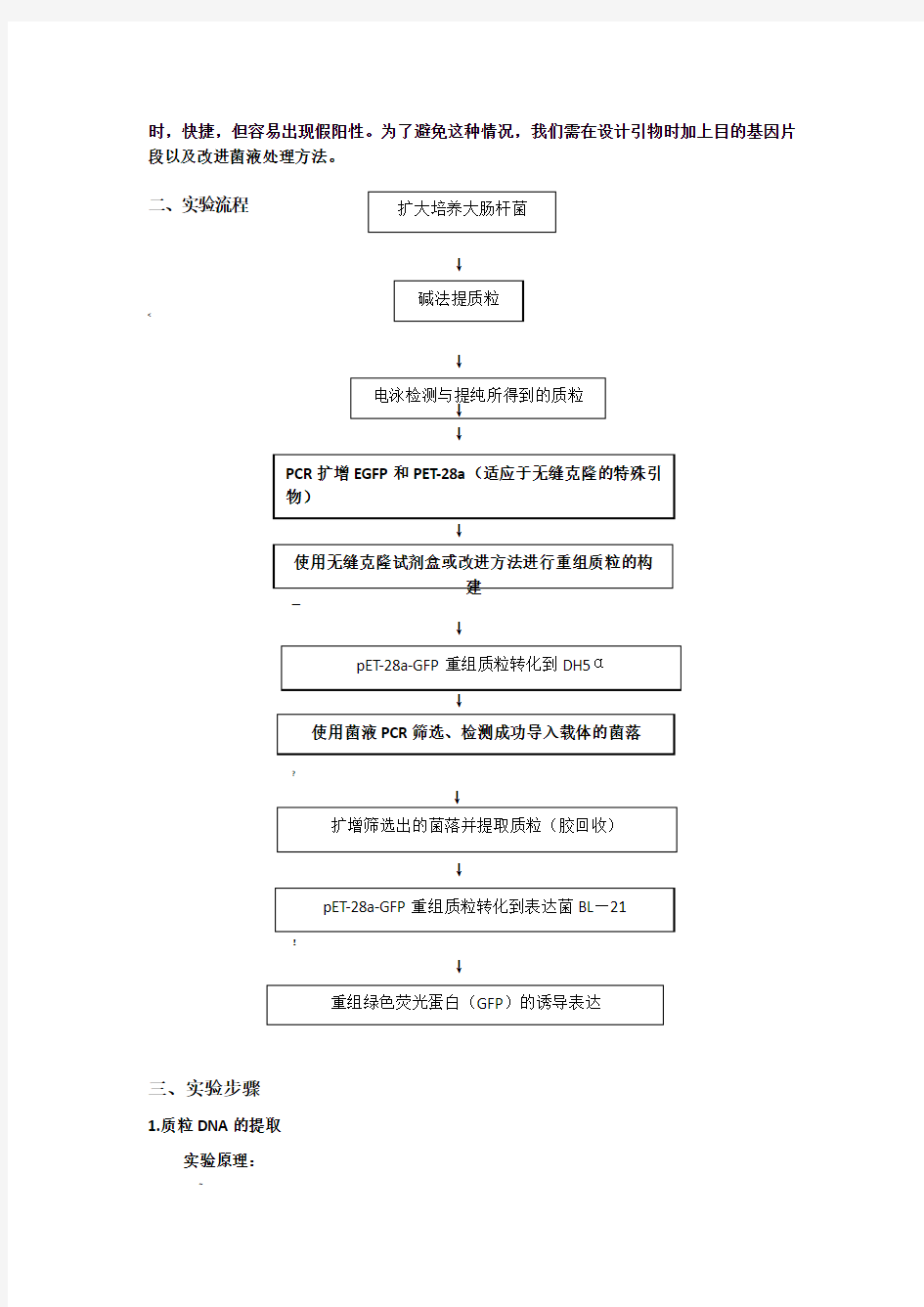
研究绿色荧光蛋白在大肠杆菌体内的基因克隆和表达。通过质粒重组形成所需要的重组质粒pET-28a-GFP,将重组质粒导入大肠杆菌体内,通过酶切、PCR及用IPTG诱导检测是否在大肠杆菌体内诱导表达成功。
<
菌液PCR是直接用菌液作为模板进行PCR的一种选取成功导入载体的菌落的方法,省
时,快捷,但容易出现假阳性。为了避免这种情况,我们需在设计引物时加上目的基因片段以及改进菌液处理方法。
三、实验步骤
1.质粒DNA的提取
实验原理:
~
质粒:一种染色体外的遗传因子,大小在1kb~200kb之间,是具有双链闭合环状结构的DNA分子,主要发现于细菌、放线菌和真菌细胞中。质粒具有自主复制能力,,常常编码一些对宿主有利的酶的基因,包括抗生素、修饰酶等。
载体:要把一个有用的外源基因通过基因工程手段,转化到细胞中去进行繁殖和表达,需要运载工具,携带外源基因进入受体细胞的这种工具就叫载体。目前除了大肠杆菌中的质粒、λ噬菌体、M13噬菌体、噬菌粒外,还有酵母人工染色体载体以及动、植物病毒载体等。
碱变性法抽提质粒DNA:基本原理是根据染色体DNA和质粒DNA分子量的巨大差异而达到分离的。在实验时,我们先分散细胞,通过螯合金属离子使酶失活,并防止DNA的降解。随后细胞在NaOH和SDS溶液中裂解时,蛋白质与染色体DNA发生变性,在酸性条件下质粒DNA复性,同时留在上清液中。而大肠杆菌DNA和蛋白质-SDS复合物等发生沉淀。由此达到分离的效果
实验目的:掌握碱法抽提质粒DNA的原理和方法。
实验材料:
材料:含pET- 28a质粒的大肠杆菌,含pEGFP-N3质粒的大肠杆菌
试剂:
@
1)溶液Ⅰ50 mL, 葡萄糖, 50 mmol/L , 25 mmol/L Tris-HCl (pH , 10 mmol/L EDTA (pH , 用前加溶菌酶4mg/℃高压灭菌15 min后置于0~4℃贮存;
2)溶液Ⅱ100 mL, mol/L NaOH ,SDS, 1% (W/V)
用时由母液2 mol/L NaOH、10%(W/V) SDS稀释现配;
3)溶液Ⅲ100 mL, 60 mL KOAc (5 mol/L), mL冰乙酸, mL H2O .该溶液钾离子浓度为3 mol/L, 醋酸根离子浓度为5mol/L;
4)酚-氯仿(1:1,V:V);
5)TE缓冲液:10mmol/LTris-HCl , 1mmol/LEDTA , 其中RNAase20ug/mL.
6)70%乙醇,无水乙醇;
7)液体LB培养基(), 10 g/L胰蛋白胨, 5 g/L酵母提取物, 10 g/L NaCl , 1 mL/L NaOH(1 mol/L);
>
8)氨苄青霉素25mg/mL贮备液
仪器设备:恒温水浴锅,高压灭菌锅,超净工作台,移液器,高速离心机,制冰机,漩涡振荡器
实验方法:
1 将含pET- 28a质粒的大肠杆菌,含pEGFP-N3质粒的大肠杆菌分别接种在液体培养基中(加卡那霉素50μg/mL),37℃培养过夜。
2 取培养菌液置Eppendorf小管中,10000rpm离心2min,弃上清液。
3 加入100μL溶液I,漩涡器上充分混匀,在室温下放置10 min。
4 加入200μL新配制的溶液Ⅱ,轻轻翻转2~3次,使之混匀,冰上放置
5 min。
5 加入150μL冰冷的溶液Ⅲ,盖紧后,温和颠倒3-5次使混匀,产生白色絮状物,冰上放置15 min。
&
6 10000rpm离心5 min,取上清液于另一干净的离心管中。
7 向上清液中加入等体积(约400μL)酚:氯仿/异戊醇(25:24:1,v/v),振荡混匀,
10000rpm 离心10 min,将上清液转移至新的离心管中。
8 加入等体积(约370μL)氯仿/异戊醇(24:1),混匀,离心2min ,取上清液于新离心
管中。
9 向上清液加入2倍体积无水乙醇,混匀,-20℃放置1h。
10 12000rpm 离心5 min,倒去上清液,把离心管倒扣在吸水纸上,吸干液体。
11 加800μL70%乙醇,12000rpm离心1 min,倒尽上清液,室温自然干燥30min。
2.琼脂糖凝胶电泳与胶回收提纯所提DNA
*
实验原理
电泳:带电荷的物质,在电场中的趋向运动称为电泳。DNA的琼脂糖凝胶电泳可以分离长度为200bp至近50kb的DNA分子。DNA的迁移率(U)的对数与凝胶浓度(T)之间存在反平行线性关系。因此,要有效地分离不同大小的DNA段,选用适当的琼脂糖凝胶浓度是非常重要的。在质粒提取的过程中,由于操作原因,提取的质粒可能有三种:线性DNA、开环DNA 、闭环超螺旋DNA 。当提取的质粒DNA电泳时,同一质粒DNA泳动速度:闭环超螺旋〉线状〉开环。但有时也会出现相反情况,因为与琼脂糖浓度、电流强度、离子强度及核酸染料含量有关。琼脂糖是由琼脂分离制备的链状多糖。其结构单元是d-半乳糖和3,6-脱水-L-半乳糖。许多琼脂糖链依氢键及其它力的作用使其互相盘绕形成绳状琼脂糖束,构成大网孔型凝胶。影响DNA在琼脂糖凝胶中迁移速率的因素主要有:(1)DNA分子的大小。分子越大,迁移的越慢,摩擦阻力越大,大分子通过凝胶孔径的效率低于较小的分子。(2)琼脂糖浓度。(3)DNA的构象。(4)所用的电压。低电压时,DN**段迁移率与所用的电压成正比。电场强度升高时,高分子量片段的迁移率遂不成比例的增加。要获得大于2kb DN**段的良好分辨率,所用电压不应高于5-8V/cm。(5)电泳缓冲。缺乏离子则电导率降低,DNA 或者不动或者迁移很慢。高离子强度时DNA变性可能会因为产热变性。
实验目的:检验是否获得所需的质粒
实验材料:
材料和仪器
电泳仪,电泳槽,样品槽模板(梳子),有机玻璃内槽,微波炉,水平仪,10、100、1000μL微量取液器各一支,凝胶成像系统,提取的pET- 28a和pEGFP-N3质粒,琼脂糖,锥形瓶,一次性手套,胶铲,封口膜,剪刀,取液器吸头。
试剂:
Gold view(DNA染料);×TBE缓冲液:Tris 、硼酸、EDTA·Na2·2H2O ,定容至200ml,使用时稀释10倍;6×上样缓冲液:溴酚蓝%、蔗糖40%;DNA marker。
!
实验方法:
琼脂糖凝胶板的制备
称取琼脂糖,置于锥形瓶中,加入×TBE缓冲液,微波炉加热直至琼脂糖溶解。
待胶液冷却到65℃(不烫手为宜),加入1μL Gold view混匀,排除气泡,缓慢倒入事先准备的制胶有机玻璃槽(插入样品槽模板梳子)内。静置30min左右,轻轻晃动拔出梳子。
加样
胶板放入电泳槽中(样品孔位于负极),注入×TBE缓冲液淹没过胶板5mm,用取液器将提取的质粒DNA5μL与1μL6×上样缓冲液混匀,加入胶板的样品小槽内。
电泳
将电泳槽与电泳仪接通,调节电压为100V左右,直至缓冲液的蓝色指示剂移动到距离胶板边沿约1~2cm处,停止电泳。
{
观察和拍照
将胶板小心取出,放入凝胶成像系统中,调节位置居中,波长为254nm的紫外灯下,观察凝胶中DNA存在处显示出荧光条带。
用胶回收试剂盒回收质粒
3.PCR扩增EGFP和PET-28a(适应于无缝克隆的特殊引物)并进行质粒重组
实验原理
无缝克隆和组装技术是一种新的、快速、简洁的克隆方法,旨在克服传统方法费时费力,过程繁冗的缺陷,它可以在质粒的任何位点进行一个或多个目标DNA的片断的插入,而不需要任何限制性内切酶和连接酶。突破传统的双酶切再加上连接,只需要一步重组法,即可得到高效率克隆的重组载体
基因克隆的引物设计及目的DNA片段的扩增,与常规PCR法是相同的。唯一的差异在于载体末端和引物末端应具有15-20个同源碱基,由此得到的PCR产物两端便分别带上了15-20个与载体序列同源性的碱基。
在没有dNTP存在的情况下用T4 DNA聚合酶中的3’5’外切酶活性22℃处理,产生含有同源序列的5’端突起.突出端的长度可以通过控制3’5’外切酶活性的作用时间来实现,加入dCTP即可终止外切酶活性。将各片段的酶切产物按等摩尔比混合,在T4 D NA连接缓冲液中于3 7℃完成突出单链的复性重组,形成含有大的缺口的环状中间体.用此中间体转化大肠杆菌,利用大肠杆菌本身的修复作用完成中间体的修复。
。
而本次实验更是采用无缝克隆法的改进方法,利用混合酶切法提高效率。同时使用特殊的退火液与退火程序消除在无缝克隆中由于延伸的单链太长而产生的二级结构从而对DNA链的连接造成的影响。
T4 DNA聚合酶在没有dNTP时表现很强的核酸外切酶活性(3’5’),产生5 ‘ssD NA突出端,单链区的长度可以通过控制反应时间来实现,若同源臂为30bp,则在22℃下处理4 5 min,2 0 bp时则处理3 0 min.待重组的DNA片段以分别单独酶切处理或所有DNA片段等摩尔体积混合再酶切处理。。
实验目的:得到pET-28a-GFP重组质粒
实验材料:PET-28a质粒,PEGFP-N3质粒,引物,Taq酶,dNTP,buffer,退火缓冲液,
实验步骤
设计引物
根据EGFP基因序列与PET-28a序列再加上前面15-20nt的特异性序列设计出引物。
:
PCR
利用设计好的引物对EGFG基因序列与PET-28a进行PCR扩增
.将引物,模板,taq酶放入PCR仪中
.设置反应程序:
①94 ℃预变性5min;
②94 ℃变性30S;
③55 ℃退火30S;
④72 ℃延伸60S;
\
⑤重复步骤②~④30次;
⑥72 ℃延伸10min。
T4 DNA聚合酶处理
T4 DNA聚合酶反应系统:1μg DNA片段、4μL 5×T4 DNA聚合酶缓冲液、μL T4 DNA 聚合酶(5 U/μL),加ddH2O至20μL.
酶切完成后,在每个反应体系中迅速加入2μL dCTP(1 0 mmol/L),混匀终止反应并放于冰上.
退火和转化
混合酶切产物的退火体系:混合片段酶切液4μL(0.2μg DNA)
1 0×退火缓冲液1μL ,1 mol/L NaCl,10mmol/L EDTA).
】
ddH20 5μL
采用的退火程序是:75℃10 min,每8S降低℃,至室温。
得到重组的质粒
重组质粒转化到DH5α中进行扩增
实验原理:
细菌处于容易吸收外源DNA的生理状态叫感受态。转化是指质粒DNA或以它为载体构建的重组DNA分子导入细菌的过程。其原理是细菌处于0℃时,在有CaCl2存在的低渗溶液中,细菌细胞膨胀成球形。转化混合物中的DNA形成抗DNA酶的羟基-钙磷酸复合物粘附于细胞表面,经42℃短时间热击处理,促进细胞吸收DNA复合物。将细菌放置在非选择性培养基中保温一段时间,促使在转化过程中获得的新的表型(如抗卡那霉素等)得到表达。而大肠杆菌DH5α是以扩增质粒为主要功能,所以利用此
性质来扩增重组质粒。
实验目的:将pET-28a-GFP重组质粒转化到DH5α中进行扩增
实验材料:
¥
材料和仪器:大肠杆菌DH5α、重组质粒、EP管、10μL、200μL和1000μL微量移液器、培养皿、接种环、高压蒸汽灭菌锅、台式高速冷冻离心机、制冰机、恒温摇床、三角瓶、超净工作台
试剂:LCaCl2溶液:高压蒸汽灭菌后4℃冰箱保存。
LB液体培养基:蛋白胨1克,酵母提取物克,NaCl 1克,去离子水100ml。高压蒸汽灭菌20min。
LB固体培养基:蛋白胨1克,酵母提取物克,NaCl 1克,琼脂糖~3克,去离子水100ml,高压蒸汽灭菌20min。
实验方法:
感受态细胞的制备( CaCl2 法)
从新活化的大肠杆菌DH5α平板上挑取一个单菌落,接种于2mlLB液体培养基试管中,放入恒温摇床,37℃振荡培养过夜,转速为200r/min。
取菌液2ml转接到一个装有50mlLB液体培养基的三角瓶中,37℃振荡培养2~3小时,按1:100扩大培养。
*
取菌液至管中,冰上放置10分钟,以免处于对数生长期的细菌继续繁殖。
置高速冷冻离心机中,4℃4000r/min离心10min,弃上清,回收菌体细胞。
用预冷的L的CaCl2 溶液1ml悬浮沉淀,用移液器轻轻吹打沉淀,使菌体细胞均匀悬浮,并立即冰浴15min,使菌体细胞完全冷下来。
再次置高速冷冻离心机中,4℃4000r/min离心10min,弃上清,回收菌体细胞。
用预冷的L的CaCl2 溶液重悬沉淀,此细胞为感受态细胞。
转化
取出感受态细胞DH5α让其在冰上自然解冻,每200μL解冻的感受态细胞加5μL pET-28a-EGFP质粒,混匀后冰浴30min。
420C热冲击90秒,立即置于冰浴5min。
/
每管加LB液体培养基800μL,轻轻混匀,在37℃,150r/min摇床中温育40min,让细菌复苏,使进入细菌中的质粒表达抗生素抗性蛋白。
26000r/min 离心3min,去掉上清(约留200μL的培养液),摇匀菌块。
用玻璃涂布器将溶液均匀涂布在含卡那霉素的LB固体培养基上,37℃培养箱中倒置培养皿过夜。
第二天早上挑取单菌落,在LB液体培养基中培养扩增。
4、1h 后,6000r/min 离心5min,去掉上清(约留200μL的培养液),摇匀菌块。
5、用玻璃涂布器将溶液均匀涂布在含IPTG和Kana的LB固体培养基上,正置培养皿半小时后,370C培养箱中倒置培养皿过夜。
6、第二天早上观察结果。
5.菌液PCR筛选成功表达的菌落
】
实验原理
菌落PCR是直接利用菌液经过处理以后作为PCR的模板进行目的基因的扩增以检测目的基因是否成功导入的方法。此方法方便快捷,但需设计好引物以及处理好菌液,以免出现假阳性。
实验目的
筛选、检测出成功导入目的基因的菌落
实验材料
菌落、dNTP、引物、Taq酶、buffer 电泳液
PCR仪电泳槽
实验步骤
…
设计引物:
选择一个载体上的特异性序列和一个目的基因上的特异性序列作为引物,如此可避免未与载体连接上的插入片段也被扩增出来呈假阳性。
菌落的处理
挑取长出的1-2μl菌液稀释为100倍体积,沸水浴10-15mins
PCR体系:20或25μl
模板的量:1-2ul
退火温度:62°C
循环数28-35
$
电泳检测
6.扩增培养选出的菌落,并提取质粒(胶回收)
同实验一
在液体培养基扩增培养后用碱法提取质粒并用电泳+胶回收的方法提纯
重组质粒转化到表达菌BL-21
实验原理:BL21是用于表达质粒的高效工程菌,将提纯后的重组质粒转化到其中可得到大量的表达。
实验目的:将pET-28a-GFP重组质粒转化到表达菌BL-21上
:
实验材料:
材料和仪器:大肠杆菌BL21、重组质粒、EP管、10μL、200μL和1000μL微量移液器、培养皿、接种环、高压蒸汽灭菌锅、台式高速冷冻离心机、制冰机、恒温摇床、三角瓶、超净工作台
试剂:LCaCl2溶液:高压蒸汽灭菌后4℃冰箱保存。
LB液体培养基:蛋白胨1克,酵母提取物克,NaCl 1克,去离子水100ml。高压蒸汽灭菌20min。
LB固体培养基:蛋白胨1克,酵母提取物克,NaCl 1克,琼脂糖~3克,去离子水100ml,高压蒸汽灭菌20min。
实验方法:
感受态细胞的制备( CaCl2 法)
从新活化的大肠杆菌DH5α平板上挑取一个单菌落,接种于2mlLB液体培养基试管中,放入恒温摇床,37℃振荡培养过夜,转速为200r/min。
取菌液2ml转接到一个装有50mlLB液体培养基的三角瓶中,37℃振荡培养2~3小时,按1:100扩大培养。A600约为时即可停止培养,此时菌体出于对数生长期前期。
取菌液至管中,冰上放置10分钟,以免处于对数生长期的细菌继续繁殖。
置高速冷冻离心机中,4℃4000r/min离心10min,弃上清,回收菌体细胞。
用预冷的L的CaCl2 溶液1ml悬浮沉淀,用移液器轻轻吹打沉淀,使菌体细胞均匀悬浮,并立即冰浴15min,使菌体细胞完全冷下来。
再次置高速冷冻离心机中,4℃4000r/min离心10min,弃上清,回收菌体细胞。
用预冷的L的CaCl2 溶液重悬沉淀,此细胞为感受态细胞。
重组DNA的转化
取出感受态细胞让其在冰上自然解冻,每200μL解冻的感受态细胞加入重组质粒1μL,混匀后冰浴30min。
42℃热冲击90秒,勿摇动EP管,置于冰上2min。
每管加LB液体培养基800μL,轻轻混匀,在37℃,150r/min摇床中温育40min,让细菌复苏.
将细菌涂布于LB固体培养基上,培养
紫外灯照射,筛选出成功转入荧光蛋白的菌落
四、参考文献
[1] 梁国栋. 最新分子生物学实验技术[ M] . 北京: 科学技术出版社, 2001, 174.
[2] NovoRec? PCR一步定向克隆(无缝克隆)技术手册
[3] 不依赖基因序列和连接反应克隆(SLIC)方法的改进王广琚,何川,张义正
[4] 菌液PCR的经验丁香园
[5] 无缝克隆试剂盒指南上海西宝生物科技有限公司主页
[6] 魏春红,李毅主编原核细胞中外源基因的表达和初步纯化,现代分子生物学技术,高
等教育出版社,:95-9
[7] 林宏辉,赵云,王茂林,陈放主编现代生物学基础实验指导,四川大学出版社
[8] 实验技术手册生物治疗国家重点实验室