受体酪氨酸激酶通路
- 格式:ppt
- 大小:7.92 MB
- 文档页数:37
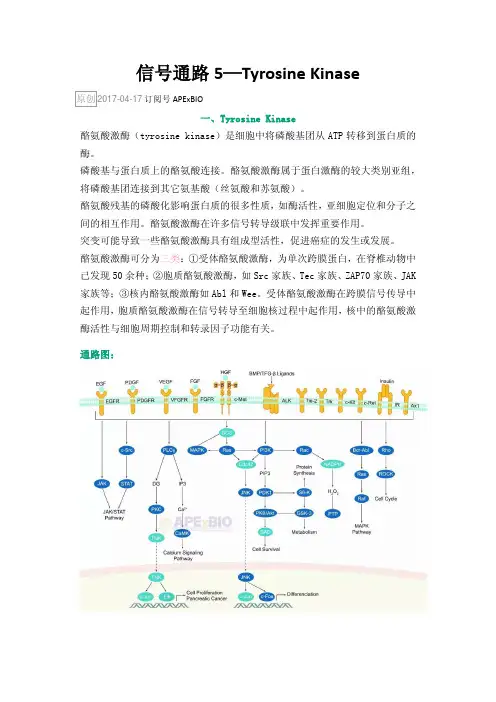
信号通路5—Tyrosine KinaseAPExBIO一、Tyrosine Kinase酪氨酸激酶(tyrosine kinase)是细胞中将磷酸基团从ATP转移到蛋白质的酶。
磷酸基与蛋白质上的酪氨酸连接。
酪氨酸激酶属于蛋白激酶的较大类别亚组,将磷酸基团连接到其它氨基酸(丝氨酸和苏氨酸)。
酪氨酸残基的磷酸化影响蛋白质的很多性质,如酶活性,亚细胞定位和分子之间的相互作用。
酪氨酸激酶在许多信号转导级联中发挥重要作用。
突变可能导致一些酪氨酸激酶具有组成型活性,促进癌症的发生或发展。
酪氨酸激酶可分为三类:①受体酪氨酸激酶,为单次跨膜蛋白,在脊椎动物中已发现50余种;②胞质酪氨酸激酶,如Src家族、Tec家族、ZAP70家族、JAK 家族等;③核内酪氨酸激酶如Abl和Wee。
受体酪氨酸激酶在跨膜信号传导中起作用,胞质酪氨酸激酶在信号转导至细胞核过程中起作用,核中的酪氨酸激酶活性与细胞周期控制和转录因子功能有关。
通路图:二、相关蛋白或基因1. Bcl-AblBcl-Abl是组成型激活的嵌合酪氨酸激酶。
Bcr-Abl酪氨酸激酶失活导致慢性粒细胞白血病(CML)。
Bcr-Abl酪氨酸激酶抑制剂用于大多数CML患者的一线治疗。
2. GSK-3Glycogen synthase kinase 3,糖原合成酶激酶3。
GSK-3是一种丝氨酸/苏氨酸激酶,主要作用是使糖原合成酶发生磷酸化而失活。
GSK-3基因家族包括GSK-3α和 GSK-3β。
胰岛素引起的Akt激活,上皮生长因子、血小板衍化生长因子等引起的Ras/Raf/ERK/p90Rsk1激活以及p90Rsk、P70S6K均能引起GSK-3α和 GSK-3β磷酸化使其失去活性,参与调节多种疾病的生理过程,包括II型糖尿病,阿尔茨海默病,炎症,癌症和双相情感障碍。
3. SykSpleen tyrosine kinase,脾脏酪氨酸激酶。
Syk是非受体细胞质酪氨酸激酶家族,在各种细胞表面受体(包括CD74,Fc受体和整合素)信号传导中起作用。


概述受体酪氨酸激酶介导的信号通路的组成、特点及其主要功能。
1.引言1.1 概述受体酪氨酸激酶介导的信号通路是细胞内一种重要的信号传导机制,它通过调控各种生物化学反应来参与细胞内的多种生理过程。
该信号通路的组成主要包括受体酪氨酸激酶、底物和下游的信号分子等。
受体酪氨酸激酶是一类能够磷酸化酪氨酸残基的酶,它能够通过与外界的信号分子结合,诱导其自身的激活,进而引发一系列的生物效应。
受体酪氨酸激酶可以被分为两种类型:单体型和受体型。
单体型受体酪氨酸激酶主要包括一些细胞内酪氨酸激酶;受体型受体酪氨酸激酶则包括一些质膜上的受体酪氨酸激酶。
这些受体酪氨酸激酶在结构上存在一定的相似性,但在功能上却可能有所差异。
受体酪氨酸激酶介导的信号通路具有一些特点。
首先,它是一种高度调控的信号传导网络,可以根据不同的外界刺激改变其活性。
其次,受体酪氨酸激酶的激活往往能够启动多条平行的信号通路,从而实现更为复杂的细胞反应。
此外,该信号通路具有信号传导速度快、反应机制多样等特点。
受体酪氨酸激酶介导的信号通路在许多生理过程中发挥着重要的功能。
例如,它参与了细胞的生长、增殖和分化过程;调节了细胞的凋亡和存活等。
此外,该信号通路还与多种疾病的发生和发展密切相关,如癌症、炎症和神经系统疾病等。
综上所述,受体酪氨酸激酶介导的信号通路具有复杂的组成、多样的特点和重要的功能。
深入了解该信号通路的组成、特点及其主要功能,对于揭示细胞信号传导的机制,以及发展相关疾病的治疗策略具有重要意义。
文章结构部分应该介绍文章的组织结构和各个章节的内容概述。
以下是一种可能的写作方式:1.2 文章结构本文将分为三个主要部分进行论述,分别是引言、正文和结论。
引言部分将首先对受体酪氨酸激酶介导的信号通路进行概述,包括其在细胞内的重要性和作用机制。
接着,本部分将介绍文章的结构和各个章节的内容。
正文部分将详细探讨受体酪氨酸激酶的组成、特点和主要功能。
在2.1小节中,将介绍受体酪氨酸激酶的组成成分,包括受体酪氨酸激酶本身和相关的配体和受体。

概述受体酪氨酸激酶介导的信号通路的特点和主要功能
受体酪氨酸激酶(RTKS)是细胞表面一大类重要受体家族,当配体与受体结合,导致受体二聚化,激活受体的酪氨酸蛋白激酶活性,随即引起一系列磷酸化级联反应,终至细胞生理和基因表达的改变.RTK-Ras信号通路是这类受体所介导的重要信号通路.其基本模式为:配体→RTK→接头蛋白→GEF →Ras →Raf (MAPKKK) →MAPKK →MAPK →进入细胞核→其他激酶或基因调控蛋白(转录因子)的磷酸化修饰,对基因表达产生多种效应.\x0d组成:该受体家族包括6个亚族.其胞外配体为可溶性或膜结合的多肽或蛋白类激素.还有RTK-Ras 信号通路中各种因子.\x0d特点:(1)激活机制为受体之间的二聚化、自磷酸化、活化自身;(2)没有特定的二级信使,要求信号有特定的结构域;(3)有Ras分子开关的参与;(4)介导下游MAPK的激活\x0d功能:RTKS信号通路主要参与控制细胞生长、分化过程.RTK-Ras信号通路具有广泛的功能,包括调节细胞的增殖分化,促进细胞存活,以及细胞代谢的调节与校正。

表皮生长因子受体酪氨酸激酶抑制剂的研究进展一、本文概述表皮生长因子受体(EGFR)酪氨酸激酶抑制剂(TKIs)是一类针对EGFR信号通路的关键药物,广泛应用于非小细胞肺癌、结直肠癌、头颈癌等多种癌症的治疗。
本文旨在综述近年来EGFR TKIs的研究进展,包括其作用机制、药物研发、临床应用以及面临的挑战等方面。
通过深入了解EGFR TKIs的研究现状和发展趋势,有望为癌症治疗提供新的思路和方法,进一步改善患者的生活质量和预后。
本文将从EGFR TKIs的作用机制出发,阐述其如何通过抑制EGFR 的酪氨酸激酶活性来阻断癌细胞的增殖和转移。
接着,我们将回顾EGFR TKIs的药物研发历程,介绍目前市场上主流的EGFR TKIs药物及其特点。
在此基础上,我们将重点关注EGFR TKIs在临床试验中的应用情况,包括其疗效、安全性以及耐药性等问题。
我们将探讨EGFR TKIs面临的挑战和未来发展方向,包括如何克服耐药性、提高治疗效果以及拓展新的适应症等。
通过本文的综述,我们希望能够为相关领域的研究者和临床医生提供有价值的参考信息,推动EGFR TKIs在癌症治疗中的进一步应用和发展。
二、EGFR-TK抑制剂的分类与机制表皮生长因子受体酪氨酸激酶抑制剂(EGFR-TK抑制剂)是近年来癌症治疗领域的重要突破,其通过抑制表皮生长因子受体(EGFR)的酪氨酸激酶活性,从而阻断细胞的生长、增殖和转移过程。
根据药物的作用机制和化学结构,EGFR-TK抑制剂主要分为两大类:可逆性抑制剂和不可逆性抑制剂。
可逆性抑制剂,如吉非替尼和厄洛替尼,能够与EGFR的ATP结合位点形成可逆性结合,从而竞争性地抑制酪氨酸激酶的活性。
这类药物对于EGFR敏感突变的非小细胞肺癌具有较好的疗效,但在长期治疗过程中,患者往往会出现耐药现象。
不可逆性抑制剂,如阿法替尼和奥希替尼,能够与EGFR的ATP 结合位点形成共价键,导致EGFR的永久性失活。

酪氨酸激酶信号通路及其在癌症中的作用
随着越来越多的人患上癌症,癌症的治疗也变得越来越重要。
目前,许多研究人员正在寻找治疗癌症的新途径,其中酪氨酸激酶信号通路被认为是一种很有潜力的治疗手段。
酪氨酸激酶信号通路包括多个酪氨酸激酶受体,这些受体可以被活化,并向下游分子释放信号,以控制细胞的增殖、分化、凋亡和迁移。
这些酪氨酸激酶信号通路在正常代谢和生理过程中都扮演着重要的角色。
然而,在癌症中,这些通路的异常激活可以导致细胞的异常增殖和凋亡,从而促进癌症的发展和转移。
最近的研究表明,酪氨酸激酶信号通路在许多不同类型的癌症中都发挥着关键的作用。
例如,它在乳腺癌、结直肠癌、肺癌和胰腺癌等癌症中的作用已经得到广泛的研究。
更进一步的研究还发现,某些药物可以阻止这些信号通路的活化,从而抑制癌细胞的生长和转移。
虽然酪氨酸激酶信号通路已经被证明是一种有效的治疗手段,但是在临床实践中它的应用还处于初期阶段。
许多研究人员仍在继续研究这个信号通路的生物学机制和治疗应用。
尽管这个领域还有很多未知的问题,但我们可以确定一点:酪氨酸激酶信号通路是一个非常有前途的治疗手段,它有望改善许多癌症患者的生命质量,为人类带来更好的未来。

概述受体酪氨酸激酶介导的信号通路的组成、特点及其主要功能1. 引言1.1 概述受体酪氨酸激酶介导的信号通路是细胞内重要的信号传递机制,它参与调控多种生物过程,如细胞增殖、分化、命运决定和免疫应答等。
该信号通路在维持细胞正常功能以及疾病的发生和发展中起着关键作用。
1.2 文章结构本文将从以下几个方面对受体酪氨酸激酶介导的信号通路进行阐述:受体酪氨酸激酶的组成、特点及其调节机制;信号通路的特点,包括蛋白质相互作用网络、多样性和复杂性;以及该信号通路中一些重要分子的功能和调控机制。
此外,我们还将重点讨论该信号通路在细胞增殖与生长调控、细胞分化和命运决定以及免疫应答调节等方面的主要功能。
1.3 目的本文旨在全面了解受体酪氨酸激酶介导的信号通路在生物体内扮演的角色,以及其对细胞功能和疾病发生发展的影响。
通过深入了解和探讨该信号通路的组成、特点及其主要功能,我们可以加深对细胞信号传递机制的认识,并为相关疾病的治疗和预防提供理论依据。
请注意,本文中的“受体酪氨酸激酶”是指一类特定的酶分子,其底下涵盖了多种具体类型的受体酪氨酸激酶。
2. 受体酪氨酸激酶介导的信号通路的组成:受体酪氨酸激酶是一种重要的信号传导分子,在细胞内起到了关键的调节作用。
它通过与特定的配体结合,激活其自身内在的激酶活性,并进而启动一系列下游信号通路。
这些信号通路可以干预各种细胞过程,并参与调控细胞增殖、生长、分化以及免疫应答等功能。
受体酪氨酸激酶主要由以下几个组成部分构成:2.1 受体酪氨酸激酶的定义和分类:受体酪氨酸激酶是一类膜上受体分子,能够感知和传递外界信息。
根据其结构和功能特点,受体酪氨酸激酶可被分为单个蛋白链型(RTKs)和多个蛋白链复合物型(RTKc)。
RTKs主要包括表皮生长因子受体(EGFR)、血小板衍生生长因子受体(PDGFR)等。
RTKc则由多个蛋白链聚集而成,其中一条链包含激酶结构域,如胞浆性酪氨酸激酶之类的。
2.2 受体酪氨酸激酶的结构特点:受体酪氨酸激酶通常由外部区、跨膜区和胞浆性区组成。

细胞生物学:受体酪氨酸激酶/Ras途径2007-8-12 14:27【大中小】【我要纠错】受体酪氨酸激酶,简称RTKs(receptor tyrosine kinase)是最大的一类酶联受体;Ras是原癌基因c-ras表达的产物,RTKs/Ras是目前研究得比较清楚的一条主要的信号转导途径。
■受体的结构特点及类型● 结构特点所有的RTKs都是由三个部分组成的:含有配体结合位点的细胞外结构域、单次跨膜的疏水α螺旋区、含有酪氨酸蛋白激酶(RTK)活性的细胞内结构域(图5-47)。
● 已发现50多种不同的RTKs,主要的几种类型包括:表皮生长因子受体、血小板生长因子受体、胰岛素和胰岛素样生长因子-1 受体等。
图5-47 几种主要的酪氨酸激酶受体■受体酪氨酸激酶的激活受体酪氨酸激酶的激活是一个相当复杂的过程,大多数受体都要先由两个单体形成一个二聚体,并在细胞内结构域的尾部磷酸化,然后在二聚体的细胞内结构域装配成一个信号转导复合物(图5-48)。
图5-48 受体酪氨酸激酶的激活及细胞内信号转导复合物的形成受体酪氨酸激酶是如何被激活的?■胰岛素受体信号转导途径● 受体结构胰岛素受体(insulin receptor)是一个四聚体,由两个α亚基和两个β亚基通过二硫键连接。
● 激活当胰岛素与受体的α亚基结合并改变了β亚基的构型后,酪氨酸蛋白激酶才被激活,激活后可催化两个反应∶①使四聚体复合物中β亚基的特异位点酪氨酸残基磷酸化,这种过程称为自我磷酸化(autophosphorylation);②使胰岛素受体底物(insulin receptor substrate,IRSs)上具有重要作用的十几个酪氨酸残基磷酸化(图5-49),磷酸化的IRSs能够与那些具有SH2结构域的蛋白结合,引起进一步的反应。
图5-49 胰岛素受体与配体结合反应胰岛素受体是由两个α亚基和两个β亚基组成的四聚体,胰岛素与α亚基结合引起β亚基构型改变,激活了β亚基的酪氨酸激酶。

常见的几种信号通路1 JAK-STAT信号通路1) JAK与STAT蛋白JAK—STAT信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程.与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK 和转录因子STAT。
(1) 酪氨酸激酶相关受体(tyrosine kinase associated receptor)许多细胞因子和生长因子通过JAK—STAT信号通路来传导信号,这包括白介素2?7(IL-2?7)、GM-CSF(粒细胞/巨噬细胞集落刺激因子)、GH(生长激素)、EGF(表皮生长因子)、PDGF (血小板衍生因子)以及IFN(干扰素)等等。
这些细胞因子和生长因子在细胞膜上有相应的受体。
这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK的结合位点。
受体与配体结合后,通过与之相结合的JAK的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递。
(2) 酪氨酸激酶JAK(Janus kinase)很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体(receptortyrosine kinase, RTK),而JAK却是一类非跨膜型的酪氨酸激酶。
JAK是英文Janus kinase 的缩写,Janus在罗马神话中是掌管开始和终结的两面神.之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定SH2结构域的信号分子。
JAK蛋白家族共包括4个成员:JAK1、JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAK homology domain, JH),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域.(3)转录因子STAT(signal transducer and activator of transcription)STAT被称为“信号转导子和转录激活子"。


细胞表面受体是一类位于细胞膜上的蛋白质分子,能够与外界的生物学信号分子结合,并将这些信号传递到细胞内部,触发特定的生物学效应。
细胞表面受体三大家族参与信号通路的特征,是近年来细胞生物学研究的热点之一。
本文将对细胞表面受体三大家族的特征、结构和信号通路等方面进行详细的探讨,并结合个人观点,希望能为读者提供一份全面深入的文章。
一、细胞表面受体三大家族的概念和分类1. G蛋白偶联受体(GPCRs)家族G蛋白偶联受体是一类广泛存在于哺乳动物细胞膜上的受体蛋白,其活性主要通过G蛋白介导的信号传导。
这一家族的受体能够响应多种信号分子,包括化学传感器、嗅覚和视觉受体等。
2. 酪氨酸激酶受体(RTKs)家族酪氨酸激酶受体是一类能够与细胞外的生长因子结合,并通过激酶活化的受体蛋白。
这一家族的受体在调控细胞生长、分化和代谢等生理过程中起着重要作用。
3. 离子通道受体家族离子通道受体是一类能够通过细胞外的化学或物理刺激,控制细胞内离子通道开启或关闭状态的受体蛋白。
这一家族的受体在神经元兴奋性调节、肌肉收缩和细胞外钙导入等过程中扮演重要角色。
二、细胞表面受体三大家族的结构特征1. 七次跨膜结构G蛋白偶联受体家族的受体蛋白在细胞膜内外都具有结构域。
细胞外的N端含有配体结合位点,细胞内的C端含有G蛋白结合位点。
2. 蛋白激酶结构酪氨酸激酶受体家族的受体蛋白包含一个独特的酪氨酸激酶结构域,当生长因子结合后,激酶活化,进而启动下游信号通路。
3. 离子通道结构离子通道受体家族的受体蛋白含有跨膜的离子通道结构域,受到细胞外的化学或物理刺激时,离子通道开启或关闭,传递特定的离子信号。
三、细胞表面受体三大家族在信号通路中的作用1. 信号转导G蛋白偶联受体家族通过激活腺苷酸环化酶、磷脂酰肌醇-磷酸激酶C等下游蛋白,调节细胞内二信号分子的生成和代谢。
2. 细胞增殖酪氨酸激酶受体家族的激活,能够通过调控MAPK信号通路促进细胞增殖。
3. 神经递质释放离子通道受体家族的开启或关闭,能够调节细胞内钙离子浓度,进而影响神经递质的释放。
布鲁顿酪氨酸激酶(BTK)信号通路与疾病陈潇ꎬ龚国清(中国药科大学药理学研究室ꎬ江苏南京211198)摘要:布鲁顿酪氨酸激酶(BTK)是B细胞受体(BCR)信号传导的关键组分ꎬ并且作为体内B细胞增殖和细胞存活的重要调节剂起作用ꎮ本文综述了布鲁顿酪氨酸激酶信号通路的传导过程ꎬ包括主要的B细胞受体信号通路与其他通路ꎮ布鲁顿酪氨酸激酶基因缺陷会引发免疫缺陷X连锁性丙种球蛋白血症(XLA)ꎬ而布鲁顿酪氨酸激酶抑制剂如依鲁替尼最近在患有各种B细胞恶性肿瘤的患者的临床研究中显示出令人印象深刻的抗肿瘤活性ꎬ在狼疮和类风湿性关节炎的实验动物模型中也观察到一定治疗作用ꎮ关键词:布鲁顿酪氨酸激酶ꎻB细胞受体ꎻ免疫缺陷X连锁性丙种球蛋白血症(XLA)ꎻ依鲁替尼ꎻB细胞恶性肿瘤ꎻ自身免疫疾病中图分类号:R967㊀文献标识码:A㊀文章编号:2095-5375(2020)03-0169-007doi:10.13506/j.cnki.jpr.2020.03.011Brutonᶄstyrosinekinase(BTK)signalingpathwayanddiseaseCHENXiaoꎬGONGGuoqing(DepartmentofPharmacologyꎬChinaPharmaceuticalUniversityꎬNanjing211198ꎬChina)Abstract:Brutonᶄstyrosinekinase(BTK)isakeycomponentofBcellreceptor(BCR)signalingandfunctionsasanimportantregulatorofcellproliferationandcellsurvivalinBcellsinvivo.ThepaperfocusonthesignalingprocessesofBTKꎬbothinBCRsignalingpathwayꎬwhichisthemajorsignalingpathwayꎬandtheothersignalingpathways.BTK-deficientgenewouldcauseimmunodeficiencyX-linkedgammaglobulinemia(XLA)ꎬwhileinhibitorsofBTKsuchasIbrutinibhaveveryrecentlyshownimpressiveanti-tumoractivityinclinicalstudiesinpatientswithvariousBcellmalignanciesꎬandprom ̄isingeffectsofBTKinhibitionwerealsoseeninexperimentalanimalmodelsforlupusandrheumatoidarthritis.Keywords:BTKꎻBCRꎻXLAꎻIbrutinibꎻBcellmalignanciesꎻAutoimmunedisease㊀㊀生化分析和体内基因靶向实验已经暗示酪氨酸激酶是B细胞发育和功能中细胞分化ꎬ增殖ꎬ存活和迁移的关键调节剂ꎮ其中一种ꎬ布鲁顿酪氨酸激酶(BTK)ꎬ在人类的原发性免疫缺陷X连锁性丙种球蛋白血症(XLA)和小鼠的X连锁免疫缺陷(Xid)中发生突变ꎮXLA和Xid的特征在于分别在前B细胞阶段几乎完全阻止B细胞发育和成熟B细胞的分化缺陷[1-2]ꎮ自1993年鉴定BTK作为该疾病的缺陷基因以来ꎬ我们对其发病机制的认识已经大大深入ꎮ在其发现后不久ꎬ证明了BTK在成熟B细胞中与B细胞受体(BCR)信号传导密切相关ꎮ除T细胞和NK细胞外ꎬBTK几乎在造血系统的所有细胞中表达ꎬ并且还参与B细胞中的许多其他信号传导途径ꎬ包括Toll样受体(TLR)和趋化因子和Fc受体信号传导ꎮ在BTK被成功鉴别后ꎬ很快BTK小分子抑制剂便被开发出来ꎬ并且在体外白血病细胞中显示出具有抗肿瘤活性[3]ꎮ2007年ꎬ一种不可逆BTK抑制剂依鲁替尼(Ibrutinibꎬ代号PCI-32765)的发现[4]ꎬ是抑制B细胞恶性肿瘤中的信号转导治疗的里程碑ꎮ依鲁替尼在2013年被美国食品药品监督管理局(FDA)批准为用于治疗套细胞淋巴瘤(mantlecelllymphomaꎬMCL)和慢性淋巴细胞白血病(CLL)[5]ꎬ2015年ꎬ获得FDA和欧洲药品管理局(EMA)的批准ꎬ用于治疗有症状的Waldenström巨球蛋白血症(WM)患者[6]ꎮ临床前研究以及临床试验现已提供证据表明BTK抑制剂的抗肿瘤活性在很大程度上取决于其在BCR信号传导之外的作用ꎬ例如B细胞中的趋化因子和TLR信号传导或骨髓微环境中破骨细胞或其他细胞产生肿瘤支持因子[7]ꎮ此外ꎬ小鼠中的各种研究证明了适当调节BTK活性的㊀作者简介:陈潇ꎬ女ꎬ研究方向:生化药理ꎬE-mail:chenxiao_cpu@163.com㊀通信作者:龚国清ꎬ男ꎬ博士研究生ꎬ副教授ꎬ研究方向:生化药理ꎬTel:025-86185307ꎬE-mail:gonggq@hotmail.com至关重要性ꎮ例如ꎬB细胞中BTK的转基因过表达与自发生发中心(GC)形成ꎬ自身抗体产生和系统性红斑狼疮(SLE)样自身免疫病理学相关[8]ꎮ相反ꎬ在患有胶原诱导的关节炎(CIA)的小鼠中或在患有SLE样疾病的小鼠中的依鲁替尼治疗降低了自身免疫症状[9-10]ꎮ在这篇综述中ꎬ我们讨论了BTK在BCR信号传导途径与其他信号通路中的功能ꎬ以及BTK缺陷对正常生理功能造成的影响与BTK抑制对疾病的治疗意义ꎮ1㊀BTK结构与BCR信号1.1㊀BTK的结构与活性调节㊀BTK是由659个氨基酸组成的蛋白质ꎬ与在肝细胞癌中表达的酪氨酸激酶(TEC)㊁诱导型T细胞激酶(ITK)㊁静息淋巴细胞激酶(RLK)和骨髓表达激酶(BMX)4个成员一起ꎬ属于非受体酪氨酸激酶的Tec家族ꎬ在整个进化过程中都非常保守ꎬ高度表达于造血细胞[11]ꎮBTK的结构域结构类似于SRC家族激酶的结构域结构ꎬ并且它包含SRC同源结构域(SH结构域)SH2和SH3ꎬ以及催化结构域ꎮ然而ꎬ与SRC家族激酶不同ꎬBTK具有氨基末端的pleckstrin同源(PH)结构域和富含脯氨酸的TEC同源(TH)结构域ꎬ其含有锌指基序ꎬ这对于最佳活性和稳定性是重要的[12]ꎮSRC家族激酶组成性地与膜结合ꎬ而BTK在细胞质中ꎬ并且仅会在其PH结构域与由PI3K产生的磷脂酰肌醇-3ꎬ4ꎬ5-三磷酸(PIP3)结合时短暂地募集到膜上[13]ꎮ在B细胞中ꎬBTK活化由PH结构域介导的质膜结合引发ꎬ其能够通过脾酪氨酸激酶(SYK)或SRC家族激酶如LYN在激酶结构域的残基Y551处磷酸化ꎮ这促进了BTK的催化活性ꎬ并导致其在SH3结构域中的位置Y223处的自身磷酸化[13-14]ꎮB细胞谱系中Y223F-BTK的转基因表达基本上校正了BTK缺陷Xid小鼠的表型[15]ꎬ证明了在体内前B细胞或B细胞中BTK功能不需要Y223F自磷酸化ꎮ因此ꎬSH3结构域ꎬ特别是SH3结构域Y223自身磷酸化位点的功能意义仍不清楚ꎬY223的磷酸化似乎不影响BTK活性[15]ꎮ在BCR刺激后ꎬ成熟B细胞中BTK水平增加ꎬ但所涉及的转录后机制十分复杂ꎬ并且至今为止仅有部分得到研究证实ꎮ包括microRNA-185(miR-185)调节BTK表达[16]ꎬBCR与Toll样受体(TLR)相互作用ꎬBTK通过涉及核因子-κB(NF-κB)的途径刺激其自身启动子的转录ꎬ启动蛋白酶体依赖性阳性自动调节反馈环[17-18]ꎮ1.2㊀BCR信号激活BTK㊀BCR复合物与二硫键连接的Igα-Igβ异二聚体非共价结合ꎮ在抗原与BCR结合后ꎬSRC家族蛋白酪氨酸激酶LYN磷酸化Igα和Igβ免疫受体酪氨酸激活基序(ITAM)ꎬ从而产生SYK的停靠位点[19]ꎮ同时ꎬLYN磷酸化BCR共受体CD19的细胞质尾部中的酪氨酸残基ꎬ这使得能够结合和激活PI3K和VAV[20]ꎮ除了CD19ꎬ用于PI3K的细胞质B细胞衔接子也可以募集PI3K并激活ꎮPI3K产生PIP3ꎬ其是用于激活下游途径的重要第二信使ꎮPI3K通过PIP3-PH结构域相互作用将BTK吸引至细胞膜ꎬ其允许SYK和LYN通过Y551的反式磷酸化完全激活BTK[21]ꎮBTK活化可以通过磷酸酶PTEN和含有SH2结构域的肌醇5ᶄ磷酸酶1(SHIP1)调节ꎬ其使PIP3去磷酸化从而抑制BTK膜结合ꎮ一旦SYK被激活ꎬ通过募集和磷酸化65kDa的含有SH2结构域的白细胞蛋白(SLP65)将信号传导至下游效应子ꎬ其作为各种信号分子的支架ꎬ包括SYK㊁BTK及其关键底物磷脂酶C-γ2(PLCγ2)[22]ꎮ1.3㊀BTK的下游信号传导㊀SLP65介导的BTK和PLCγ2的募集完成了所谓的微信号体的形成ꎬ其包括VAVꎬPI3KꎬBTKꎬSLP65和PLCγ2ꎮBTK主要负责Y753和Y759位置的PLCγ2磷酸化ꎬ这对PLCγ2的脂肪酶活性至关重要[23]ꎮ此外ꎬBTK不依赖于其激酶活性ꎬ可以募集磷脂酰肌醇-4-磷酸-5-激酶(PIP5K)ꎬ从而刺激产生磷脂酰肌醇-4ꎬ5-二磷酸(PIP2)的正反馈环ꎬPIP2可作为PI3K和PLCγ2的底物[24]ꎮ被BTK活化后ꎬPLCγ2切割PIP2以产生两个第二信使ꎬ肌醇三磷酸(iIP3)和二酰基甘油(DAG)ꎬ其激活分叉和部分重叠的信号传导途径[25]ꎮDAG介导蛋白激酶C(PKC)β的激活ꎬ其诱导细胞外信号调节激酶ERK1和ERK2的RAS信号传导依赖性磷酸化ꎮ与ERK1和ERK2不同ꎬ其他丝裂原活化蛋白激酶(MAPK)如p38ꎬ可由PLCγ2诱导ꎬ无须通过RAS进行中间信号传导[26]ꎮ重要的是ꎬPKCβ还通过支架复合物激活NF-κB途径ꎬ该支架复合物包括含有半胱天冬酶募集结构域的蛋白11(CARD11)ꎬBCL10和黏膜相关淋巴组织淋巴瘤易位蛋白1(MALT1)[27]ꎮ1.4㊀BTK在pre-BCR信号传导中的作用㊀由于人类pre-BCR结合后形成由酪氨酸磷酸化的LYN㊁SYK㊁SLP65㊁PI3K㊁BTK㊁VAV和PLCγ2组成的筏相关Ca2+信号传导模块ꎬ所以可以假设由pre-BCR和BCR介导的信号通路是相似的[28]ꎮ在小鼠中ꎬ在pre-BCR信号在功能上与白细胞介素7受体(IL-7R)信号传导途径相互作用ꎬ除了Janus激酶3(JAK3)-信号转导和转录激活因子5(STAT5)信号传导之外ꎬ它还激活SRC家族激酶和ERK途径ꎮIL-7R和pre-BCR信号传导协同诱导前B细胞的增殖[29]ꎮ缺乏关键的pre-BCR分子的小鼠ꎬ例如SYK㊁SLP65㊁BTK和PLCγ2ꎬ仅在前B细胞阶段显示出一些发育阻滞ꎮSLP65缺陷的白血病前B细胞在细胞表面表达高水平的pre-BCRꎬ但这不可能有助于它们在体内的强增殖能力ꎬ因为SLC的转基因过表达不诱导或增加白血病的发展[30]ꎮ在正常的前B细胞中ꎬSLP65通过直接抑制JAK3-STAT5信号传导途径下调IL-7介导的前B细胞增殖和存活[31]ꎮBtk缺陷型小鼠不会发生前B细胞白血病ꎬ然而ꎬ与其激酶活性无关ꎬBTK确实与SLP65合作调节IL-7介导的前B细胞增殖和存活ꎬ这表明BTK可以作为肿瘤抑制剂起作用[32]ꎮ2㊀BTK在其他信号通路中的作用2.1㊀BTK与趋化因子受体信号传导㊀生化分析和体外黏附和迁移测定已经确定BTK参与趋化因子受体途径ꎬ其对于B细胞运输ꎬ组织归巢和体内平衡是必需的ꎮBTK是趋化因子受体CXCR4和CXCR5的关键信号传导因子[33]ꎮ由骨髓和生发中心的基质细胞高度表达的CXC-趋化因子配体12(CXCL12)诱导BTK活化ꎬ最可能是通过BTK和CXCR4连接的异源三聚体G蛋白亚基之间的直接相互作用ꎮGα和Gβγ亚基都可以直接结合BTK的PH结构域和相邻的TH结构域ꎮ因为在缺乏LYN或SYK的B细胞中发现CXCL12控制的迁移减少ꎬ所以可以推测这些激酶在CXCR4连接后激活BTK[34]ꎮ在SYK抑制剂的存在下ꎬ在Y551诱导CXCL12诱导BTK磷酸化的发现证实了这一点ꎮ在体外实验中ꎬ依鲁替尼可以抑制MCL和CLL细胞中CXCL12诱导和CXCL13诱导的PLCγ2㊁ERK1㊁ERK2㊁JNK和AKT的磷酸化ꎬ以及细胞黏附和迁移ꎬ这与BTK在趋化因子受体信号传导中起关键作用的观点一致[35]ꎮBTK在体内趋化因子信号传导中的作用首先通过小鼠中BTK缺陷型B细胞的过继转移实验证明ꎬ其显示B细胞归巢至淋巴结特别受影响[34]ꎮ2.2㊀BTK和TLR信号通路㊀BTK在TLR信号中的作用首先通过发现BTK缺陷型B细胞的增殖响应于TLR4配体细菌脂多糖(LPS)而降低ꎮTLR信号传导诱导下游转录因子NF-κBꎬ激活蛋白1(AP1)和干扰素调节因子3(IRF3)其在B细胞中导致活化标志物的上调㊁增殖㊁抗体分泌㊁类别转换重组和产生促炎细胞因子ꎮBTK可与大多数TLR的细胞质Toll/IL-1受体(TIR)结构域以及下游衔接子MyD88ꎬTRIF和MyD88衔接子样蛋白(MAL)和IL-1R相关激酶1(IRAK1)直接相互作用[36]ꎮTLR9和BCR刺激可以协同诱导IL-6的产生ꎬ因此BTK是自噬体样区室中TLR9和BCR共定位所必需的ꎮ由于BCR信号传导在细胞表面开始并且当其传递至细胞内区室时继续激活MAPKꎬ可以推测TLR和BCR信号传导通过BTK互连[37]ꎮ3㊀BTK缺陷病3.1㊀X连锁无丙种球蛋白血症(XLA)㊀XLA是原发性免疫缺陷病(PIDs)的遗传形式之一ꎮ它是由BTK基因突变引起的ꎬ导致骨髓内B细胞的发育和成熟缺陷ꎬ外周血中成熟B细胞显著减少或完全缺失ꎮXLA患者中的前B细胞体积小ꎬ与BTK在诱导或其增殖中的基本功能一致[38-39]ꎮ由于缺乏成熟的B细胞ꎬXLA患者血清中所有主要免疫球蛋白(Ig)的水平显著降低ꎬ因此会遭受严重和慢性细菌感染ꎮXLA患者在其循环中具有<1%的正常B细胞数量ꎬ导致明显缺乏浆细胞ꎬ因此外周血中所有亚类的Igs水平非常低[40]ꎮMirsafian等[41]利用深度高通量RNA测序(RNA-Seq)方法ꎬ发现BTK突变可能导致先天免疫系统的失调ꎬ并增加XLA患者单核细胞中细胞凋亡的易感性ꎮ通过分析该疾病的雌性携带者中循环B细胞的X染色体失活状态ꎬ显示XLA的发育缺陷是B细胞谱系固有的ꎮ载体表现出突变体BTK基因对X染色体的单侧失活ꎬ表明在活性X染色体上具有缺陷BTK等位基因的B谱系细胞具有选择性缺陷ꎮ因此ꎬXLA的女性携带者是健康的并且没有表现出免疫异常[42-43]ꎮ在母体给予的Ig消失后ꎬ男孩患者通常在上呼吸道或下呼吸道中出现严重或复发的包囊细菌感染ꎬ主要是中耳炎㊁鼻窦炎和肺炎ꎬ少部分会患脑膜炎[44-45]ꎮ病毒感染通常不会引起XLA的异常问题ꎬ因为T细胞和NK细胞功能正常ꎮXLA患者终生且完全依赖抗生素和静脉或皮下Ig替代疗法[46]ꎮ3.2㊀小鼠的X连锁免疫缺陷(Xid)㊀Xid表型的CBA/N小鼠的免疫缺陷是由单个保守蛋白酪氨酸激酶BTK结构改变引起的遗传疾病ꎬ其特征是骨髓中B细胞发育前的部分阻断ꎬ外周成熟B细胞数量的中度减少ꎬ淋巴器官IgM和IgG3的循环抗体滴度低[47]ꎮ与XLA相反ꎬ表达R28C-BTK突变的Xid小鼠或在BTK基因中具有靶向破坏的小鼠仅表现出轻度B细胞病症ꎮ与野生型小鼠的B细胞相比ꎬBTK缺陷型小鼠中的外周B细胞具有明显的存活劣势[48-49]ꎮBTK缺陷型小鼠对T细胞非依赖性Ⅱ型抗原无反应ꎬ与野生型同窝小鼠相比ꎬ抗原特异性血清IgM和IgG3水平显著降低ꎬ对T细胞依赖性抗原的初级反应也受到损害[50]ꎮXid小鼠对PR8流感感染的免疫反应相对正常ꎮX-31流感病毒鼻内感染BTK缺陷小鼠和同窝野生型小鼠ꎬ发现二者病毒清除率相当ꎮ在流感感染后ꎬ野生型小鼠的肺中B-1细胞数量增加ꎬ而在幼稚BTK缺陷小鼠中ꎬ肺和胸膜腔中不存在B-1细胞ꎬ并且在流感感染后不诱导B-1细胞[51]ꎮBTK缺陷的Xid表型还可导致对各种病原体的易感性降低ꎬ与WT对照相比ꎬ感染肺炎支原体的Xid小鼠显示出存活率增加和组织损伤减少ꎬ可能是因为它们未能产生破坏性抗原-抗体反应[52]ꎮ4㊀BTK抑制对疾病的治疗4.1㊀Waldenström巨球蛋白血症(WM)㊀WM以其发现者命名ꎬ是一种罕见的淋巴增生性疾病ꎬ其特征是骨髓和其他器官中淋巴浆细胞淋巴瘤细胞的恶性积聚ꎬ以及血清中单克隆IgM副蛋白的存在[53]ꎮWM是一种异质性疾病ꎬ可出现血细胞减少㊁淋巴结肿大㊁肝脾肿大㊁高粘血症㊁神经病变㊁冷球蛋白血症或淀粉样变性ꎮ尽管WM患者往往存活数年ꎬ甚至数十年ꎬ病情无法治愈ꎬ并且绝大多数患者的疾病过程表现为影响患者生活质量和日常生活活动的症状性疾病复发[54]ꎮ在WM患者中ꎬMyD88中常见的L265P点突变导致MyD88的组成型活性形式ꎬ且BTK也具有诱导活性ꎬ尽管所涉及的机制仍不清楚ꎮ最近显示MyD88L265P与WM细胞中的磷酸化BTK结合ꎬ这表明BTK可能在这些细胞中的MyD88L265P中具有直接作用[55]ꎮ依鲁替尼治疗WM细胞可消除MyD88L265P-BTK相关性并降低NF-κB活化ꎮ在两种WM细胞系和原代WM细胞中ꎬ当IRAK1和IRAK4也被抑制时ꎬ依鲁替尼诱导细胞凋亡并增加细胞凋亡水平ꎮ因此ꎬ依鲁替尼治疗MyD88L265P突变WM细胞的功效可能完全依赖于NF-κB活化的消除[56]ꎮ4.2㊀慢性淋巴细胞白血病(CLL)㊀CLL的特征在于血液中非增殖性单克隆CD5+成熟B细胞的积累ꎮCLLB细胞通常具有低表面IgM表达并显示对BCR连接的无反应性反应ꎬ这表明慢性BCR内化和信号转导[57]ꎮ一些发现表明BTK信号传导基本上有助于CLL的启动或维持ꎮ在CLL小鼠模型中ꎬBTK缺乏使肿瘤形成消失ꎬ而转基因BTK过表达增加肿瘤发生率和总体死亡率[8]ꎮ体外用依鲁替尼治疗ꎬ可以减少CLL细胞的存活和增殖ꎬ消除了BCR刺激的AKT和ERK磷酸化ꎬ以及VCAM1介导的黏附和淋巴细胞胞质蛋白1(LCP1)的表达ꎬ是F肌动蛋白交联分子ꎬ对CXCL12介导的迁移至关重要[35]ꎮBTK的抑制干扰多种途径ꎬ这些途径对于CLL细胞存活ꎬ增殖和体内迁移可能是重要的ꎮBTK信号传导可能支持CLL细胞向淋巴结中的增殖中心迁移ꎬ因为体外伊布鲁尼治疗CLL细胞有效阻断CXCL12诱导的和CXCL13诱导的迁移ꎮ此外ꎬ当与B细胞激活因子(BAFF)㊁肿瘤坏死因子(TNF)㊁IL-6㊁IL-4和CD40L一起培养时ꎬ依鲁替尼处理的CLL细胞在体外显示出降低的活力ꎬ这表明BTK抑制可能抵消促存活因子在CLL微环境中的作用ꎮ当CLL细胞与护士细胞(NLCs)共培养时ꎬCLL细胞存活ꎬ增殖和CCL3和CCL4产生的减少也表明了破坏淋巴结微环境中共刺激反馈的可能性[58]ꎮ4.3㊀套细胞淋巴瘤(MCL)㊀MCL显著偏向的BCR谱表表明抗原刺激在其发病过程中起着至关重要的作用ꎬ但所涉及的抗原是未知的ꎮ在有限的患者组中观察到LYN㊁SLP65㊁SYK和PKCβ的组成型磷酸化ꎬ提示BCR信号传导的促存活作用ꎮ此外ꎬ发现SYK基因和SYK蛋白过表达ꎬ并且MCL细胞显示NF-κB和AKT的组成性激活ꎬ这可能反映BCR或TLR信号的激活[59]ꎮBTK在MCL中显示出高表达ꎬ并且在未刺激的原代MCL细胞中观察到Y223处的BTK自身磷酸化增加ꎮ依鲁替尼治疗原代MCL细胞或细胞系导致活力降低ꎬ以及在BCR活化或响应CXCR4或CXCR5时黏连和迁移受损ꎮ接受间歇性或连续依鲁替尼治疗的MCL患者分别有复发性淋巴细胞增多症或单次淋巴细胞增多症[60]ꎮ4.4㊀多发性骨髓瘤㊀多发性骨髓瘤细胞来源于不再在其细胞表面表达BCR的浆细胞ꎮ为了存活和增殖ꎬ多发性骨髓瘤细胞似乎依赖于由增加的破骨细胞活性和骨重建产生的信号ꎮ骨髓基质细胞ꎬ破骨细胞和成骨细胞为多发性骨髓瘤细胞提供关键的激活和归巢信号ꎬ例如增殖诱导配体(A ̄PRIL)㊁IL-6和CXCL12[61]ꎮ在小鼠中ꎬBTK和TEC对于成骨细胞形成是必不可少的ꎬ其由NF-κB配体的受体活化剂(RANKL)诱导ꎮ与此观察结果一致ꎬ依鲁替尼阻断RANKL诱导的BTK和下游PLCγ2的磷酸化ꎬ并通过骨吸收活性测量体外抑制人破骨细胞功能ꎮ在来自患有多发性骨髓瘤的患者的破骨细胞或骨髓基质细胞中ꎬ依鲁替尼下调肿瘤支持因子的产生ꎬ包括CCL3㊁转化生长因子β㊁APRIL和CXCL12ꎮ依鲁替尼阻断了CXCL12诱导的多发性骨髓瘤细胞的黏附和迁移ꎬ并减少了由IL-6引发的多发性骨髓瘤细胞生长和存活[62]ꎮ依鲁替尼还抑制了多发性骨髓瘤样细胞在体外形成集落的潜力ꎬ可以想象这种药物也可能破坏多发性骨髓瘤细胞中的BTK信号传导ꎮ多发性骨髓瘤细胞中的BTK抑制也可能阻断涉及疾病进展的其他途径ꎬ因为多发性骨髓瘤中的TLR信号传导可能增加疾病进展[63]ꎮ4.5㊀自身免疫性疾病㊀类风湿性关节炎(RA)是一种使人衰弱的全身性自身免疫性疾病ꎬ其特征在于在受影响的关节中循环自身抗体㊁滑膜炎症㊁血管翳形成以及软骨和骨质破坏[64]ꎮ已经进行了几项研究以研究BTK抑制在动物模型中的自身免疫中的功效ꎮ依鲁替尼在人B细胞中选择性地阻断BCR信号传导ꎬ但不影响T细胞受体(TCR)信号传导ꎮChang等[65]通过对依鲁替尼在体内疾病模型中的作用研究了其在关节炎中的作用机制ꎮ依鲁替尼治疗在CIA模型和CAIA模型中降低了在该模型中起重要作用的促炎细胞因子和趋化因子ꎬ包括滑膜和血清中IL-6㊁IL-1β㊁IL-17㊁TNF-α㊁KC和IFN-γ的水平ꎻ对关节滑膜炎㊁血管翳形成㊁软骨和骨破坏的有效抑制ꎬ并且观察到BCR介导的B淋巴细胞增殖和功能的显著抑制ꎮRN486是BTK的选择性可逆抑制剂[66]ꎬ在体外处理限制了人和鼠B细胞中CD69的上调以及向浆细胞的分化ꎮ它还限制了PBMC-B细胞共培养系统中体外IL-6和IL-2的产生ꎮ这表明BTK抑制也可以影响B细胞的细胞因子产生ꎬ尽管所涉及的途径仍有待阐明ꎮ除类风湿性关节炎外ꎬ还在系统性红斑狼疮(SLE)模型中研究了BTK抑制剂的功效ꎮ在患有狼疮的MRL.lpr/lpr小鼠中ꎬ用依鲁替尼治疗限制了蛋白尿和血尿素氮水平的升高ꎬ说明肾功能障碍或损伤得到了保护ꎮ在该模型中ꎬ自身抗体和免疫复合物的形成对于疾病是必需的ꎬ并且在依鲁替尼治疗后血清抗dsDNA抗体水平降低[9]ꎮ依鲁替尼在B6.Sle1/B6.Sle1.Sle3狼疮易感小鼠发病前给药ꎬ降低了其自身反应性IgG但不降低IgM的水平ꎬ小鼠脾脏大小和脾细胞数量显著降低ꎮ这是由于活化的B细胞㊁GCB细胞和血浆母细胞ꎬ间接限制了CD4+和CD8+T细胞的活化ꎬ且巨噬细胞ꎬ树突细胞和嗜中性粒细胞未受影响ꎬ这表明依鲁替尼单独在B细胞中的作用足以抑制T细胞ꎮ与未处理的对照相比ꎬ处理的小鼠表现出肾损伤明显减少[10]ꎮRN486在NZBˑNZW狼疮小鼠疾病发作后ꎬ与未给药治疗的对照相比ꎬ停止了蛋白尿的进展并且降低了肾脏中IgM㊁IgG和C3以及巨噬细胞浸润的沉积ꎮRN486抑制了B细胞活化ꎬ并且dsDNA特异性IgG浆细胞的数量减少ꎬ导致抗dsDNA抗体的血清水平降低ꎬ而不影响总浆细胞数量[67]ꎮ以上数据表明ꎬ通过靶向不同病理途径抑制BTKꎬ可以抑制或预防不同自身免疫模型中的疾病症状ꎮ5 结论与展望在这篇综述中ꎬ我们概述了我们目前对BTK信号通路的研究进展ꎬ以及BTK缺陷疾病与BTK抑制剂能够有效治疗的相关疾病ꎮ近年来在确定BTK抑制剂的作用机制方面取得了很大进展ꎬBTK参与不同的病理机制ꎬ现在看来很可能在很多情况下BTK抑制对肿瘤进展的影响是由B细胞上的各种受体和对微环境刺激的反应产生的信号的复杂相互作用的结果ꎮ因此ꎬ对恶性肿瘤中基因组畸变背景下BTK信号传导的致癌作用的新见解对于优化BTK靶向治疗剂的使用是至关重要的ꎮ需要更多的研究来了解哪些患者从哪种特定化合物中获益最多ꎮ高通量组合筛查策略应有助于确定可以优先用于临床研究的依鲁替尼组合ꎮ预期这种组合方案通过阻止对BTK抑制的抗性的发展和避免终身用抑制剂治疗而产生持久的反应ꎮ我们已经讨论了BTK在自身免疫病理学中的潜在意义ꎮ然而ꎬ目前尚不清楚BTK蛋白是否在各种外周血B细胞亚群中差异表达ꎬ或者在自身免疫的情况下其在B细胞中的表达或活性是否增加ꎮ来自自身免疫动物模型中BTK抑制的有希望的数据表明ꎬBTK的药理学调节可以为治疗B细胞分化中的自身免疫BTK信号传导和RA和SLE等自身免疫疾病提供有效的新治疗策略ꎮ参考文献:[1]㊀CONLEYMEꎬDOBBSAKꎬFARMERDMꎬetal.PrimaryBcellimmunodeficiencies:comparisonsandcontrasts[J].AnnuRevIm ̄munolꎬ2009(27):199-227.[2]TSUKADASꎬSAFFRANDCꎬRAWLINGSDJꎬetal.Deficientex ̄pressionofaBcellcytoplasmictyrosinekinaseinhumanX-linkedagammaglobulinemia[J].Cellꎬ1993ꎬ72(2):279-290.[3]MAHAJANSꎬGHOSHSꎬSUDBECKEAꎬetal.Rationaldesignandsynthesisofanovelanti-leukemicagenttargetingBrutonᶄstyrosinekinase(BTK)ꎬLFM-A13[alpha-cyano-beta-hydroxy-beta-methyl-N-(2ꎬ5-dibromophenyl)propenamide][J].JBiolChemꎬ1999ꎬ274(14):9587-9599.[4]PANZꎬSCHEERENSHꎬLISJꎬetal.Discoveryofselectiveirre ̄versibleinhibitorsforBrutonᶄstyrosinekinase[J].ChemMedChemꎬ2007ꎬ2(1):58-61.[5]FENGYꎬDUANWꎬCUXꎬetal.Brutonᶄstyrosinekinase(BTK)inhibitorsintreatingcancer:apatentreview(2010-2018)[J].Ex ̄pertOpinTherPatꎬ2019ꎬ29(4):217-241.[6]CASTILLOJJꎬPALOMBAMLꎬADVANIRꎬetal.IbrutinibinWaldenstrommacroglobulinemia:latestevidenceandclinicalexpe ̄rience[J].TherAdvHematolꎬ2016ꎬ7(4):179-186.[7]HENDRIKSRWꎬSARAVANANYꎬKILLP.TargetingBrutonᶄstyrosinekinaseinBcellmalignancies[J].NatRevCancerꎬ2014ꎬ14(4):219-232.[8]KILLPꎬDEBRUIJNMJꎬVANHULSTJAꎬetal.Brutonᶄstyrosinekinasemediatedsignalingenhancesleukemogenesisinamousemodelforchroniclymphocyticleukemia[J].AmJBloodReseꎬ2013ꎬ3(1):71-83.[9]HONIGBERGLAꎬSMITHAMꎬMINTSꎬetal.TheBrutontyrosinekinaseinhibitorPCI-32765blocksB-cellactivationandiseffica ̄ciousinmodelsofautoimmunediseaseandB-cellmalignancy[J].ProcNatlAcadSciUSAꎬ2010ꎬ107(29):13075-13080.[10]HUTCHESONJꎬVANARSAKꎬBASHMAKOVAꎬetal.ModulatingproximalcellsignalingbytargetingBtkameliorateshumoralautoim ̄munityandend-organdiseaseinmurinelupus[J].ArthritisResT ̄herꎬ2012ꎬ14(6):R243.[11]KHANWN.RegulationofBlymphocytedevelopmentandactivationbyBrutonᶄstyrosinekinase[J].ImmunolResꎬ2001ꎬ23(2-3):147-156. [12]BRADSHAWJM.TheSrcꎬSykꎬandTecfamilykinases:Distincttypesofmolecularswitches[J].CellSignalꎬ2010ꎬ22(8):1175-1184.[13]MOHAMEDAJꎬYULꎬBÄCKESJÖCMꎬetal.Brutonᶄstyrosinekinase(Btk):functionꎬregulationꎬandtransformationwithspecialemphasisonthePHdomain[J].ImmunolRevꎬ2009ꎬ228(1):58-73.[14]RAWLINGSDJꎬSCHARENBERGAMꎬPARKHꎬetal.ActivationofBTKbyaphosphorylationmechanisminitiatedbySRCfamilyki ̄nases[J].Scienceꎬ1996ꎬ271(5250):822-825.[15]MIDDENDORPSꎬDINGJANGMꎬMAASAꎬetal.FunctionofBru ̄tonᶄstyrosinekinaseduringBcelldevelopmentispartiallyinde ̄pendentofitscatalyticactivity[J].JImmunolꎬ2003ꎬ171(11):5988-5996.[16]BELVERLꎬDEYéBENESVGꎬRAMIROAR.MicroRNAspreventthegenerationofautoreactiveantibodies[J].Immunityꎬ2010ꎬ33(5):713-722.[17]DACUNHA-BANGCꎬNIEMANNCU.TargetingBrutonᶄsTyrosineKinaseAcrossB-CellMalignancies[J].Drugsꎬ2018ꎬ78(16):1653-1663.[18]RIPJꎬDEBRUIJNMJWꎬAPPELMANMKꎬetal.Toll-LikeRe ̄ceptorSignalingDrivesBtk-MediatedAutoimmuneDisease[J].FrontImmunolꎬ2019(10):95.[19]KIMMKꎬPANXQꎬHUANGZYꎬetal.FcγReceptorsDifferinTheirStructuralRequirementsforInteractionwiththeTyrosineKi ̄naseSykintheInitialStepsofSignalingforPhagocytosis[J].ClinImmunolꎬ2001ꎬ98(1):125-132.[20]SATTERTHWAITEABꎬCHEROUTREHꎬKHANWNꎬetal.BtkdosagedeterminessensitivitytoBcellantigenreceptorcross-linking[J].ProcNatlAcadSciUSAꎬ1997ꎬ94(24):13152-13157.[21]VALLAKꎬFLOWERSCRꎬKOFFJL.TargetingtheBcellreceptorpathwayinnon-Hodgkinlymphoma[J].ExpertOpinInvestigDrugsꎬ2018ꎬ27(6):513-522.[22]JONGSTRA-BILENJꎬPUIGCANOAꎬHASIJAMꎬetal.DualfunctionsofBrutonᶄstyrosinekinaseandTeckinaseduringFcgam ̄mareceptor-inducedsignalingandphagocytosis[J].JImmunolꎬ2008ꎬ181(1):288-298.[23]WEBERMꎬTREANORBꎬDEPOILDꎬetal.PhospholipaseC-gam ̄ma2andVavcooperatewithinsignalingmicroclusterstopropagateBcellspreadinginresponsetomembrane-boundantigen[J].JExpMedꎬ2008ꎬ205(4):853-868.[24]COPPOLINOMGꎬDIERCKMANRꎬLOIJENSJꎬetal.Inhibitionofphosphatidylinositol-4-phosphate5-kinaseIalphaimpairslocalizedactinremodelingandsuppressesphagocytosis[J].JBiolChemꎬ2002ꎬ277(46):43849-43857.[25]KIMYJꎬSEKIYAFꎬPOULINBꎬetal.MechanismofB-cellreceptor-inducedphosphorylationandactivationofphospholipaseC-gamma2[J].MolCellBiolꎬ2004ꎬ24(22):9986-9999.[26]SPAARGARENMꎬBEULINGEAꎬRURUPMLꎬetal.TheBcellantigenreceptorcontrolsintegrinactivitythroughBtkandPLCgam ̄ma2[J].JExpMedꎬ2003ꎬ198(10):1539-1550.[27]QUCꎬLIUYꎬKUNKALLAKꎬetal.TrimericGprotein-CARMA1axislinkssmoothenedꎬthehedgehogreceptortransducerꎬtoNF-kappaBactivationindiffuselargeB-celllymphoma[J].Bloodꎬ2013ꎬ121(23):4718-4728.[28]GUOBꎬKATORMꎬGARCIA-LLORETMꎬetal.EngagementoftheHumanPre-BCellReceptorGeneratesaLipidRaft–De ̄pendentCalciumSignalingComplex[J].Immunityꎬ2000ꎬ13(2):243-253.[29]HENDRIKSRWꎬMIDDENDORPS.Thepre-BCRcheckpointasacell-autonomousproliferationswitch[J].TrendsImmunolꎬ2004ꎬ25(5):249-256.[30]KERSSEBOOMRꎬMIDDENDORPSꎬDINGJANGMꎬetal.BrutonᶄstyrosinekinasecooperateswiththeBcelllinkerproteinSLP-65asatumorsuppressorinPre-Bcells[J].JExpMedꎬ2003ꎬ198(1):91-98.[31]FLEMMINGAꎬBRUMMERTꎬRETHMꎬetal.TheadaptorproteinSLP-65actsasatumorsuppressorthatlimitspre-Bcellexpansion[J].NatImmunolꎬ2003ꎬ4(1):38-43.[32]YINSꎬGAMBERꎬSUNJꎬetal.AMurineModelofChronicLym ̄phocyticLeukemiaBasedonBCell-RestrictedExpressionofSf3b1MutationandAtmDeletion[J].CancerCellꎬ2019ꎬ35(2):283-296.[33]UMEZAWAYꎬAKIYAMAHꎬOKADAKꎬetal.Molecularmecha ̄nismsforenhancementofstromalcell-derivedfactor1-inducedchemotaxisbyplateletendothelialcelladhesionmolecule1(PE ̄CAM-1)[J].JBiolChemꎬ2017ꎬ292(48):19639-19655. [34]DEGORTERDJꎬBEULINGEAꎬKERSSEBOOMRꎬetal.BrutonᶄstyrosinekinaseandphospholipaseCgamma2mediatechemokine-controlledBcellmigrationandhoming[J].Immunityꎬ2007ꎬ26(1):93-104.[35]DEROOIJMFꎬKUILAꎬGEESTCRꎬetal.TheclinicallyactiveBTKinhibitorPCI-32765targetsB-cellreceptor-andchemokine-controlledadhesionandmigrationinchroniclymphocyticleukemia[J].Bloodꎬ2012ꎬ119(11):2590-2594.[36]KELLYPNꎬROMERODLꎬYANGYꎬetal.Selectiveinterleukin-1receptor-associatedkinase4inhibitorsforthetreatmentofauto ̄immunedisordersandlymphoidmalignancy[J].JExpMedꎬ2015ꎬ212(13):2189-2201.[37]KENNYEFꎬQUINNSRꎬDOYLESLꎬetal.BrutonᶄsTyrosineKi ̄naseMediatestheSynergisticSignallingbetweenTLR9andtheBCellReceptorbyRegulatingCalciumandCalmodulin[J].PLoSOneꎬ2013ꎬ8(8):e74103.[38]CARRILLO-TAPIAEꎬGARCIA-GARCIAEꎬHERRERA-GONZALEZNEꎬetal.DelayeddiagnosisinX-linkedagamma ̄globulinemiaanditsrelationshiptotheoccurrenceofmutationsinBTKnon-kinasedomains[J].ExpertRevClinImmunolꎬ2018ꎬ14(1):83-93.[39]NOMURAKꎬKANEGANEHꎬKARASUYAMAHꎬetal.GeneticdefectinhumanX-linkedagammaglobulinemiaimpedesamatura ̄tionalevolutionofpro-Bcellsintoalaterstageofpre-BcellsintheB-celldifferentiationpathway[J].Bloodꎬ2000ꎬ96(2):610-617.[40]VANZELMMCꎬPUMARMꎬSHUTTLEWORTHPꎬetal.FunctionalAntibodyResponsesFollowingAllogeneicStemCellTransplantationforTP53Mutantpre-B-ALLinaPatientWithX-LinkedAgammaglobu ̄linemia[J].FrontImmunolꎬ2019(10):895.[41]MIRSAFIANHꎬRIPENAMꎬLEONGWMꎬetal.TranscriptomeprofilingofmonocytesfromXLApatientsrevealedtheinnateimmunefunctiondysregulationduetotheBTKgeneexpressionde ̄ficiency[J].SciRepꎬ2017ꎬ7(1):6836.[42]CONLEYMEꎬBROWNPꎬPICKARDARꎬetal.ExpressionofthegenedefectinX-linkedagammaglobulinemia[J].NEnglJMedꎬ1986ꎬ315(9):564-567.[43]FEARONERꎬWINKELSTEINJAꎬCIVINCIꎬetal.Carrierdetec ̄tioninX-linkedagammaglobulinemiabyanalysisofX-chromosomeinactivation[J].NEnglJMedꎬ1987ꎬ316(8):427-431.[44]EL-SAYEDZAꎬABRAMOVAIꎬALDAVEJCꎬetal.X-linkedagammaglobulinemia(XLA):Phenotypeꎬdiagnosisꎬandtherapeuticchallengesaroundtheworld[J].WorldAllergyOrganJꎬ2019ꎬ12(3):100018.[45]WINKELSTEINJAꎬMARINOMCꎬLEDERMANHMꎬetal.X-linkedagammaglobulinemia:reportonaUnitedStatesregistryof201patients[J].Medicine(Baltimore)ꎬ2006ꎬ85(4):193-202. [46]HENDRIKSRWꎬBREDIUSRGꎬPIKE-OVERZETKꎬetal.BiologyandnoveltreatmentoptionsforXLAꎬthemostcommonmonogeneticimmunodeficiencyinman[J].ExpertOpinTherTar ̄getsꎬ2011ꎬ15(8):1003-1021.[47]TARAKHOVSKYA.XidandXid-likeimmunodeficienciesfromasignalingpointofview[J].CurrOpinImmunolꎬ1997ꎬ9(3):319-323.[48]TANWARSꎬDHARAꎬVARANASIVꎬetal.MediationoftransitionalBcellmaturationintheabsenceoffunctionalBrutonᶄstyrosinekinase[J].SciRepꎬ2017(7):46029.[49]CORNETHOBJꎬWOLTERINKRGJKꎬHENDRIKSRW.BTKSignalinginBCellDifferentiationandAutoimmunity[J].BCellReceptorSignalingꎬ2015(393):67-105.[50]MAURYANꎬGUJARRꎬGUPTAMꎬetal.ImmunoregulationofdendriticcellsbythereceptorTcellIgandmucinprotein-3viaBrutonᶄstyrosinekinaseandc-Src[J].JImmunolꎬ2014ꎬ193(7):3417-3425.[51]BAUMGARTHN.ThedoublelifeofaB-1cell:self-reactivityse ̄lectsforprotectiveeffectorfunctions[J].NatRevImmunolꎬ2011ꎬ11(1):34-46.[52]SANDSTEDTKꎬBERGLöFAꎬFEINSTEINRꎬetal.DifferentialsusceptibilitytoMycoplasmapulmonisintranasalinfectioninX-linkedimmunodeficient(xid)ꎬseverecombinedimmunodeficient(scid)ꎬandimmunocompetentmice[J].ClinExpImmunolꎬ1997ꎬ108(3):490-496.[53]KRAUSEJR.WHOClassificationofTumoursofHaematopoieticandLymphoidTissues:AnOverview[J].CriticalValuesꎬ2009ꎬ2(2):30-32.[54]TREONSP.HowItreatWaldenströmmacroglobulinemia[J].Bloodꎬ2009ꎬ114(12):2375-2385.[55]NAKAMURAAꎬOHWADACꎬTAKEUCHIMꎬetal.DetectionofMYD88L265Pmutationbynext-generationdeepsequencinginperipheralbloodmononuclearcellsofWaldenströmᶄsmacroglobu ̄linemiaandIgMmonoclonalgammopathyofundeterminedsignifi ̄cance[J].PLoSOneꎬ2019ꎬ14(9):e0221941.[56]YANGGꎬZHOUYꎬLIUXꎬetal.AmutationinMYD88(L265P)supportsthesurvivaloflymphoplasmacyticcellsbyactivationofBrutontyrosinekinaseinWaldenstrommacroglobulinemia[J].Bloodꎬ2013ꎬ122(7):1222-1232.[57]MUZIOMꎬAPOLLONIOBꎬSCIELZOCꎬetal.ConstitutiveactivationofdistinctBCR-signalingpathwaysinasubsetofCLLpatients:amolecularsignatureofanergy[J].Bloodꎬ2008ꎬ112(1):188-195.[58]PONADERSꎬCHENSSꎬBUGGYJJꎬetal.TheBrutontyrosineki ̄naseinhibitorPCI-32765thwartschroniclymphocyticleukemiacellsurvivalandtissuehominginvitroandinvivo[J].Bloodꎬ2012ꎬ119(5):1182-1189.[59]JARESPꎬCOLOMERDꎬCAMPOE.Molecularpathogenesisofmantlecelllymphoma[J].JClinInvestꎬ2012ꎬ122(10):3416-3423.[60]PALSINGHSꎬDAMMEIJERFꎬHENDRIKSRW.RoleofBrutonᶄstyrosinekinaseinBcellsandmalignancies[J].MolCancerꎬ2018ꎬ17(1):57.[61]WANGWꎬWEIRꎬLIUSꎬetal.BTKinducesCAM-DRthroughregulationofCXCR4degradationinmultiplemyeloma[J].AmJTranslResꎬ2019ꎬ11(7):4139-4150.[62]TAIYTꎬCHANGBYꎬKONGSYꎬetal.Brutontyrosinekinasein ̄hibitionisanoveltherapeuticstrategytargetingtumorinthebonemarrowmicroenvironmentinmultiplemyeloma[J].Bloodꎬ2012ꎬ120(9):1877-1887.[63]NAYMAGONLꎬABDUL-HAYM.Novelagentsinthetreatmentofmultiplemyeloma:areviewaboutthefuture[J].JHematolOncolꎬ2016ꎬ9(1):52.[64]LINDSTROMTMꎬROBINSONWH.AMultitudeofKinases WhicharetheBestTargetsinTreatingRheumatoidArthritis?[J].RheumDisClinNorthAmꎬ2010ꎬ36(2):367-383.[65]CHANGBYꎬMINMHꎬFRANCESCOMꎬetal.TheBrutontyrosinekinaseinhibitorPCI-32765amelioratesautoimmunearthritisbyinhibitionofmultipleeffectorcells[J].ArthritisResT ̄herꎬ2011ꎬ13(4):R115.[66]XUDꎬKIMYꎬPOSTELNEKJꎬetal.RN486ꎬaselectiveBrutonᶄstyrosinekinaseinhibitorꎬabrogatesimmunehypersensitivityresponsesandarthritisinrodents[J].JPharmacolExpTherꎬ2012ꎬ341(1):90-103.[67]MINA-OSORIOPꎬLASTANTJꎬKEIRSTEADNꎬetal.Suppressionofglomerulonephritisinlupus-proneNZBˑNZWmicebyRN486ꎬaselectiveinhibitorofBrutonᶄstyrosinekinase[J].ArthritisRheumꎬ2013ꎬ65(9):2380-2391.(上接第129页)验中3种植物激素对于熏倒牛种子萌发的影响强度ꎬ表现为GA3>IAA>NAAꎮ统计不同处理条件下熏倒牛种子的发芽率和发芽势等发现ꎬ采用植物激素并不能彻底打破熏倒牛种子的休眠ꎬ原因概括如下:①应用外源萌发促进物质处理熏倒牛种子仅能解除部分休眠ꎬ而达不到层积处理过程中种子内部发生的一系列反应ꎻ②种子中存在其他抑制种子萌发的物质ꎬ植物激素并不能消除其影响[13-14]ꎮ因此ꎬ针对熏倒牛种子休眠机制的分析和萌发特性还应进一步深入研究ꎮ参考文献:[1]㊀王晶晶ꎬ景明ꎬ李炀ꎬ等.藏药熏倒牛质量标准研究[J].中国中医药信息杂志ꎬ2013ꎬ20(4):48-49.[2]王维恩ꎬ张晓峰ꎬ沈建伟ꎬ等.藏药熏倒牛化学成分研究[J].天然产物研究与开发ꎬ2009ꎬ21(2):199-202. [3]中国科学院植物志委员会.中国植物志:第43卷[M].北京:北京科学出版社ꎬ1998:87.[4]刘越敏ꎬ柳娜ꎬ寇亮ꎬ等.藏药熏倒牛的研究现状与进展[J].药学研究ꎬ2019ꎬ38(11):648-651.[5]廖腾飞ꎬ雷家军.尖萼耧斗菜种子萌发特性研究[J].种子ꎬ2011ꎬ30(1):92-93.[6]薛正帅ꎬ傅竞ꎬ崔成文ꎬ等.鹿藿种子萌发特性研究[J].长江大学学报(自科版)ꎬ2018ꎬ15(10):34-36ꎬ40. [7]杨霞艳ꎬ左经会ꎬ吴承梦ꎬ等.甘肃耧斗菜种子萌发特性研究[J].六盘水师范学院学报ꎬ2019ꎬ31(3):1-5. [8]叶青雷ꎬ李志ꎬ王立志ꎬ等.不同外源激素对桑树种子发芽的影响[J].黑龙江农业科学ꎬ2015(11):78-80. [9]杨锋利ꎬ汪茜.温度和不同激素及其浓度和浸种时间对美人蕉种子萌发的影响[J].中国农学通报ꎬ2015ꎬ31(13):126-129.[10]呼凤兰.5种植物激素对黑豆种子萌发的影响[J].种子ꎬ2019ꎬ38(6):95-99.[11]付婷婷ꎬ程红焱ꎬ宋松泉.种子休眠的研究进展[J].植物学报ꎬ2009ꎬ44(5):629-641.[12]刘华ꎬ蒋齐ꎬ李如来ꎬ等.不同植物激素对甘草种子萌发的影响研究[J].宁夏农林科技ꎬ2013ꎬ54(2):19-21. [13]唐安军ꎬ龙春林ꎬ刀志灵.种子休眠机理研究概述[J].植物分类与资源学报ꎬ2004ꎬ26(3):241-251.[14]杨期和ꎬ叶万辉ꎬ宋松泉ꎬ等.植物种子休眠的原因及休眠的多形性[J].西北植物学报ꎬ2003ꎬ23(5):837-843.。
受体酪氨酸激酶在细胞信号传导中的作用在细胞运作的过程中,不同种类的分子之间的相互作用是必不可少的。
细胞信号传导是维持正常机体生理功能的基础,而受体酪氨酸激酶作为重要的调节因子,在细胞信号传导中扮演着至关重要的角色。
本文将从分子结构、酪氨酸激酶信号通路以及生物学功能三个方面展开探讨,深入阐述受体酪氨酸激酶在细胞信号传导中的重要作用。
一、分子结构受体酪氨酸激酶又被称为RTK(Receptor Tyrosine Kinase,受体酪氨酸激酶)或TK(Tyrosine Kinase,酪氨酸激酶)。
在所有的RTK家族成员中,酪氨酸激酶是最普遍的一类。
它们的筛选方式是:细胞膜表面有大量的RTK,这些RTK呈多聚态,在接受到激活因子(如细胞外的生长因子)后,RTK发生跨膜自聚合,并激活其内部的酪氨酸激酶活性,从而启动细胞内信号转导途径。
其主要结构包括N 端、胞外域、跨膜域、细胞内域,依次由N末端向C末端排列。
其中胞外域具有多类结构,有Ig样折叠、Fibronectin样结构、肝素结构等,各自对应不同的生长因子家族。
跨膜域长度一般为20~30个氨基酸残基,通过疏水相互作用,在质膜上形成单个的α螺旋。
而最重要的部分是C末端的酪氨酸激酶结构域,其由两个相互作用的叶状蛋白质构成,每个叶状蛋白质包含N和C两翼。
N翼包含胞内氨基酸序列、ATP结合位点和碳水化合物酰基转移酶结构域,C翼则含有底物结合位点和酪氨酸磷酸化作用位点。
通过这些区域的协同作用,酪氨酸激酶能够将底物(主要是自身)上的酪氨酸残基磷酸化。
二、酪氨酸激酶信号通路细胞在接受到封装在信号分子中的指令时,便会立刻启动其内部信号传递过程,从而作出相应的反应。
对于酪氨酸激酶而言,其信号传递可以大致分为以下几个过程:1.生长因子结合生长因子被RTK的胞外域捕获后,RTK就会在膜上 dimerize(自聚合)。
这样其相互作用的胞内域就会形成一个略微扭曲的结构,尽管不一定将底物磷酸化。
1 JAK—STAT信号通路1) JAK与STAT蛋白JAK-STAT信号通路是近年来发现的一条由细胞因子刺激的信号转导通路,参与细胞的增殖、分化、凋亡以及免疫调节等许多重要的生物学过程.与其它信号通路相比,这条信号通路的传递过程相对简单,它主要由三个成分组成,即酪氨酸激酶相关受体、酪氨酸激酶JAK和转录因子STAT。
(1) 酪氨酸激酶相关受体(tyrosine kinase associated receptor)7)、GM—CSF(粒细胞/巨噬细胞集落刺激因子)、GH(生长激素)、EGF(表皮生长因子)、PDGF7(IL—2许多细胞因子和生长因子通过JAK—STAT信号通路来传导信号,这包括白介素2 (血小板衍生因子)以及IFN (干扰素)等等。
这些细胞因子和生长因子在细胞膜上有相应的受体。
这些受体的共同特点是受体本身不具有激酶活性,但胞内段具有酪氨酸激酶JAK的结合位点。
受体与配体结合后,通过与之相结合的JAK的活化,来磷酸化各种靶蛋白的酪氨酸残基以实现信号从胞外到胞内的转递.(2) 酪氨酸激酶JAK(Janus kinase)很多酪氨酸激酶都是细胞膜受体,它们统称为酪氨酸激酶受体(receptor tyrosine kinase, RTK),而JAK 却是一类非跨膜型的酪氨酸激酶。
JAK是英文Janus kinase的缩写,Janus在罗马神话中是掌管开始和终结的两面神.之所以称为两面神激酶,是因为JAK既能磷酸化与其相结合的细胞因子受体,又能磷酸化多个含特定SH2结构域的信号分子。
JAK蛋白家族共包括4个成员:JAK1、JAK2、JAK3以及Tyk2,它们在结构上有7个JAK同源结构域(JAK homology domain, JH),其中JH1结构域为激酶区、JH2结构域是“假”激酶区、JH6和JH7是受体结合区域(如图4).(3) 转录因子STAT(signal transducer and activator of transcription)STAT被称为“信号转导子和转录激活子"。
受体酪氨酸激酶分类受体酪氨酸激酶(RTKs)是一种重要的细胞膜受体家族,在细胞信号传导中发挥着关键作用。
RTKs的分类通常基于其结构和序列相似性,不同类型的RTKs在结构和功能上有所区别。
本文将对RTKs进行分类,并对每个分类进行简要介绍。
1.基本的RTKs类基本的RTKs类是最常见的RTKs类型,包括一系列重要的受体如胰岛素受体、表皮生长因子受体(EGFR)和血小板源生长因子受体(PDGFR)等。
它们共同特点是具有胞外结构域、跨膜区和细胞内酪氨酸激酶结构域。
这些受体在各种生物过程中起到关键作用,如细胞增殖、分化和存活等。
2.钠离子依赖的RTKs类钠离子依赖的RTKs类是一类特殊的RTKs,其结构域中含有钠离子结合位点。
它们包括血红蛋白酶受体和肝细胞生长因子受体等。
这些受体在胚胎发育和细胞分化中扮演重要角色。
3.间充质受体酪氨酸激酶类间充质受体酪氨酸激酶(STK)类是一类结构上与RTKs相似的受体,但其酪氨酸激酶结构域与传统RTKs不同。
这类受体包括TGF-β受体和BMP受体等。
它们通过激活SMAD信号通路调节细胞增殖、分化和胚胎发育过程。
4.精索激酶受体类精索激酶受体类是一类包含特殊激酶结构域的RTKs。
精索激酶受体包括ROR受体家族和DYRK家族等。
这些受体在细胞增殖、发育和免疫调节中起到重要作用。
5.类钛素激酶受体类类钛素激酶受体类是一类与RTKs结构上相似但功能上不同的受体。
这些受体包括RET受体和ROS受体等。
它们在神经系统发育和甲状腺发育中发挥着重要作用。
总结起来,RTKs根据结构和功能的差异可以分为基本的RTKs类、钠离子依赖的RTKs类、间充质受体酪氨酸激酶类、精索激酶受体类和类钛素激酶受体类等。
这些不同类型的RTKs在细胞信号传导过程中起到关键作用,对于理解细胞生物学和疾病发生发展具有重要意义。
通过深入研究各类RTKs,我们能够更好地理解细胞信号网络的调控机制,为疾病的治疗提供新的靶点和策略。