新药Tucatinib(图卡替尼)合成检索总结报告
- 格式:pdf
- 大小:500.96 KB
- 文档页数:5
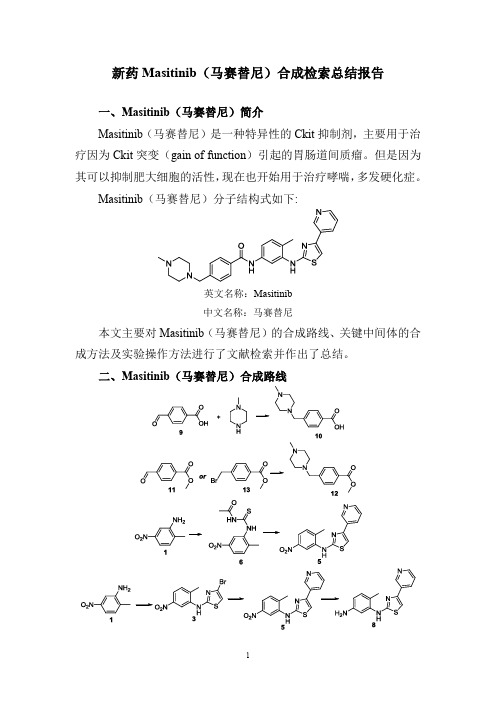
新药Masitinib(马赛替尼)合成检索总结报告
一、Masitinib(马赛替尼)简介
Masitinib(马赛替尼)是一种特异性的Ckit抑制剂,主要用于治疗因为Ckit突变(gain of function)引起的胃肠道间质瘤。
但是因为其可以抑制肥大细胞的活性,现在也开始用于治疗哮喘,多发硬化症。
Masitinib(马赛替尼)分子结构式如下:
英文名称:Masitinib
中文名称:马赛替尼
本文主要对Masitinib(马赛替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Masitinib(马赛替尼)合成路线
三、Masitinib(马赛替尼)合成检索总结报告(一) Masitinib(马赛替尼)中间体3的合成
(二) Masitinib(马赛替尼)中间体5的合成方法一
(三) Masitinib(马赛替尼)中间体5的合成方法二
①Masitinib(马赛替尼)中间体6的合成
②Masitinib(马赛替尼)中间体5的合成
(四) Masitinib(马赛替尼)中间体8的合成。
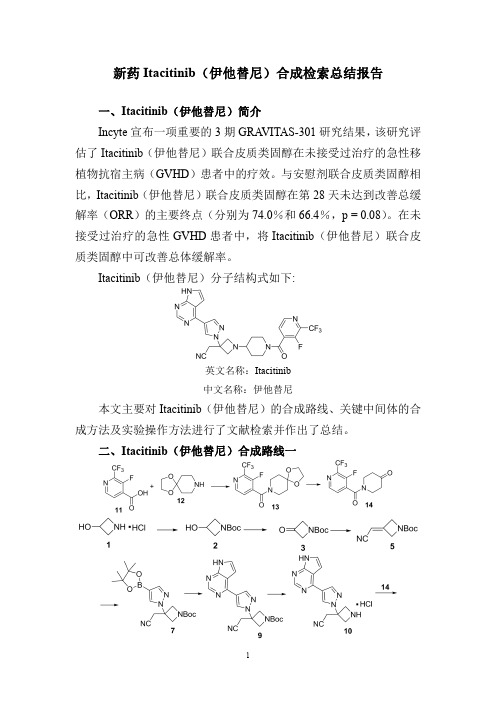
药物Fostamatinib(福坦替尼)合成检索总结报告一、Fostamatinib(福坦替尼)简介Fostamatinib(福坦替尼)于2018年4月在美国上市,主要用于对之前疗法效果不佳的慢性免疫性血小板减少症成年患者的治疗。
Fostamatinib(福坦替尼)是一种脾脏酪氨酸激酶(SYK)抑制剂,通过阻止血小板的破坏来应对疾病的潜在自身免疫作用。
Fostamatinib(福坦替尼)不良反应:腹泻,高血压,恶心,呼吸道感染,头晕,皮疹,腹痛,疲劳,胸痛和中性粒细胞减少。
Fostamatinib(福坦替尼)分子结构式如下:CAS:901119-35-5CAS:1025687-58-4英文名称:Fostamatinib英文名称:Fostamatinib disodium 中文名称:福坦替尼中文名称:福坦替尼二钠盐二、Fostamatinib(福坦替尼)合成路线三、Fostamatinib (福坦替尼)合成检索总结报告(一)Fostamatinib (福坦替尼)中间体2的合成合成方法实验步骤参考文献操作方法一3-Hydroxy-2-nitropyridine 1(20g,0.143mole)was dissolved in methanol (400mL)and a solution of 25%sodium methoxide in methanol (33mL,0.13mole)was added at room temperature.The mixture was stirred for 30min,then was cooled to 0o C,and bromine (7.2mL,0.14mole)was added slowly.The reaction was stirred at 00C for 30min,then was quenched with glacial AcOH (2.5mL).The solvent was removed in vacuo to afford material 2(30g,96%),which was used without further purification.WO2006/81178;(2006);(A2);WO2006/81179;(2006);(A1);WO2006/81264;(2006);(A1);WO2006/81289;(2006);(A2);US2008/194547;(2008);(A1)操作方法二3-Hydroxy-2-nitropyridine 1(50g,357mmol)was dissolved in 900mL of N,N-dimethylformamide.N-bromosuccinimide (82.57g,464mmol)was slowly added thereto and stirred at room temperature for 12hours.After the reaction was completed,the solution was concentrated using a rotary evaporator,and then extracted twice with distilled water and dichloromethane.The precipitate was formed using methyl alcohol and distilled water and filtered to obtain a mixture of compound 2and dibrominated compound (67.5g,Y =86.4%)in an 8:2ratio.KR2017/50048;(2017);(A)KoreanA solution of 2-nitropyridin-3-ol (1)(16.0g,114mmol)in dimethylformamide (120mL)was cooled to 0°C,and then N-bromosuccinimide (26.4g,1.48mmol)was gradually操作方法三added thereto.The solution wasthen stirred at 0°C for 1hour,and at room temperature for another 2hours.The reaction solution was concentrated in vacuo,and then the residue was washed with diethyl ether.The filtrate was poured to aqueous saturated sodium bicarbonate,followed by extraction with diethyl ether.The organic layer was washed sequentially with water and saturated brine,then dried with anhydrous magnesium sulfate,and concentrated in vacuo to yield the subject compound (2)(15.5g,62%)as a white powder.EP2341052;(2011);(A1)English 操作方法四A solution of 2-nitropyridin-3-ol 1(15g,104mmol)in DMF (189mL)was cooled to 0°C then N-bromosuccinimide (24g,135mmol)was added gradually over 25minutes.The solution was stirred at 0°C for an additional 15minutes and then the mixture was allowed to warm to RT,and stirred over night.The reaction mixture was concentrated in vacuo and purified by column chromatography using 100%CH 2Cl 2to afford 6-bromo-2-nitro-pyridin-3-ol 2(13.67g,56.2mmol,54.1%yield)as a yellow solid.WO2017/97728;(2017);(A1)English(二)Fostamatinib (福坦替尼)中间体3的合成合成方法实验步骤参考文献操作方法一A mixture of 6-bromo-2-nitropyridin-3-ol 2(3.28g,15mmol),ethyl 2-bromo-2-methylpropanoate (3.51g,18mmol),and K 2CO 3in dimethyl formamide (30mL)was stirred at room temp for 48h.Water was added and the mixture was extracted with EtOAc.The combined organic layers were washed with water and brine,dried,concentrated and purified with column chromatography,eluting with petroleum ether :EtOAc to afford ethyl 2-((6-bromo-2-nitropyridin-3-yl)oxy)-2-methylpropanoate 3(1.3g,36%)as a yellow solid.WO2011/59839;(2011);(A1)English(三)Fostamatinib (福坦替尼)中间体4的合成方法一。

新药Fisogatinib(非索替尼)合成检索总结报告一、Fisogatinib(非索替尼)简介Fisogatinib(非索替尼)是由Blueprint Medicines开发的一款在研的强效、高选择性成纤维细胞生长因子受体-4(FGFR4)抑制剂,用于治疗FGFR4驱动的晚期或转移性肝细胞癌HCC。
Fisogatinib(非索替尼)具有成为首个分子水平生物标记物驱动的HCC靶向药物的潜力。
Fisogatinib(非索替尼)分子结构式如下:英文名称:Fisogatinib中文名称:非索替尼本文主要对Fisogatinib(非索替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Fisogatinib(非索替尼)合成路线三、Fisogatinib (非索替尼)合成检索总结报告(一)Fisogatinib (非索替尼)中间体3的合成合成方法实验步骤参考文献操作方法一1(5.00g,20.5mmol)and 2(3.73g,20.5mmol)were dissolved in tetrahydrofuran (30ml),added with a solution of cesium carbonate (20.00g,61.5mmol)in water (30ml),and added with a catalytic amount of Pd(PPh 3)Cl 2.Theresulting mixture was heated to reflux for 4h under nitrogen atmosphere.The reaction solution was concentrated to dryness and extracted with ethyl acetate.The organic phase was washed once with saturated sodium chloride,dried over anhydrous sodium sulfate,and concentrated under reduced pressure.The resulting crude product was subjected tocolumn chromatography (200-300mesh silica gel,petroleum ether/ethyl acetate=10/1)to obtain intermediate 3(3.80g,yield of 62%)as a pale yellow solid.US2019/209564;(2019);(A1)English;CN110386921;(2019);(A).操作方法二A mixture of 6-bromo-2-chloroquinazoline (1)(5.0g,20.5mmol),3,5-dimethoxyphenylboronic acid (2)(3.7g,20.5mmol),Cs 2CO 3(20.0g,61.5mmol)and Pd(PPh 3)2Cl 2(1.4g,2.1mmol)in THF (50mL),dioxane (50mL)and water (10mL)was degassed with N 2three times,and stirred at 80°C for 3hours.An aliquot of the reaction mixture was analyzed by both TLC and LCMS,which indicated that the reaction had proceeded to completion.The mixture was cooled to room temperature,and extracted with EtOAc (3×200mL).The combined organic layers were washed with water and brine,dried over sodium sulfate,filtered and concentrated.The residue was purified by silica gel chromatography (petroleum ether/EtOAc =8:1)to obtain 2-chloro-6-(3,5-dimethoxyphenyl)quinazoline (3)as a light yellow solid (2.4g,38%).WO2014/11900;(2014);(A2)English;US2015/197519;(2015);(A1)English;US2017/9695165;(2017);(B2)English(二)Fisogatinib(非索替尼)中间体4的合成合成方法实验步骤参考文献操作方法一To a solution of2-chloro-6-(3,5-dimethoxyphenyl)quinazoline(3)(2.7g,8.9mmol)indry THF(80mL)wasadded dropwise sulfuryl chloride(3.0g,22.3mmol)at-20°C,and the reactionmixture was stirred for an additionalhour.An aliquot of the reaction mixture was analyzed byboth TLC and LCMS,which indicated that the reaction hadproceeded to completion.The reaction mixture wasquenched with water(1mL),and the solvents were removedunder reduced pressure.The precipitate was washed withCH3CN and dried to obtain2-chloro-6-(2,6-dichloro-3,5-dimethoxyphenyl)quinazoline(4)(2.6g,79%)as a whitesolid.WO2014/11900;(2014);(A2)English;US2015/197519;(2015);(A1)English;US2017/9695165;(2017);(B2)English操作方法二3(3.80g,12.6mmol)was dissolved in tetrahydrofuran(100ml),under nitrogen cooled to-20~30o C,was addeddropwise sulfuryl chloride(5.11g,37.9mmol),the resultingmixture at the same temperature reaction for2hours.Thereaction mixture was slowly raised to ambient temperature,was added acetonitrile(100ml),stirred for10minutes,theresulting solid was collected by filtration.Drying,to giveintermediate4(2.80g,60%yield)as a pale yellow solid.US2019/209564;(2019);(A1)English;CN110386921;(2019);(A).(三)Fisogatinib(非索替尼)中间体6的合成合成方法实验步骤参考文献操作方法一(3R,4R)-3-(((S)-1-phenylethyl)amino)tetrahydro-2H-pyran-4-ol5(2.0g,9.04mmol)was taken up in methanol(10ml)followed by addition of Et3N(1.260ml,9.04mmol)andBOC-anhydride(2.308ml,9.94mmol).The reaction mixturewas stirred at room temperature overnight.The solvents werethen removed in vacuo and the residue was taken up in DCM(10ml)and hexane(20ml)and heated to80°C.until thesolvent level was reduced by half.The reaction mixture wasremoved from heat and cooled to room temperature whilestirring.5ml of ether was then added and the reaction wasstirred at room temperature for2hours.The reaction mixtureUS2015/119405;(2015);(A1)English。

抗乳腺癌新药Tucatinib的新合成路线简介Tucatinib(Tukysa)妥卡替尼 /图卡替尼 (ONT-380)是一种小分子口服酪氨酸激酶(TKI)抑制剂,对HER2具有高度特异性的靶向选择性。
2020年04月17日,美国FDA批准Seattle Genetics公司开发的HER2特异性抑制剂Tukysa(tucatinib)与曲妥珠单抗和卡培他滨联用,用于治疗局部晚期、不可切除性或转移性(包括伴有脑转移)的HER2阳性乳腺癌成人患者。
接受Tukysa、曲妥珠单抗和卡培他滨联合治疗的患者的中位无进展生存期(PFS)为7.8个月,总生存期(OS)为21.9个月;而接受安慰剂、曲妥珠单抗和卡培他滨联合治疗的患者为5.6个月,总生存期(OS)为17.4个月。
在脑转移患者中,Tucatinib联合组1年无进展生存率为24.9%,安慰剂联合组为0%。
该研究成果参见:10.1056/NEJMoa1914609近期,Mao团队报道了新的路线,用于高效合成Tucatinib。
该成果发表在Synthesis上(DOI: 10.1055/s-0037-1610706)。
此前,Tucatinib的合成报道路线由Array BioPharma公司公开在一份专利文献中( WO 2007059257, 2007)。
专利报道的合成路线如下图所示:以4-硝基-2-氰基苯胺为原料,第一步与DMF-DMA缩合制备亚胺3(收率87%);随后经钯碳催化加氢还原硝基,得到胺4(90%产率);接着与1,1'-硫代羰基二咪唑(TCDI)和氨基醇发生缩合,制备得到硫脲衍生物5(收率仅34%);进一步与中间体6发生关环反应,得到关键中间体7(收率62%);最后,在对甲基苯磺酸作用下,发生分子内脱水关环形成恶唑啉,完成目标化合物tucatinib的合成。
作者将Tucatinib从a和b两处断键,分别拆分为三个片段:硫醚恶唑啉17、硝基苯3以及原研路线关键片段6。
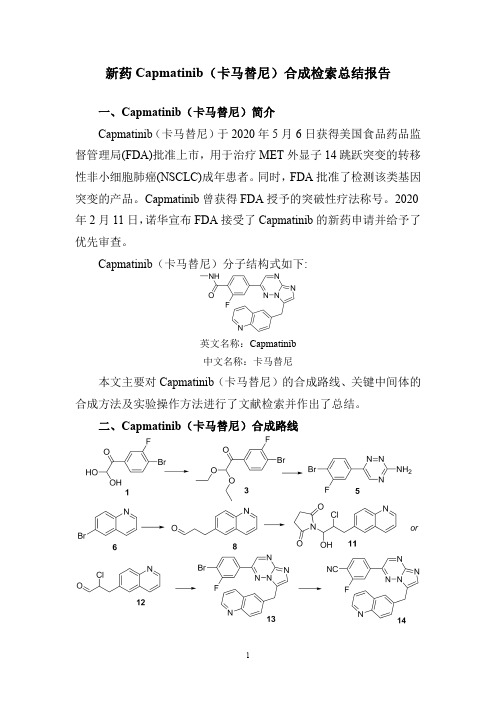
新药Capmatinib(卡马替尼)合成检索总结报告一、Capmatinib(卡马替尼)简介Capmatinib(卡马替尼)于2020年5月6日获得美国食品药品监督管理局(FDA)批准上市,用于治疗MET外显子14跳跃突变的转移性非小细胞肺癌(NSCLC)成年患者。
同时,FDA批准了检测该类基因突变的产品。
Capmatinib曾获得FDA授予的突破性疗法称号。
2020年2月11日,诺华宣布FDA接受了Capmatinib的新药申请并给予了优先审查。
Capmatinib(卡马替尼)分子结构式如下:英文名称:Capmatinib中文名称:卡马替尼本文主要对Capmatinib(卡马替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Capmatinib(卡马替尼)合成路线三、Capmatinib (卡马替尼)合成检索总结报告(一)Capmatinib (卡马替尼)中间体3的合成合成方法实验步骤参考文献操作方法一To a solution of 1-(4-bromo-3-fluorophenyl)-2,2-dihydroxy-ethanone 1(3.5g,0.014mol)in toluene (30mL)was added ethyl orthoformate 2(5.8mL,0.035mol)and p-toluene-sulfonic acid (100mg).The reaction was refluxed for 4h.After cooled to RT,the mixture was diluted with ethyl acetate,washed with saturated NaHCO 3solution,water,and brine,dried over MgSO 4,filtered and concentrated to give the product 3(4.0g,93%)which was used in the next step without further 2008/39457;(2008);(A1)English 操作方法二A 22L flask was charged with the hydrate of (4-bromo-3-fluorophenyl)-2-oxoacetaldehyde 1(1020g,4.41mol),toluene (7.5L),triethyl orthoformate 2(1633g,1.8L,11.04mol,2.5equiv),para-toluene sulfonic acid (33.5g,0.176mol,0.4equiv)at room temperature,and the resulting reaction mixture was heated to 110°C.and stirred at 110°C.for 6h.When HPLC showed that the reaction was complete,the reaction mixture was cooled down to room temperature before being poured into a 50L separation funnel along with ethyl acetate (7.5L)and the saturated aqueous sodium bicarbonate solution (NaHCO 3,3L).The mixture was stirred and the layers were separated.The aqueous layer was extracted with ethyl acetate (2L).The combined organic layers were washed with brine (4L),dried with sodium sulfate (Na 2SO 4),and concentrated under the reduced pressure to afford crude 1-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone 3(1240g,1345.7g theoretical,92.1%yield)which was used in the subsequent reaction without further 2009/291956;(2009);(A1)English(二)Capmatinib (卡马替尼)中间体5的合成合成方法实验步骤参考文献操作方法一A mixture of l-(4-bromo-3-fluorophenyl)-2,2-diethoxy-ethanone3(15.2g,50mmol),aminoguanidine bicarbonate4(10.2g,75mmol)and potassium hydroxide(6.6g,100mmol)in ethanol(200mL)and water(4mL)was refluxedovernight.The solvent was evaporated under reducedpressure and the residue was washed with acetonitrile andfiltered.The filtrate was concentrated under reducedpressure.The residue was dissolved in dichloromethane(100mL),washed with water,brine,and concentrated underreduced pressure.The residue was dissolved in ethanol(50mL).To the solution was added0.2N hydrochloric acid(50mL).The resultant mixture was heated to110o C for8h,andcooled with an ice-water bath.The precipitate that formedwas collected by filtration and washed with isopropanol togive the desired product5.(5.5g,41%)WO2008/64157;(2008);(A1)English操作方法二A22L flask was charged with1-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone3(1240g,4.07mol),ethanol(11L),water(1.4L),potassium hydroxide(KOH,910g,16.3mol,4.0equiv),and aminoguanidine bicarbonate4(1105g,8.13mol,2.0equiv)at room temperature.The resulting reactionmixture was then heated to75°C.for14h.When HPLCshowed the condensation reaction was deemed complete,thereaction mixture was cooled down to room temperaturebefore being filtered.The filtrate was then concentratedunder the reduced pressure to remove the most of thesolvents.The residual aqueous solution was extracted withethyl acetate(EtOAc,3×6L).The organic layers werecombined and concentrated under the reduced pressure togive a dark brown solid.This solid was dissolved in ethanol(4L)and the resulting solution was treated with a solution of0.2M aqueous hydrochloric acid solution(4L).Theresulting slurry was subsequently heated to50°C.for6hbefore being allowed to cool down to room temperature.Asolution of saturated aqueous sodium bicarbonate solution(NaHCO3,2L)was slowly added to the slurry and theresulting mixture was then concentrated under the reducedpressure to remove most of the solvents.The aqueousresidue was then treated with ethyl acetate(20L)to dissolveUS2009/291956;(2009);(A1)Englishthe solids.The two layers were separated andthe aqueous layer was extracted with ethyl acetate (2×2L).The combined organic layers were concentrated under the reduced pressure.The dark brown solids were treated with methyl tert-butyl ether (MTBE,4L)and the resulting slurry was heated to 30°C.and stirred at 30°C.for 30min.The mixture was filtered and the solids (green to orange in color)were collected (save the filtrate)and washed with methyl tert-butyl ether (MTBE,2L)to give the first crop of the crude desired product (5).The filtrate was evaporated under the reduced pressure,and the resulting dark brown solids were treated with methyl tert-butyl ether (MTBE,2L).The resulting slurry was heated to 30°C.and stirred at 30°C.for 30min.The mixture was filtered to give the second crop of the crude desired product (5)which was washed with MTBE (1L).The combined solids were dried in vacuum at 40-45°C.to afford 6-(4-bromo-3-fluorophenyl)-1,2,4-triazin -3-amine 5(585g,1095.1g theoretical,53.4%yield)which was used in the subsequent reaction without further purification.(三)Capmatinib (卡马替尼)中间体8的合成方法一合成方法实验步骤参考文献操作方法一Tris(dibenzylideneacetone)dipalladium (480mg,0.52mmol)and tri-tert-butyl-phosphonium tetrafluoroborate (300mg,1.0mmol)in a flask was evacuated and refilled with nitrogen (2times).1,4-dioxane (31mL)was added followed by consecutive addition of 6-bromoquinoline 6(7.2g,35mmol),2-propen-l-ol 7(4.7mL,69mmol)and N-cyclohexyl -N-methyl-cyclohexanamine (8.9mL,42mmol).The reaction vessel was evacuated and refilled with nitrogen (2times).The reaction mixture was stirred at 30o C for 24h.Diethyl ether (30mL)was added to the reaction mixture and then filtered and washed with diethyl ether.The organic extract was concentrated under reduced pressure.The residue was purified by flash chromatography eluting with ethyl acetate in hexanes (0-50%)to afford the desired product 8.(55%).WO2008/64157;(2008);(A1)English;US2011/212967;(2011);(A1)English.A 22L flask was charged with tris(dibenzylideneacetone)-dipalladium(0)(70.0g,0.076mol,0.015equiv),tri-tert-butylphosphonium tetrafluoroborate (44g,0.152mol,0.03。
新药Tepotinib(特泊替尼)合成检索总结报告一、Tepotinib(特泊替尼)简介2019年09月12日,美国食品和药物管理局已授予其靶向抗癌药MET抑制剂Tepotinib(特泊替尼)突破性药物资格,用于治疗接受含铂化疗后病情进展、携带MET基因第14号外显子跳跃突变的转移性非小细胞肺癌患者。
2018年3月,Tepotinib(特泊替尼)被日本卫生劳动福利部授予了治疗携带MET基因第14号外显子跳跃突变的晚期NSCLC患者的SAKIGAKE资格(创新药物)。
Tepotinib(特泊替尼)分子结构式如下:英文名称:Tepotinib中文名称:特泊替尼本文主要对Tepotinib(特泊替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Tepotinib(特泊替尼)合成路线三、Tepotinib (特泊替尼)合成检索总结报告(一)Tepotinib (特泊替尼)中间体3的合成合成方法实验步骤参考文献操作方法一2.2L of a freshly prepared 1.5M sodium methoxide solution are added dropwise with stirring to a suspension of 259g (1.09mol)of 3-methoxycarbonylbenzamidinium acetate 1and 528g (1.08mol)of ({2-dimethylamino-1-[dimethyli-mmoniomethyl]vinylamino}methylene)dimethyl-ammonium dihexafluorophosphate 2(“a minoreductone precursor”,prepared in accordance with C.B.Dousson et al.,Synthesis 2005,1817)in 1L of methanol.The reaction mixture is then warmed to 60°C.over the course of 40min and held at this temperature for 30min.The reaction mixture is then cooled to room temperature,diluted with 10L of dichloromethane and washed three times with 5L of water each time.The organic phase is dried over sodium sulfate and evaporated.The residue is recrystallised from ethyl acetate:methyl 3-[5-(dimethylaminomethyleneamino)pyrimidin-2-yl]-benzo ate 3as beige crystals;m.p.146°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1)English 操作方法二100g of 3-hydroxymethylbenzamidinium acetate 1(419.75mmol)and 204.93g of a minoreductone precursor 2(419.74mmol)are suspended in 1000ml of dried MeOH in an N 2-flushed 2L three-necked flask,and a freshly prepared solution of 28.99g of sodium in 300ml of MeOH is added dropwise with stirring,and the mixture is subsequently stirred at 60°C.for 30min,giving a clear solution.For work-up,the reaction batch is cooled,diluted with dichloromethane,washed 2×with water,dried over sodium sulfate and evaporated to dryness in a rotary evaporator.The residue 3is crystallised from a little methanol and diethyl 2010/311733;(2010);(A1)English(二)Tepotinib (特泊替尼)中间体4的合成合成方法实验步骤参考文献操作方法一160ml(2.88mol)of concentrated sulfuric acid are added to a suspension of 103.5g (364mmol)of methyl 3-[5-(di-methylaminomethyleneamino)-pyrimidin-2-yl]benzoate 3in 1.3L of water,and the mixture is heated at the boil for 4hours.The reaction mixture is cooled to room temperature,diluted with water and filtered with suction.The residue is washed with water and dried in vacuo:3-(5-hydroxypyri-midin-2-yl)benzoic acid as brownish crystals;m.p.293-295°2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1);US2011/269765;(2011);(A1);US2011/269756;(2011);(A1).操作方法二103.5g of methyl 3-[5-(dimethylaminomethylenamino)-pyrimidin-2-yl]benzoate 3(364.04mmol)are suspended in 1300ml of water in a 2l single-necked flask,and 160ml of conc.sulfuric acid (95-97%)(2.88mol)are subsequently added,and the reaction batch is warmed at 130°C.(oil-bath temperature)for 4h.For work-up,the reaction batch is cooled,and the precipitate formed is filtered off,washed with water and dried at 50°C.in a vacuum drying cabinet.Yield:78.9g (364.5mmol)of 3-(5-hydroxypyrimidin-2-yl)-benzoic acid 2010/311733;(2010);(A1)English(三)Tepotinib (特泊替尼)中间体5的合成合成方法实验步骤参考文献操作方法一32.7ml (445mmol)of thionyl chloride are added to a suspension of 88.0g (366mmol)of 3-(5-hydroxypyrimidin -2-yl)benzoic acid 4in 1.4l of methanol,and the mixture is heated at 80°C.for 2hours.20ml (276mmol)of thionyl chloride and,after 2hours,a further 10ml (138mmol)of thionyl chloride are then added.After each addition,the reaction mixture is stirred at 80°C.for 2hours.The reaction mixture is concentrated to a volume of about 300ml in vacuo.The resultant precipitate is filtered off and dried in vacuo:methyl 3-(5-hydroxypyrimidin-2-yl)benzoate 5as brownish crystals;m.p.219-223°C.US2010/280030;(2010);(A1)English;US2010/273796;(2010);(A1)English;US2011/269765;(2011);(A1)English;US2011/269756;(2011);(A1).78.8g of 3-(5-hydroxypyrimidin-2-yl)benzoic acid 4are suspended in 1.4l of absolute methanol,and 32.7ml of。
新药Cabozantinib(卡博替尼)合成检索总结报告一、Cabozantinib(卡博替尼)简介Cabozantinib(卡博替尼)是一个多靶点小分子酪氨酸激酶抑制剂。
Cabozantinib(卡博替尼)的靶点包括MET、ROS1、RET、AXL、NTRK、KIT等九大靶点。
目前,Cabozantinib(卡博替尼)已经在甲状腺髓样癌、肾癌、非小细胞肺癌、肝癌、软组织肉瘤、前列腺癌、乳腺癌、卵巢癌、肠癌等多种实体瘤中,证实了较好的治疗效果,对于骨转移的控制效果尤其突出。
Cabozantinib(卡博替尼)分子结构式如下:英文名称:Cabozantinib中文名称:卡博替尼本文主要对Cabozantinib(卡博替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Cabozantinib(卡博替尼)合成路线三、Cabozantinib(卡博替尼)合成检索总结报告(一)Cabozantinib (卡博替尼)中间体3的合成合成方法实验步骤参考文献操作方法一The intermediate 1(45.0g,0.2mol)was dissolved in chlorobenzene (450mL,10v/w),4-nitrophenol 2(70.1g,0.50mol)and N,N-diisopropylethylamine (51.7g,0.4mol)were sequentially added.After stirring at 140o C for 14h,the reaction mixture was cooled to r.t.,The precipitates were collected by filtration and washed with appropriatepetroleum ether to afford compound 3as a yellow solid in 88.2%yield.Bioorganic and Medicinal ChemistryLetters ;vol.29;nb.19;(2019);Art.No:126630.操作方法二To chloroquinoline 1(0.24g,1.1mmol)in diphenyl ether (20mL)was added 4-nitrophenol 2(0.30g,2.2mmol)and the resulting mixture was heated to 170°C.for 24h.The mixture was partitioned between ethyl acetate and 1M NaOH(aq).The organic phase was collected,washed with water and brine,dried (MgSO 4),filtered and concentrated.The residue was purified by flash column chromatography (90%ethyl acetate/hexanes-ethyl acetate)to afford 3(0.25g,69%)as a colorless solid.US2008/4273(2008);(A1)English操作方法三4-Nitrophenol 2(12.5g,89.6mmol)was added into a suspension of 4-chloro-6,7-dimethoxyquinoline 1(10.0g,44.8mmol)in PhCl (80mL).The resulting mixture was stirred at reflux for 16h.The solvent was evaporated under reduced pressure,and the residue was dissolved in CH 2Cl 2(150mL)The solution was washed by 10%NaOH aqueous solution (3×30mL),water (30mL)and dried (MgSO 4),and evaporated to obtain the title compound 3as a yellow solid (9.6g,65.7%)without further purification.Bioorganic and MedicinalChemistry ;vol.27;nb.17;(2019);p.3825–3835.操作方法四4-chloro-6,7-dimethoxyquinoline 1(671mg,3.0mmol)and 4-nitrophenol 2(500mg,3.6mmol)were placed in 7mL of chlorobenzene.Heat slowly to 140°C and continue to react at this temperature for 20h.Then the heating was stopped,cooled to room temperature,the solvent was evaporated under reduced pressure,and the residue was dissolved in dichloromethane.Then washed successively with saturated potassium carbonate solution,washed with water,dried over anhydrous sodium sulfate,and concentrated under reduced pressure,purified by silica gel column chromatography (PE /CN109988110;(2019);(A)Chinese.EA =3:1),to give apale yellow solid (3)620mg,63%yield.操作方法五To a suspension of 4-chloro-6,7-dimethoxyquinoline 1(40g,0.18mol)and 4-nitrophenol 2(26.2g,0.19mol)in toluene (60mL)was added DIPEA (27.8g,0.22mol).The reaction was heated to 115°C for 24hours and then concentrated in vacuo.The residue was washed with EtOH (40mL)to give the title compound 3as a pale yellow solid (28g,47.8%).WO2013/180949;(2013);(A1)English操作方法六A reactor was sequentially charged with 4-chloro-6,7-dimethoxy-quinoline 1(8.0kg),4-nitrophenol 2(7.0kg),4-dimethylaminopyridine (0.9kg),and 2,6-lutidine (40.0kg).The reactor contents were heated to approximately147°C.When the reaction was complete (less than 5percent starting material remaining as determined by in process HPLC analysis,approximately 20hours),the reactor contents were allowed to cool to approximately 25°C.Methanol (26.0kg)was added,followed by potassiumcarbonate (3.0kg)dissolved in water (50.0kg).The reactor contents were stirred for approximately 2hours.Theresulting solid precipitate was filtered,washed with water (67.0kg),and dried at 25°C for approximately 12hours to afford the title compound 3(4.0kg).WO2015/164869;(2015);(A1)English;EP2017/2758057(2017);(B1)English操作方法七523g of p-nitrophenol 2(3.76mol)was dissolved in 600ml (6.45mol)of N,N-dimethylacetamide,(4.3mol)of potassium t-butoxide and 800g (3.58mol)of4-chloro-6,7-dimethoxyquinoline 1and 1.5L (16.1mol)of N,N-Dimethylacetamide solution,and the reaction solution was heated to 100°C to 120°C and reacted for 2hours.The reaction solution was cooled to room temperature,poured into 3.5L ice water,stirred for 1to 2hours and then filtered.The filter cake was washed twice with 2L of water and then dried in vacuo at 35°C,To give 6,7-dimethoxy-4-(4-nitro-phenoxy)quinoline 3as a pale yellowish white powder 918.2g,the molar yield was 78.6%.CN103664778;(2017);(B)Chinese(二)Cabozantinib (卡博替尼)中间体4的合成合成方法实验步骤参考文献To 3(0.25g,0.77mmol)in 1:1MeOH/THF (50mL)was added Zn dust (0.55g,8.4mmol)and ammonium chloride操作方法一(0.085g,1.6mmol)in water(5mL).The resulting mixturewas heated to reflux for2h,then filtered through celite and concentrated.The residue was dissolved in dichloromethane,washed with water,brine,dried(MgSO4),filtered andconcentrated to provide crude4(0.25g,>100%)which wasused without further purification.US2008/4273(2008);(A1)English操作方法二500ml of methanol was added to the autoclave,and100g of intermediate3and25g of Raney nickel were added.Raisethe temperature at30°C for10hours;Press filtration,concentration,crystallization,filtration,and drying gave86gof Intermediate4in a yield of95%.CN108264482;(2018);(A)ChiNese;CN110240563;(2019);(A).操作方法三Conc hydrochloric acid(11mL,0.2v/w)and iron powder(56.7g,1.0mol)were sequentially added into90%ethanol(550mL,10v/w)under stirring for10min.Then thereaction mixture was heated to60o C,the intermediate3(55.0g,0.17mol)was added.After refluxing for1h,activatedcarbon(1.5g)was added and then reflux for30min.Themixture was filtered while hot,the filtrate was cooled toroom temperature,adjusted to pH12with10N NaOH,poured into water and stirred for2h.The precipitates werecollected by filtration and washed with water until the filtratewas nearly neutral to obtain compound4as a pale yellowsolid in90.7%yield.m.p.:196.4-197.2o C.Bioorganic andMedicinalChemistryLetters;vol.29;nb.19;(2019);Art.No:126630.操作方法四A mixture of3(9.6g,29.4mmol),Fe(8.2g,0.15mol)andAcOH(0.5mL)in90%EtOH(100mL)was refluxed withvigorous agitation for4h.The hot solution was filteredthrough celite and the filter cake was washed with hot EtOH(20mL).The combined filtrate was concentrated underreduced pressure to afford a dark brown solid,which wasrecrystallized from EtOH to afford4as yellow solid(7.2g,82.9%).Bioorganic andMedicinalChemistry;vol.27;nb.17;(2019);p.3825-3835操作方法五6,7-dimethoxy-4-(4-nitrophenyloxy)quinoline3(620mg,1.9mmol)was dissolved in ethanol(40mL).After beingdissolved by stirring,tin(II)chloride dihydrate(1.25g,4.9mmol)was added in portions.After the addition wascompleted,slowly increase to70°C for6h.After thereaction was completed,the reaction solution was cooled toroom temperature and diluted with a1N NaOH(10mL)aqueous solution.It was extracted with ethyl acetate(3×15mL),and the organic layer was combined and washedsequentially with1N aqueous NaOH,water and saturatedaqueous sodium chloride.Dried over anhydrous sodiumsulfate,filtered,and concentrated under reduced pressure toyield a yellow solid355mg,60%yield.CN109988110;(2019);(A)Chinese。
上市新药恩曲替尼(entrectinib)合成检索总结报告一、恩曲替尼(entrectinib)简介恩曲替尼(entrectinib)由基因泰克公司研发,于2019年8月在美国上市,主要用于用于治疗C-ROS癌基因(ROS1)呈阳性的转移性非小细胞肺癌(NSCLC)的成年患者,也可用于治疗12岁及以上具有神经营养性酪氨酸受体激酶(NTRK)基因融合蛋白突变的实体瘤患者。
恩曲替尼(entrectinib)的作用机制::抑制NTRK、ROS1和间变性淋巴瘤激酶(ALK)受体。
恩曲替尼(entrectinib)的不良反应:疲劳,便秘,消化不良,浮肿,头晕,腹泻,恶心,感觉异常,呼吸困难,肌痛,认知障碍,体质量增加,咳嗽,呕吐,发热,关节痛和视力障碍。
恩曲替尼(entrectinib)分子结构式如下:CAS:1108743-60-7英文名称:entrectinib中文名称:恩曲替尼二、恩曲替尼(entrectinib)合成路线三、恩曲替尼(entrectinib )合成检索总结报告(一)恩曲替尼(entrectinib )中间体2的合成序号实验步骤参考文献1With magnetic stirring,Toluene (50mL)was added to the 250mL three-necked flask of the pound 1(2.5g,15.15mmol),boronic acid (3.9g,16.65mmol)and K 3PO 4(6.4g,30.30mmol).Pd(PPh 3)4(385mg,0.33mmol)was added,vacuumed again and replaced with nitrogen three times.The temperature was raised to 110°C,and the reaction was stirred for 5hours with heat.After cooling to room temperature,the solid formed by adding ethyl acetate (100mL)was filtered.The filtrate is concentrated,The residue was passed through a silica gel column to give 2.7g of a white solidCN108623576;(2018);(A)Chinese。
药物Neratinib(来那替尼)合成检索总结报告一、Neratinib(来那替尼)简介Neratinib(来那替尼)于2017年7月在美国上市,主要用于阳性乳腺癌成人患者的延长辅助治疗。
Neratinib(来那替尼)常见的不良反应有腹泻、恶心、腹痛、乏力、呕吐、皮疹等等。
Neratinib(来那替尼)分子结构式如下:CAS:698387-09-6CAS:915942-22-2英文名称:Neratinib英文名称:Neratinib Maleate中文名称:来那替尼中文名称:来那替尼马来酸盐本文主要对Neratinib(来那替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Neratinib(来那替尼)合成路线(中间体其他合成路线请参考总结报告)三、Neratinib (来那替尼)合成检索总结报告(一)Neratinib (来那替尼)中间体3的合成合成方法实验步骤参考文献操作方法一50mmolof 3-aminoacrylonitrile 2,50mmol solid base catalyst ZrO 2-Cr 2O 3and 50mL of tetrahydrofuran was added to the reaction flask,Stir at room temperature,a mixture of 55mmol of methyl 4-ethoxy-2-chloro-5-nitrobenzoate 1and 15mL of tetrahydrofuran was added dropwise to the above reaction flask,after the reaction was stirred at room temperature for 2h,Then reflux 2h,After cooling the catalyst was filtered,The catalyst can be reused after drying.The solvent was distilled off under reduced pressure,To the residue was added 400mL of dichloromethane,Wash with 50mL of distilled water three times,The combined organic layers,After drying over anhydrous sodium sulfate,the solvent was distilled off under reduced pressure,The compound of formula (3).Yield 87%.CN106905234;(2017);(A)Chinese (二)Neratinib (来那替尼)中间体4的合成合成方法实验步骤参考文献40mmol of compound 3,40mmol anhydrous potassium carbonate and 40mL DMF was added to the reaction flask,CN106905234;操作方法一The reaction was stirred at50~60o C for 4h,cool to room temperature,Add 60mL of water,Stirring 0.5h,The precipitated solid was filtered,The crude solid was recrystallized from ethanol,Drying under reduced pressure to obtain compound of formula (4).Yield 90%.(2017);(A)Chinese(三)Neratinib (来那替尼)中间体5的合成合成方法实验步骤参考文献操作方法一A mixtureof 20g (77mmol)of 4-hydroxy-7-ethoxy-6-nitroquinoline-3-carbonitrile 4was refluxed with 120mL of POCl 3under N 2for 2hours.The mixture was cooled to room temperature and excess POCl 3was removed in vacuo.The residue was then cooled in an ice bath and 500mL of CH 2Cl 2was slowly added to dissolve the residue.The resulting solution was added to a flask containing 250mL of ice-cold saturated K 2CO 3and stirred for 30minutes.The organic layer was extracted,washed,dried over MgSO 4and evaporated to give 18g (86%)of the title compound 5as an off-white solid mp 200-202°2018/208584;(2018);(A1)English 操作方法二A mixture of 3.45g (13mmol)of 7-Ethoxy-4-hydroxy-6-nitro-quinoline-3-carbonitrile 4,5.55g (26mmol)of phosphorous pentachloride,and 10ml of phosphorous oxychloride was refluxed for 3hours.The mixture was diluted with hexane and the solid was collected.The solid was dissolved in 500ml of ethyl acetate and washed with cold diluted sodium hydroxide solution.The solution was dried over magnesium sulfate and filtered through a pad of silica gel.The solvent was removed giving 2.1g of beige solid 5.EP1950201;(2008);(A1)English;EP1117659;(2003);(B1)English;EP973746;(2003);(B1)English(四)Neratinib (来那替尼)中间体6的合成合成方法实验步骤参考文献50mmol of compound of formula (5),100mL ethanol and 100mL distilled water was added to the reaction flask,The system temperature was raised to 50o C,Followed by adding。