Direct catalytic upgrading of biomass pyrolysis vapors by a dual function Ru_TiO2 catalyst
- 格式:pdf
- 大小:478.09 KB
- 文档页数:11
生物炭的制备与应用研究作者:孙晓鹏来源:《乡村科技》2019年第02期[摘要] 近年来,生物炭得到各界的广泛认识,有关学者对于生物炭的研究越来越多。
本文简述生物炭的原料来源与制备过程,着重对其在各领域的应用做出阐述,并分析生物炭利用中存在的问题,为后续生物炭的研究与利用提供依据。
[关键词] 生物炭;制备;应用[中图分类号] TQ127.11 [文献标识码] A [文章编号] 1674-7909(2019)02-115-2生物炭(Biochar)是由生物质在完全或部分缺氧的环境下,通过高温热解产生的物化特性稳定、富含碳素的固态物。
当前,国内外对生物炭的研究成为一大热点。
全球对于生物炭的研究源于对亚马逊盆地的黑土[1]的认识,早期科学家并未对其引起重视。
随着环境问题的不断恶化,科学家在研究中发现生物炭在诸多方面的应用都有重要意义。
我国最早对生物炭的研究与应用是在纺织业和污水处理方面。
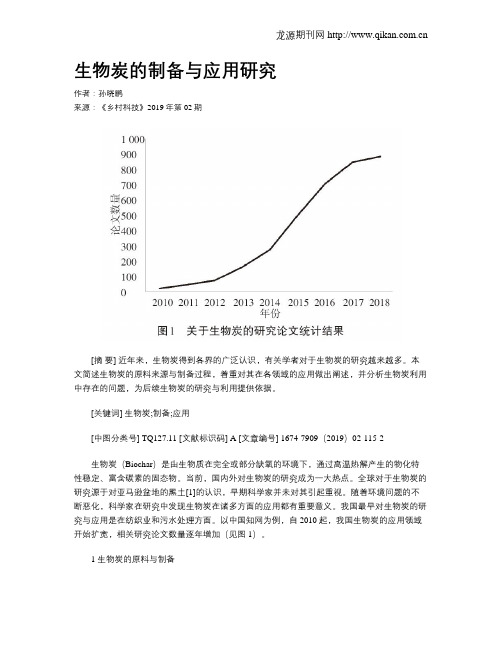
以中国知网为例,自2010起,我国生物炭的应用领域开始扩宽,相关研究论文数量逐年增加(见图1)。
1 生物炭的原料与制备生物炭的制备原料非常多,如花生壳、玉米芯、玉米秸秆、甜菜根、稻谷壳和果皮等,都可被用于制备生物炭。
生物炭制备通常使用的方法为高温裂解法和水热碳化法。
其中,高温裂解法即将生物质原料置于缺氧或氧含量极低的环境下,对其有控制地进行高温分解制备得到生物炭的方法,其又可分为慢速热解、中速热解和快速热解[2];水热碳化法是一种将生物质置于一定温度的水中,并在一定压力的条件下得到生物炭的一种制备方法[3]。
此外,由于因为材料、热解方式、温度等的不同,不同方法得到的生物炭性质存在差异。
2 生物炭的性质生物炭主要是由烷基和芳香结构组成,富含大量的碳。
不同原料制备的生物炭的钾、钙、镁等元素的含量不同,受温度、pH值等的影响,生物炭中的某些元素含量会发生变化。
在pH 值方面,生物炭通常呈碱性。
已有研究显示,在制备生物炭时,热解的温度越高,生物炭的pH值也会越高[4],同时,在原料方面,粪便类原料的碱性较高,豆科植物的碱性较高[5]。


热解温度、热解工艺、原料类型对生物炭的理化特性的影响摘要:许多研究表明热解温度、热解工艺、原料类型是影响生物炭的理化特性的重要因素。
然而目前尚没有确定在特定热解温度、热解工艺以及原材料下制备的生物炭性态特征的研究。
为此,本文旨在综合分析以上三种因素对生物炭性态特征影响,以帮助学者、生物炭生产商和相关从业人员从不同出发点选择合适的生物炭制备参数。
为特定条件下生物炭特性预测提供了理论依据和技术支撑。
研究结果对解决特定环境问题和促进作物生长的功能性生物炭产品的设计与生产提供了良好的理论支撑。
关键词:生物炭;热解温度;热解工艺Effects of pyrolysis temperature, pyrolysis process and raw material type on physicochemical properties of biocharAbstract:Various studies have established that feedstock choice, pyrolysis temperature, and pyrolysis type influence final biochar physicochemical characteristics. However, overarching analyses of pre-biochar creation choices and correlations to biochar characteristics are severely lacking. Thus, the objective of this work was to help researchers, biochar-stakeholders, and prac- titioners make more well-informed choices in terms of how these three major parameters influence the final biochar product. Results can be used to createdesigner biochars to help solve environmental issues and supply avariety of plant- available nutrients for crop growth.Key words: Biochar; Pyrolysis temperature; pyrolysis technology1.引言生物炭是在限氧条件下,在相对较低的温度(300-700°C)下通过生物质热解产生的富含碳的一种环境友好型材料[1]。

第49卷第2期2021年1月广㊀州㊀化㊀工Guangzhou Chemical IndustryVol.49No.2Jan.2021生物质制5-HMF 及其非均相催化剂-溶剂体系研究进展唐玉梅(西南科技大学环境与资源学院,四川㊀绵阳㊀621000)摘㊀要:5-羟甲基糠醛(5-HMF)是一种重要的生物质基平台分子,是制备多种精细化合物的中间体㊂近年来,有关5-HMF制备及影响因素的探究都得到了不断的扩展㊂本文简单介绍了生物质转化为5-HMF 的反应机理,描述了以各种生物质资源底物(果糖㊁葡萄糖㊁纤维素)制备5-HMF 的反应路径以及难点,阐述了制备过程中主要非均相催化剂 溶剂体系对的影响,总结了当前研究的进展㊂关键词:5-羟甲基糠醛;生物质;非均相催化剂;溶剂体系㊀中图分类号:X712㊀㊀㊀㊀文献标志码:A㊀㊀㊀文章编号:1001-9677(2021)02-0016-03㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀㊀作者简介:唐玉梅(1999-),女,本科,研究方向:固体废物㊂Research Progress on 5-Hydroxymethylfurfural Preparation from Biomassin Heterogeneous Catalyst -solvent SystemTANG Yu -mei(School of Environment and Resource,Southwest University of Science and Technology,Sichuan Mianyang 621000,China)Abstract :5-Hydroxymethylfurfural (5-HMF )is an important biomass -based platform molecule and an intermediate for the preparation of many fine compounds.In recent years,the research and influencing factors on the preparation of 5-HMF have been continuously expanded.The reaction mechanism of the conversion of biomass to 5-HMF was briefly introduced.The paths and difficulties of converting various biomass resource (fructose,glucose,cellulose)substrates to 5-HMF were described,and the impact of heterogeneous catalyst -solvent systems for 5-HMF preparation was reviewed.Finally,research progress of current research was summarized.Key words :5-Hydroxymethylfurfural;biomass;heterogeneous catalyst;solvent systems5-HMF 是以纤维素㊁葡萄糖和果糖为主要原料在酸性条件下转化所得的下游化合物,是美国能源部所提出的十二种平台化合物之一㊂其制备影响因素主要是催化剂和溶剂,非均相催化剂相较于传统的均相液体催化剂有着低腐蚀性㊁易分离㊁良好的热稳定性㊁高重复利用性等特点,在工业化连续生产等方面有较大的优点㊂溶剂作为反应的介质,影响着产物的选择性,在保证高选择率的条件下,溶剂的绿色环保性也成为一个重要的考虑因素㊂通常催化剂和溶剂作为一个体系对共同影响着反应底物的转化率和反应的选择率,因此研究非均相催化剂-溶剂体系下5-HMF 的制备具有重要意义㊂1㊀生物质转化制备5-HMF 的机理与难点制备5-HMF 的生物质原料丰富,主要有纤维素㊁葡萄糖与果糖㊂生物质可以直接或者间接转换成5-HMF,前者通过设置相应的催化剂㊁溶剂体系直接生成转化物,但存在原料结构稳定难以破坏,反应体系成本高,5-HMF 转化率和选择率低下等问题,后者通过降解㊁水解㊁异构化㊁脱水等多步逐级反应生成中间物,再进一步转换为目标产物[1],相对来说具有更高的收率和更低的成本,因此也成为5-HMF 合成的主要方法㊂图1展示了果糖㊁葡萄糖和纤维素制备5-HMF 的反应路径㊂图1㊀生物质制备5-HMF 的路径图Fig.1㊀Path diagram of 5-HMF production from biomass第49卷第2期唐玉梅:生物质制5-HMF及其非均相催化剂-溶剂体系研究进展17㊀1.1㊀果糖转化制备5-HMF果糖作为最早的5-HMF生物质制备原料,被认为是合成5-HMF产率最高的一种生物质,也被选为评估生物质转化催化体系的理想模型底物[2]㊂目前果糖合成5-HMF可以通过链状结构或者环状结构进行反应,两条路径的反应本质都是果糖脱去三分子水生成5-HMF㊂果糖工业化生产5-HMF的难点在于原料成本高㊁以及生成的5-HMF在酸性水溶液的条件下很容易进一步分解为乙酰丙酸和甲酸等副产物,同时中间产物会发生交叉聚合反应生成腐殖质,从而降低5-HMF收率[3]㊂1.2㊀葡萄糖转化制备5-HMF葡萄糖属于吡喃型结构,可以直接脱去三分子水直接生成5-HMF,但该结构稳定,烯醇化程度低,直接催化葡萄糖反应难度较大,所以普遍采用将葡萄糖通过烯醇化或者1,2氢转移步骤异构为果糖,果糖再脱水合成5-HMF[4]的方法㊂但在此反应路径中,葡萄糖选择性异构化为果糖是反应的限速步骤,也是转化的难点所在㊂此前,已有研究表明葡萄糖的异构化反应需要Lewis酸酸性位点或者碱性位点的存在,而异构生成的果糖转化为5-HMF需要Bronsted酸的催化[5],这就表明在葡萄糖合成5-HMF的不同阶段可能需要通过离子交换预处理创造不同的酸性位点[6]㊂所以,研究酸碱两性催化剂,或者设计协同使用Bronsted酸和Lewis酸催化剂的反应体系,提高葡萄糖作为生物质原料制备5-HMF的选择率和转换率,是目前能源研究领域的热点也是难点㊂1.3㊀纤维素转化制备5-HMF纤维素是由D-葡萄糖以-1,4糖苷键组成的高分子化合物,分子结构稳定且复杂㊂纤维素转化为5-HMF需要在酸性条件下水解为葡萄糖,葡萄糖异构化为果糖,果糖脱水再生成HMF㊂但分子间存在的巨大的氢键作用力使得纤维素结构稳定㊁难以被破坏[4],进而对后续5-HMF的合成造成阻力㊂使用物理㊁化学㊁生物等方式对纤维素进行预处理可以破坏纤维素复杂的网络结构㊁促进纤维素降解为低分子化合物,减少额外的能源消耗和成本投入[7]㊂但是纤维素转化合成5-HMF的反应步骤多㊁反应路径长,各个反应阶段使用的催化剂-溶剂体系也不尽相同,因此各个阶段反应生成副产物的概率会增加,对产物的选择性合成也会有较大影响㊂因此,考虑纤维素的解聚效率和开发一体化性能卓越的催化反应体系,实现纤维素到5-HMF直接转换的高效制备,是利用纤维素的重点和难点㊂2㊀非均相固体酸-溶剂催化体系在5-HMF的合成反应中,固体酸催化剂材料的孔隙结构决定了反应底物能否进入催化剂内部与活性位点接触以及反应产物5-HMF能否及时转运出以防形成腐殖质造成催化剂积碳;溶剂作为反应的介质,可以起到溶解㊁稳定㊁保护㊁催化等功能㊂因此可以通过调节固体酸材料的孔隙结构和理化性质,使其在溶剂中表现出良好的催化性能,从而提升5-HMF的产率㊂2.1㊀沸石分子筛-溶剂体系沸石固体酸是一类多孔性固体,分子筛的孔隙结构㊁比表面积大小㊁形貌特征㊁表面理化性质以及酸性位点的类型与数量都会影响沸石分子筛的催化性能[8]㊂表1总结了一些沸石分子筛在各种溶剂中获得的5-HMF效率,可以看出不同沸石分子筛-溶剂体系中HMF的获得率差异较大,可能与反应时间㊁催化剂在溶剂中表现的理化性质有关㊂Nikolla E[9]在研究Sn-Beta-HCl和Ti-Beta-HCl在相同的反应溶剂中的催化性能时,在反应时间前者仅为后者的三分之二时,含Sn高硅分子筛催化获得5-HMF的收率仍然高于Ti,因此,在此溶剂体系中含Sn的分子筛具有的孔隙结构对葡萄糖异构化表现出的活性更高㊂Jia X[10]研究功能化沸石-溶剂催化体系,以溶剂作为唯一变量,探讨相同的Ypho沸石催化下5-HMF的收率,发现在DMSO/H2O溶剂介质中得到了最高的5-HMF产率㊂其原因可能在于Ypho沸石的强酸性和疏水性在高极性溶剂DMSO中更能催化果糖与酸性位点结合,发生选择性互变异构,促进果糖的脱水反应㊂表1㊀沸石分子筛-溶剂体系制备HMFTable1㊀Preparation of HMF in zeolite molecularsieve-solvent systems催化剂原料反应溶剂原料转换率/%HMF收率/% HZSM-5[6]葡萄糖NaCl-H2O/MIBK8042 Nb-Beta[11]葡萄糖NaCl-H2O/MIBK97.482.1 SBA-15-SO3H[12]果糖DMSO10096 AlSiO-20[13]葡萄糖H2O/THF/NaCl/63.1 2.2㊀杂多酸固体酸-溶剂体系杂多酸是一种过渡金属氧化物离子簇,具有超强的Bronsted酸性㊁高质子迁移率㊁能够溶于极性溶剂,酸性和氧化性兼备使得杂多酸在均相和非均相体系中表现出多功能催化㊂其具有的Keggin结构和Dawson结构,使得杂多酸催化性能可以调节[14]㊂表3呈现出来的杂多酸固体酸可以和离子液体㊁有机溶剂的水溶液多相溶剂构成相应的反应体系,在此体系下,5-HMF制备效率也可以达到一个较理想的状态,因此杂多酸-溶剂体系,也可以作为5-HMF合成的良好反应环境㊂多相溶剂中的有机溶剂作为添加剂可以有效以抑制催化剂催化碳水化合物转化反应期间的副反应,但也会使5-HMF的生成受到抑制㊂例如Song-Bai Yu[15]在以蔗糖为原料, Cs2.3H0.7PW12O40为催化剂,探究5-HMF的收率时,在DMSO 的水溶液中,5-HMF的获得率可以达到91.8%,但在THF的水溶液中,5-HMF就降为66.1%,而在DMF溶剂中,却无法获得5-HMF㊂表2㊀杂多酸固体酸-溶剂体系制备HMFTable2㊀Preparation of HMF in heteropolyacid solidacid-solvent systems催化剂原料反应溶剂原料转换率/%HMF收率/% Cs2.5H0.5PW12O40[16]果糖H2O/MIBK78.177.6 SiO2-ATS-PTA[17]葡萄糖丙酮/H2O/78.1H3PW12O40[15]蔗糖DMSO/H2O/82 [MimAM]H2PW12O40[18]葡萄糖THF/H2O-NaCl99.853.9 2.3㊀碳基固体酸-溶剂体系碳基固体酸是一种新型的酸催化剂,其制备原料来源于生物质,改性后可用于5-HMF的生产,表现出功能多样性和生产高效性,因此迅速成为研究热点㊂表4总结了碳基固体酸在溶剂体系中5-HMF的收率,研究中发现,具有更高比表面积和孔隙体积的碳基固体酸催化剂能与溶剂进行更好的协同反18㊀广㊀州㊀化㊀工2021年1月应㊂Wang Q[19]曾研究过磺化的CS固体酸,发现在DMSO溶剂中比表面积和孔隙体积最大的C2-SO3H在150ħ下反应5min,获得了相对最高的HMF产率,而且在170ħ下C2-SO3H催化反应3min就可以获得最高87%的5-HMF收率㊂碳基固体酸-DMSO溶剂反应体系是一种很好的5-HMF合成体系㊂而且在以果糖为原料时,无论何种碳基固体酸和反应溶剂,都呈现出较高的是原料转化率和5-HMF收率㊂因此,高效利用碳基固体酸高效生成5-HMF,对于实现生物质能源化利用具有重大的意义㊂表3㊀碳基固体酸-溶剂体系制备HMFTable3㊀Preparation of5-HMF in carbon-based solidacid-solvent systems 催化剂原料反应溶剂原料转换率/%HMF收率/%Glu-TsOH[20]果糖DMSO99.991碳基固体酸CS[21]果糖DMSO10090CNT-PSSA[22]果糖DMSO/89S-TsC[23]果糖GVL/H2O/93.7 CH0.94O0.37S0.027[24]果糖DMSO-[BMIM][Cl]98843㊀结㊀语作为一种重要的生物质衍生物,5-HMF可以替代石油制备多种以石油为原料的平台化合物和新型高分子材料㊂果糖作为原料的反应体系,5-HMF的收率普遍很高,但以葡萄糖和纤维素等其他多糖聚合物作为原料合成5-HMF仍然受到较大的阻力㊂高比表面积和孔隙体积以及强酸性的固体酸普遍表现出更好的催化性能;溶剂作为催化剂和生物质反应的介质,与催化剂协同作用调控催化产物的生成方向,影响反应的选择率㊂非均相催化剂-溶剂体系下,5-HMF的收率基本可以达到一个较理想的状态,其中碳基固体酸-溶剂体系则是制备5-HMF收率非常理想的一类催化剂-溶剂体系,因此,可以预测该反应体系将会是未来研究的重要方向㊂参考文献[1]㊀Peng W H,Lee Y Y,Wu C,et al.Acid-base bi-functionalized,large-pored mesoporous silica nanoparticles for cooperative catalysis of one-pot cellulose-to-HMF conversion[J].Journal of Materials Chemistry, 2012,22(43):23181-23185.[2]㊀Wang N,Yao Y,Li W,et al.Catalytic dehydration of fructose to5-hydroxymethylfurfural over a mesoscopically assembled sulfated zirconia nanoparticle catalyst in organic solvent[J].RSC Adv.,2014,4(100): 57164-57172.[3]㊀徐杰,马继平,马红.5-羟甲基糠醛的制备及其催化氧化研究进展[J].石油化工,2012,41(11):1225-1233.[4]㊀张云雷.基于糖类生物质资源转化制备5-羟甲基糠醛的多孔催化剂设计及其催化性能与机理研究[D].镇江:江苏大学,2017. [5]㊀Lima S,Dias A S,Lin Z,et al.Isomerization of d-glucose to d-fructose over metallosilicate solid bases[J].Applied Catalysis A (General),2008,339(1):21-27.[6]㊀Moreno-Recio M,Santamaría-González J,Maireles-Torres P.Brönsted and Lewis acid ZSM-5zeolites for the catalytic dehydration of glucose into5-hydroxymethylfurfural[J].Chemical Engineering Journal,2016,303:22-30.[7]㊀Li X,Xu R,Yang J,et al.Production of5-hydroxymethylfurfural andlevulinic acid from lignocellulosic biomass and catalytic upgradation [J].Industrial Crops and Products,2019,130:184-197.[8]㊀张听伟.碳基固体酸催化生物质制取糠醛㊁5-羟甲基糠醛的研究[D].合肥:中国科学技术大学,2019.[9]㊀Nikolla E,Román-Leshkov Y,Moliner M,et al. One-PotSynthesis of5-(Hydroxymethyl)furfural from Carbohydrates using Tin-Beta Zeolite[J].ACS Catalysis,2011,1(4):408-410. [10]Jia X,Yu I K M,Tsang D C W,et al.Functionalized zeolite-solventcatalytic systems for microwave-assisted dehydration of fructose to5-hydroxymethylfurfural[J].Microporous and Mesoporous Materials, 2019,284:43-52.[11]Candu N,El Fergani M,Verziu M,et al.Efficient glucose dehydrationto HMF onto Nb-BEA catalysts[J].Catalysis Today,2019,325:109-116.[12]Wang L,Zhang L,Li H,et al.High selective production of5-hydroxymethylfurfural from fructose by sulfonic acid functionalized SBA-15catalyst[J].Composites Part B(Engineering),2019,156:88-94.[13]Li X,Xia Q,Nguyen V C,et al.High yield production of HMF fromcarbohydrates over silica–alumina composite catalysts[J].Catalysis Science&Technology,2016,6(20):7586-7596.[14]Huang Y B,Fu Y.Hydrolysis of cellulose to glucose by solid acidcatalysts[J].Green Chemistry,2013,15(5):1095-1111. [15]Yu S B,Zang H J,Yang X L,et al.Highly efficient preparation of5-hydroxymethylfurfural from sucrose using ionic liquids and heteropolyacid catalysts in dimethyl sulfoxide–water mixed solvent [J].Chinese Chemical Letters,2017,28(7):1479-1484. [16]Zhao Q,Wang L,Zhao S,et al.High selective production of5-hydroymethylfurfural from fructose by a solid heteropolyacid catalyst [J].Fuel,2011,90(6):2289-2293.[17]Huang F,Su Y,Tao Y,et al.Preparation of5-hydroxymethylfurfuralfrom glucose catalyzed by silica-supported phosphotungstic acid heterogeneous catalyst[J].Fuel,2018,226:417-422. [18]Zhao P,Zhang Y,Wang Y,et al.Conversion of glucose into5-hydroxymethylfurfural catalyzed by acid-base bifunctional heteropolyacid-based ionic hybrids[J].Green Chemistry,2018,20(7):1551-1559.[19]Wang Q,Hao J,Zhao Z.Microwave-Assisted Conversion of Fructoseto5-Hydroxymethylfurfural Using Sulfonated Porous Carbon Derived from Biomass[J].Australian Journal of Chemistry,2018,71(1):24-31.[20]Wang J,Xu W,Ren J,et al.Efficient catalytic conversion of fructoseinto hydroxymethylfurfural by a novel carbon-based solid acid[J].Green Chemistry,2011,13(10):2678-2681.[21]Zhao J,Zhou C,He C,et al.Efficient dehydration of fructose to5-hydroxymethylfurfural over sulfonated carbon sphere solid acid catalysts [J].Catalysis Today,2016,264:123-130.[22]Liu R,Chen J,Huang X,et al.Conversion of fructose into5-hydroxymethylfurfural and alkyl levulinates catalyzed by sulfonic acid-functionalized carbon materials[J].Green Chemistry,2013,15(10).[23]Huang F,Li W,Liu Q,et al.Sulfonated tobacco stem carbon asefficient catalyst for dehydration of C6carbohydrate to5-hydroxymethylfurfural inγ-valerolactone/water[J].Fuel Processing Technology,2018,181:294-303.[24]Guo F,Fang Z,Zhou T J.Conversion of fructose and glucose into5-hydroxymethylfurfural with lignin-derived carbonaceous catalyst under microwave irradiation in dimethyl sulfoxide-ionic liquid mixtures[J].Bioresour Technol,2012,112:313-318.。

CHEMICAL INDUSTRY AND ENGINEERING PROGRESS 2018年第37卷第3期·938·化 工 进展生物基糠醛催化转化制备戊二醇的研究进展樊冬娜1,刘晓然2,王喜成2,于奕峰1,陈爱兵1(1河北科技大学化学与制药工程学院,河北 石家庄050018;2中国科学院青岛生物能源与过程研究所,山东 青岛266101)摘要:生物基糠醛制备戊二醇的工艺相比于传统的石油基路线,具有原料来源广泛、生产过程绿色无污染等优点。
本文总结了国内外以生物基糠醛为原料制备戊二醇的研究现状,并对应用于糠醛催化加氢制备戊二醇的铑、铱、铂、铜基催化剂分别进行了归纳整理,同时详细论述了两种糠醛氢解路线,即糠醛加氢分别以糠醇和四氢糠醇为中间体而氢解生成戊二醇的过程。
在此基础上,提出了解决目前糠醛氢解制备戊二醇过程中存在的反应物浓度低、活性差、反应压力高等问题的建议。
对未来从经济、环保等多角度出发设计并完善生物基戊二醇的生产工艺以及拓展高效利用生物基糠醛制备下游精细化工产品的方法做出了展望。
为开发在温和条件下高效、稳定的催化生物基糠醛氢解的催化剂体系提供了参考。
关键词:生物质;催化加氢;糠醛;戊二醇中图分类号:TQ223.1 文献标志码:A 文章编号:1000–6613(2018)03–0938–09 DOI :10.16085/j.issn.1000-6613.2017-0885Catalytic conversion of biomass-derived furfural into pentanediolsF AN Dongna 1,LIU Xiaoran 2,WANG Xicheng 2,YU Yifeng 1,CHEN Aibing 1(1College of Chemical and Pharmaceutical Engineering ,Hebei University of Science and Technology ,Shijiazhuang050018,Hebei ,China ;2Qingdao Institute of Biomass Energy and Bioprocess Technology ,Chinese Academy of Sciences ,Qingdao 266101,Shandong ,China )Abstract :Compared to the traditional production of pentanediols from fossil resources ,the production of pentanediols from biomass based furfural possessed various advantages such as the abundant raw materials and the green production process. In this paper ,the status of the research on the production of pentanediols from biomass based furfural was discussed. A summary on the hydrogenation of furfural to pentanediols over rhodium ,iridium ,platinum and copper based catalysts was presented. Two kinds of reaction routes of furfural hydrogenolysis were also discussed. As a consequence ,issues realated to the process of furfural hydrogenolysis ,such as low reactant concentration ,low reaction activity and quite high reaction pressure ,were also presented. To design a process that is economical and environmental ,the production techniques of pentanediols from biomass derived furfural hydrogenation were prospected in this paper. This review can be used for a reference in developing highly active catalytic system for the furfural hydrogenolysis under mild conditions. Key words :biomass ;catalytic hydrogenation ;furfural ;pentanediol近年来,利用农副产品、农林废弃物等转化的生物基化合物来高效、经济地制备高品位燃料或高附加值精细化学品的绿色化工,逐渐成为当今科学界和工业界研究的热点,也是人们面临的巨大挑 收稿日期:2017-05-15;修改稿日期:2017-06-20。

烘焙与HZSM-5催化剂联用改善柏木热解产物品质张杨;梅艳阳;杨晴;杨海平;刘捷;陈汉平【期刊名称】《农业工程学报》【年(卷),期】2015(0)23【摘要】为研究烘焙与催化剂对生物质热解产物特性的耦合影响机制,该文选用HZSM-5催化剂,对不同温度(200、230、260、290℃)烘焙后的柏木进行热解试验。
结果发现,将烘焙与 HZSM-5催化剂联用后,随着烘焙温度的升高,催化热解后积碳量呈下降趋势,最高降低了62.6%;气体产物中CO的体积分数从53.69%降低至40.84%,H2和CO2的体积分数分别增大了43.1%和35.04%,CH4的体积分数整体变化不明显;液体产物中,酸类物质大幅减少,芳香烃类产物显著增多,酚类产物发生富集;结果表明,烘焙与 HZSM-5催化剂的联用有效地改善了快速热解产物尤其是生物油的品质。
但是也要指出,烘焙温度过高时,积碳量增多、芳香烃类产物减少,因此柏木适宜的烘焙温度应该选择在230~260℃之间。
%Fast pyrolysis of biomass is a promising technology for bio-energy because of its high liquid yield and low cost. The liquid product of fast pyrolysis is called bio-oil, which is complex mixtures of water and various organic compounds. Bio-oil has many advantages such as little sulfur and nitrogen content, highly potential values, readily stored and transported properties. However, the high acidity, low heating values and high oxygen and water contents in bio-oil limit its broaden applications. Catalytic pyrolysis and catalytic upgrading of pyrolysis vapors have been used to improve the quality of bio-oil. However, there is a crucial challenge: Rapiddeactivation of catalyst caused by serious coking. The relevant studies have shown that catalyst deactivation has a direct relationship with the high content of oxygenated compounds in pyrolysis volatile caused by high oxygen content in biomass. In addition, thermal pretreatment of biomass, torrefaction, has been shown to improve the quality of bio-oil by lowering the oxygen content and enhancing the aromatic yield. So, in this paper, the couple effects of torrefaction and catalytic pyrolysis on characteristics of pyrolytic products of cedarwood have been investigated. Pretreament of cedarwood via torrefaction was performed in a tube furnace at varying reaction temperature(200, 230, 260, 290℃) with a residence time of 30min. The torrefied cedarwood were characterized by elemental analysis and proximate analysis. The results showed that increasing torrefaction temperature caused the increase of carbon content from 48.16% to 54.3%, the oxygen content decreased from 44.33%to35.65%. And the torrefied cedarwood product has a brown/black color, reduced volatile content and increased energy density:21.25 MJ/kg (after 30 min reaction time at 290℃) versus 18.13MJ/kg for untreated cedarwood. Then the torrefied cedarwood were subsequently catalytically fast pyrolyzed over HZSM-5 in a vertical tubular reactor at 550℃ with a residence time of 30 min. The gas products of pyrolysis were analyzed by chromatograph(GC), and liquid products were analyzed by gas chromatography mass spectrometry (GC-MS). Torrefaction caused deacetylation and decomposition of hemicellulose, cleavage of ester linkages and demethoxylation of lignin. And the experimental results showthat after coupled torrefaction and catalytic pyrolysis, increasing the torrefaction temperature caused the coke yield decreased; the content of CO in gas product decreased from 53.69% to 40.84%, the content of H2 and CO2 increased by 43.1%and 35.04%respectively, and the content ofCH4 had no obvious change. As for liquid products, with the increasing of torrefaction temperature, the content of acid significantly deceased, aromatic yield increased , and enrichment of phenols. The result indicated that the couple effects between torrefaction and catalytic pyrolysis are very important for upgrading of bio-oil. However, severe torrefaction can lead the coke yield increased and aromatic yield reduced. So the optimal torrefaction condition of cedarwood is 230-260℃.【总页数】6页(P208-213)【作者】张杨;梅艳阳;杨晴;杨海平;刘捷;陈汉平【作者单位】华中科技大学煤燃烧国家重点实验室,武汉 430074; 华中科技大学中欧清洁与可再生能源学院,武汉430074;华中科技大学煤燃烧国家重点实验室,武汉 430074;华中科技大学煤燃烧国家重点实验室,武汉 430074;华中科技大学煤燃烧国家重点实验室,武汉 430074;华中科技大学煤燃烧国家重点实验室,武汉 430074; 华中科技大学中欧清洁与可再生能源学院,武汉 430074;华中科技大学煤燃烧国家重点实验室,武汉 430074【正文语种】中文【中图分类】TK16【相关文献】1.半焦基催化剂裂解煤热解产物提高油气品质 [J], 王兴栋;韩江则;陆江银;高士秋;许光文2.碱处理HZSM-5分子筛对神东煤热解产物分布的影响 [J], ZHANGNina;ZHANG Zhuangzhuang;LI Gang;XU Long;LAN Tingwei;GAO Ting;MA Xiaoxun3.碱处理HZSM-5分子筛对神东煤热解产物分布的影响 [J], 张妮娜;张壮壮;李刚;徐龙;兰婷玮;高婷;马晓迅4.低碳烃在Ga/HZSM-5型催化剂上转化为芳烃的研究Ⅱ不同金属改性的Ga/HZSM-5催化剂 [J], 朱华青;翟效珍;刘盛楹;高志贤;王建国5.低碳烃在Ga/HZSM-5型催化剂上转化为芳烃的研究ⅠGa/HZSM-5催化剂芳构化过程考察 [J], 朱华青;翟效珍;刘盛楹;高志贤;王建国因版权原因,仅展示原文概要,查看原文内容请购买。
acs catalysis 模板-回复the following question :[The Role of Catalysts in Sustainable Energy Conversion Processes]Introduction:In recent years, the global pursuit of sustainable energy solutions has gained substantial momentum. As the need for renewable energy sources becomes increasingly apparent, finding efficient and cost-effective methods for energy conversion becomes paramount. One of the key factors in achieving this goal lies in the discovery and development of catalysts. Catalysts play a vital role in sustainable energy conversion processes by accelerating chemical reactions, reducing energy consumption, and minimizing environmental impacts. This article aims to explore the significance of catalysts in sustainable energy conversion processes, emphasizing their potential applications and ongoing research developments.Catalysis in Sustainable Energy Conversion:1. Definition and significance:Catalysis is a process that enables the acceleration of chemicalreactions without being consumed in the process. In the context of sustainable energy conversion, catalysts facilitate the conversion of raw materials into useful energy resources by lowering the activation energy barrier and enhancing the overall reaction rate. By utilizing catalysts, energy conversion processes can potentially become more efficient, cost-effective, and environmentally friendly.2. Applications in hydrogen production:Hydrogen is widely regarded as a promising fuel source due to its high energy density and clean combustion properties. However, its widespread utilization is hindered by the difficulty of producing hydrogen in a sustainable manner. Catalysts play a crucial role in various hydrogen production processes such as steam reforming, water splitting, and biomass reforming. For instance, catalytic steam reforming of natural gas using transition metal catalysts, such as nickel, can effectively produce hydrogen with minimum greenhouse gas emissions.3. Role in carbon dioxide reduction:Carbon dioxide (CO2) emissions are a major contributor to climate change, necessitating the development of efficient methods for reducing and utilizing CO2. Catalysts offer potential solutions byfacilitating CO2 conversion into useful products, such as methane, methanol, or formic acid. This process, known as CO2 reduction, can effectively reduce the impact of greenhouse gas emissions while simultaneously producing valuable chemical compounds.4. Catalytic conversion of biomass:Biomass, including agricultural waste, forestry residues, and dedicated energy crops, provides a renewable source of carbon for energy conversion. Catalytic processes can convert biomass into valuable chemicals and fuels, such as bioethanol and biodiesel, through various reactions, including hydrolysis, fermentation, and esterification. Catalysts, such as zeolites or metal oxides, enable the transformation of complex biomass molecules into simpler,high-energy content compounds.5. Photoelectrocatalysis for solar energy conversion:Solar energy represents an abundant and renewable energy source, with tremendous potential for sustainable power generation. Photoelectrocatalysis integrates catalysts with photovoltaic materials to harness solar energy more efficiently. By using catalysts to facilitate the conversion of sunlight into chemical energy or to generate hydrogen via water splitting,photoelectrocatalysis offers a promising route for solar energy utilization.Current Challenges and Future Perspectives:Although catalysts play a critical role in sustainable energy conversion processes, several challenges remain. These include the development of more active and selective catalysts, understanding catalytic mechanisms at the molecular level, and scaling up catalytic processes for commercial applications. Furthermore, catalysts need to be designed to minimize the use of precious metals, reduce toxicity, and enhance overall efficiency.To address these challenges, extensive research efforts are underway, focusing on the design and synthesis of novel catalyst materials, exploration of advanced catalytic mechanisms, and optimization of catalytic reaction conditions. Integrating artificial intelligence, machine learning, and computational modeling techniques are also gaining traction in catalyst development.Conclusion:Catalysts serve as the backbone of sustainable energy conversion processes, enabling efficient and clean energy production. Throughtheir role in hydrogen production, carbon dioxide reduction, biomass conversion, and solar energy utilization, catalysts offer vast opportunities for advancing sustainable energy solutions. Ongoing research and development in catalyst design and understanding catalytic mechanisms will undoubtedly lead to more efficient and cost-effective catalysts, accelerating the transition towards a greener and more sustainable future.。
CHEMICAL INDUSTRY AND ENGINEERING PROGRESS 2010年第29卷第6期·1034·化工进展生物原油化学法精炼生物质油技术综述彭锦星,范志华,陈冠益(天津大学环境科学与工程学院,天津大学内燃机燃烧学国家重点实验室,天津 300072)摘要:生物质快速热解制取的生物原油,经过精制提质,具有柴油或汽油的特点,可用于车用燃料。
生物原油制取技术发展较快,技术较可控,但其精制提质过程复杂,需要突破的技术障碍明显。
本文对国内外的生物原油精制提质研究进展与技术发展进行了系统的综述,认为生物油水相制氢和油相制油的技术路线更具发展前景,并提出了生物原油分级利用的建议。
关键词:生物原油;生物质油;精制;改性;综述中图分类号:TK 6;TQ 35文献标识码:A 文章编号:1000–6613(2010)06–1034–07Progress in upgrading or refinery of bio-oilPENG Jinxing,F AN Zhihua, CHEN Guanyi(Faculty of Environment Science and Engineering,State Key Laboratory of Engines,Tianjin University,Tianjin 300072,China)Abstract:Through upgrading, bio-oil by biomass fast pyrolysis is promising technology for diesel or gasoline substitute. Biomass to bio-oil is considered extensively a controllable technology with a fast progress. But the upgrading or refinery of bio-oil is complex. The paper summarized recent development of the technology of bio-oil upgrading or refinery, and the research way of aqueous of bio-oil for hydrogen and the unaqueous for oil is a promising scheme,and some suggestion for using bio-oil by classification is put forward.Key words:raw bio-oil;refinery of bio-oil;refinery;modified; review生物质能是唯一可替代车用油的可再生能源,因此备受关注。
BIOMOLECULAR ENGINEERING,BIOENGINEERING,BIOCHEMICALS,BIOFUELS,AND FOODDirect Catalytic Upgrading of Biomass Pyrolysis Vapors bya Dual Function Ru/TiO 2CatalystShaolong Wan,Trung Pham,Sarah Zhang,Lance Lobban,Daniel Resasco,and Richard MallinsonCenter for Biomass Refining,School of Chemical,Biological and Materials Engineering,The University ofOklahoma,Norman,OK 73019DOI 10.1002/aic.14038Published online February 22,2013in Wiley Online Library ()The results of catalytic treatment of vapors exiting a g/min pyrolysis unit before product condensation to the liquid phase using a Ru/TiO 2catalyst for oak and switchgrass pyrolysis are reported.The pyrolysis is conducted at 500 C and the catalysis at 400 C at atmospheric pressure with a hydrogen partial pressure of 0.58atm.It is found that the cata-lytic treatment provides significant conversion of light oxygenates to larger,less oxygenated,molecules and,simultane-ously,bio-oil phenolics are also converted to less oxygenated phenolics with methoxy methyl groups transferred to the ring.The activity of the catalyst gradually diminished with increasing biomass fed to the system.Untreated pyrolysis oil forms a single liquid phase with some tarry material,consistent with the literature,whereas the treated liquid product forms separate oil and aqueous phases,the latter of which is about 80%water.The oil from the treated vapors has a lower initial viscosity with only a small increase upon accelerated aging compared to the untreated product oil,which has a dramatic increase in viscosity after aging.This is indicative of poor oil stability for untreated oil that is further confirmed by large increases in molecular weight,while the treated oil has a small increase in molecular weight after accelerated aging.In an effort to understand compatibility with refinery streams,the solubility of the oils in tetralin was examined.The untreated oil was found to have very limited solubility in tetralin,whereas the treated oil phase was com-pletely soluble except for a small aqueous phase that appeared.There are a number of challenges in developing a high yield process for pyrolysis based conversion of biomass to transportation fuels.The Ru/TiO 2catalyst used here shows promise for conducting multiple types of favorable reactions in the presence of the full spectrum of primary pyrolysis products that creates significant product stability under mild conditions.This could lead to higher liquid yields of stable,refinery compatible,product oil.VC 2013American Institute of Chemical Engineers AIChE J ,59:2275–2285,2013Keywords:pyrolysis,catalytic upgrading,Ru/TiO 2,oak,switchgrassIntroductionFast pyrolysis,where biomass is exposed to high tempera-tures (usually about 500 C)at a high heating rate in an inert atmosphere,has been cited as the lowest cost pathway to convert the polymeric cellulose,hemicellulose,and lignin into smaller,liquid molecular weight range molecules to pro-duce fuels.1–3Liquid yields of up to about 70%of the bio-mass can be achieved,with the remainder converted to bio-char and gases (primarily CO and CO 2).The elemental com-position of the liquid range pyrolysis oil is only slightly dif-ferent than that of the parent biomass.A small amount of deoxygenation occurs through formation of CO,CO 2,and water,so carbon is lost at a relatively similar rate through formation of the carbon oxides and bio-char.The liquid py-rolysis oil consists of hundreds of different molecules nearly all of which remain oxygenated.These molecules include the products of carbohydrate depolymerization and fragmen-tation which can include monomeric dehydrated sugars (i.e.,levoglucosan),various furans (C 5A C 8),and light oxygenates( C 4,including acetic acid,hydroxyacetaldehyde,acetol,and C 2A C 4ketones and aldehydes).4These light oxygenates represent as much as 15–25%of the biomass carbon,depending on pyrolysis conditions and biomass pretreatment.The pyrolytic depolymerization of the lignin fraction pro-duces a mixture of phenolics most of which are substituted with methoxy groups at the 2and/or 6positions.In addition to the C 6A C 12monomeric phenolic units,the pyrolysis oil can contain about 25%phenolic oligomers most of which are dimers,trimers,and tetramers;however,molecules as large as 5000Da can be present.5,6These lignin fragments are a large contributor to the high viscosity of condensed pyrolysis oil.The pyrolysis oil is highly corrosive,with relatively low heating value and combustion properties and with many reactive oxygen moieties,and it tends to polymerize upon storage,making it more difficult to transport and to refine.Thus,it is unsuitable to use as a refined blendstock,and it is poorly suited for a refinery feedstock.To convert biomass to fungible fuels or refinery blend-stocks,biomass oxygen must be substantially removed.At the same time,the hydrogen to carbon ratio of the biomassCorrespondence concerning this article should be addressed to R.Mallinson at mallinson@.VC 2013American Institute of Chemical Engineers AIChE Journal 2275July 2013Vol.59,No.7is also somewhat low.There is no real single chemical path-way to achieve this objective and any practical conversion will be a combination of several that involve loss of H2O and CO2in addition to other reactions that are less efficient, for example,loss of CO and loss of smaller hydrocarbons, such as light alkanes or alkenes,as well as small oxygenates (acids,aldehydes,etc.).Additionally,with the loss of water and low H/C ratio of the resultant products,hydrogen will need to be added.The major problem is how to do this while meeting the requirements of low cost and environmental impact,and also maximizing fuel(or chemicals feedstocks) production.There is not likely to be one single process that will satisfy all of these requirements,for any feedstock,in any location,but there is one overarching point that should be recognized.The primary goal of making investments in biofuel/biorefinery processes(and the full value chain that includes growing the biomass)is,ultimately,to displace fos-sil resources.Within all of the constraints that must be satisfied to be successful then,is the yield of biofuel(or bio-chemicals)that displaces petroleum.The potential of these biofuel solutions is limited to the amount of biomass that can be converted.According to the USDA-DOE Billion ton study,7this would be on the order of one billion tons per year,without displacing food crops.Thus,once some bio-mass carbon is lost from the desired product stream(liquid fuel components),more feedstock cannot be obtained arbitra-rily.Note that this is true on a plant by plant basis in that one cannot build a plant based upon the assumption that bio-mass feedstocks can be obtained from any distance;so for any plant,the maximum practical distance from which feed-stocks may be transported will govern the process size,not the output of thefinal product or maximizing the economy of scale.Thus,for a similar investment and similar acreage, the product yield obtained is a critically important variable for economics and petroleum displacement.An improvement in yield translates directly to reduction in capital and operat-ing costs per unit of production.Most processes under development for catalytically upgrading pyrolysis products generally involve two catalytic functions,acids such as zeolites and/or hydrotreating at high pressures over metal-based materials.The process configura-tions are varied and include use of catalysts within the pyro-lyzer;8,9(less commonly)use of catalysts following the pyrolyzer operating in the vapor phase;10–12and,predomi-nantly,collecting the products as a liquid(pyrolysis oil)and then hydrotreating in an essentially separate unit operating at high pressure.13,14Virtually,all configurations incorporate this last step.Acidic catalysts used for this upgrading include a wide va-riety of materials,but most common are zeolites.The num-ber of zeolites that have been tested for this application is large and the zeolites have included many additives includ-ing alkali,rare earth,and noble metals.Investigations go back at least to the1980s when the crystalline zeolites became widely available.15,16HZSM5still seems to be the most useful catalyst,17but larger pore Y and related FCC type materials are also used.With HZSM5especially,aro-matics are a significant target product,produced primarily from dehydration of carbohydrate derived precursors.One of the issues is that biomass pyrolysis conditions may not be suitable for in situ zeolite upgrading,wherein the tempera-ture of the catalysis must be the same as the pyrolysis,which may not be optimal.At these conditions of high temperature, many smaller fragments may be produced which leave the reactor mostly as light olefins(with further loss of liquid yield).These olefins might then be recovered by oligomeri-zation in a separate step after separation.8Coke formation on the zeolite is typically significant and decreases the car-bon yield in the liquid product,although the coke(and char) may be burned during regeneration to supply the heat needed for the system.The coke causes rapid catalyst deactivation necessitating large catalyst to oil ratios and almost certainly continuous regeneration in something similar to an FCC type configuration.Additionally,the char/coke combustion may require additional catalysts to convert CO and the high tem-peratures and presence of the high partial pressures of water may significantly reduce catalyst(replacement)lifetime even with ultrastable versions of the catalyst(in the case of the larger pore materials).Obtaining hydrogen from this carbon also appears to discount the loss of liquid product yield and the low yield of hydrogen per carbon compared to natural gas,thereby increasing net CO2production18and possibly capital cost if a separate reformer is required.The other major avenue for upgrading the pyrolysis oil is hydrotreating.Two proposed configurations are(1)hydro-treating after condensing the liquid bio-oil13,14and(2)by high pressure pyrolysis with hydrotreating in the pyrolysis reactor,followed by high pressure hydrotreating.19In both configurations,hydrotreating is typically multiple stages with different,but high pressure,operating conditions.The oxy-gen is removed as water,but the small oxygenates leave as light alkanes,methane,ethane,propane,and so on.The loss of this carbon greatly reduces liquid yield(lost liquid yield of15–25wt%of the original biomass,depending on feed-stock).At the same time,substantial hydrogen is carried away in the light alkane products.The hydrogen may be mostly recoverable by reforming the alkanes product gas stream,but as mentioned earlier,this loss of carbon yield in the liquid has significant impacts on the net CO2reduction (since less petroleum transportation fuel is displaced)and the reforming is likely less efficient(in terms of direct CO2pro-duction)than methane steam reforming.For thefinal use of the pyrolysis oil as a transportation fuel,compatibility with existing fuels is the goal.Cost effec-tiveness suggests that the upgrading/refining conducted on the small scale with the pyrolysis should be minimized in favor of larger scale refining at consolidated sites,such as an existing petroleum refinery.Thus,the goal of distributed upgrading would be to stabilize the oil to prevent degrada-tion of properties and also to produce a product that will be compatible with insertion points of existing refineries.While stabilization criteria are relatively well known,for example, limited change in viscosity with storage for one,compatibil-ity with petroleum refinery streams is more difficult to deter-mine.What will the refiners accept?Christensen et al.20 studied the extent of conversion of the various oxygen groups in distillate fractions by hydrotreating at various lev-els of severity,but the severity levels needed by refiners are not defined.Mercader et al.21have performed studies consid-ering these issues and offer some guidance on critical param-eters.Among the important issues are solubility in the petroleum derived stream as well as tendency to coke.As an alternative,an upgrading strategy to minimize the loss of carbon while stabilizing the oil before further treat-ment may be considered.Various configurations may be envisioned,but we describe here the conversion of the pyrol-ysis vapors before their condensation to liquid pyrolysis bio-oil.Several types of chemistry may be used depending on2276DOI10.1002/aic Published on behalf of the AIChE July2013Vol.59,No.7AIChE Journalthe target molecules,including light oxygenate condensation reactions and mild deoxygenation with transalkylation.Approximately 40–60%of the carbon in nonupgraded bio-oil is in the category of light oxygenates containing four or fewer carbons.Most of this carbon would be lost from the liquid product with the approaches discussed earlier,so retaining this carbon during upgrading can represent a major improvement in overall carbon capture in the liquid product.The pathway (Figure 1)shows the condensation pathways of the light oxygenates over oxide catalysts,consisting predom-inantly of aldol condensation and ketonization reactions at moderate temperatures.The utility of these reactions is that carbon chain number is significantly increased while oxygen,particularly reactive oxygen,is reduced and no hydrogen is consumed.These reactions have been studied extensively on model systems and make use of acid and/or base properties of zeolites and metal oxides (some of which also make use of oxygen vacancies).22–31ZrO 2suppresses ketonization and its loss of carbon compared with CeO 2and their mixed oxides,but still allows conversion of acetol and acetic acid.32The analogous conversion pathways exist for the other pyrolysis products in this category,such as acetone,acetaldehyde,furfurals,and so on.The lignin fraction can vary greatly from as low as 10%for switchgrass to 401%for some woody biomass.The resulting phenolics product fraction include methoxy groups that represent 10–15%of this carbon and is typically lost upon cracking and hydrotreating,reducing alkylaromatics and increasing benzene yield.Overall carbon yield could be increased perhaps 10%by retaining this carbon by transalky-lation of methyl groups to the ring,with substantially improved fuel quality.Additionally,the phenolics are typi-cally multifunctional,with more than one oxygen.As men-tioned earlier,these multioxygenated phenolics are a key component of the high molecular weight materials that poly-merize in the liquid phase.It has been shown 33that these oligomers from lignin pyrolysis form after condensation and do not appear to be a significant product in the pyrolysisvapor,where mainly phenolic monomers are observed,also confirming much earlier work on primary products from pyrolysis by Evans and Milne.34We have recently shown that acidic zeolite catalysts can be used to retain methoxy carbon by transalkylation of the methoxy methyl group to the aromatic ring,reducing methane production.35–37Addi-tionally,under atmospheric pressure hydrogen,the incorpora-tion of a metal in the zeolite catalyzes deoxygenation without hydrogenation of the aromatic ring.Further work has shown that guaiacol,due to its two oxygen groups,is much more deactivating than anisole.Nevertheless,good ac-tivity for converting guaiacol for both transalkylation and deoxygenation has been found for metal/metal oxide dual function catalysts,with Ru/TiO 2showing the best results thus far,although the deoxygenation function is less stable,38as shown in Figure 2.With increasing time on stream,guaia-col conversion remains high,but the ability of this catalyst to promote complete deoxygenation decreases,while con-tinuing to remove one oxygen atom.We have found condi-tions in which stable conversion to mostly cresols,for example,retains the methoxy carbon and the products are much less deactivating.These single oxygen aromatics are ultimately more easily fully deoxygenated under mild hydro-treating conditions,without forming cyclohexanes.In a simi-lar fashion,we have examined the ketonization of acetic acid over a Ru/TiO 2catalyst,although under different condi-tions,and found it to be highly effective 39for this reaction,with further studies on the reactions of light oxygenates ongoing.Efforts to treat the pyrolysis vapors with metal oxide and metal/metal oxide catalysts have been made by Lu et al.12and showed the potential for upgrading the vapors coming from the pyrolysis unit.Thus far,our studies have been applied to model com-pounds and to simple mixtures to develop understanding of the chemical pathways.In this article,we report results of catalytic treatment of vapors exiting a g/min pyrolysis unit before product condensation to the liquid phase using a Ru/TiO 2dual functioncatalyst.Figure 1.Condensation reaction pathways for bio-oil light oxygenates based on model compound studies over ametal oxide catalyst.AIChE JournalJuly 2013Vol.59,No.7Published on behalf of the AIChEDOI 10.1002/aic2277Materials and Methods Biomass and catalystThe feedstocks used in this work were switchgrass and red oak sawdust,both ground to a size of 0.5–1mm.The ground samples were then dried in vacuum (0.02MPa)at 60 C overnight and then cooled.The switchgrass was an Alamo variety supplied by the Samuel Roberts Noble Foundation with a lignin,hemicellulose,cellulose,and ash wt %of 9.32,36.56,38.65,and 4.87,respectively,based on NIR correlated data.The oak was sourced locally,with an estimated ash content of 2%which was determined by the residue after ox-idation at 800o C from TGA experiment.Typical red oak heartwood compositions can be found in the literature with lignin,hemicellulose,and cellulose wt %of 21.3,46.9,and 27.2,respectively.40The dry switchgrass had an elemental composition of 46.0C%,6.0H%,43.1O%(O by differ-ence)and oak sawdust of 43.0C%,5.8H%,49.2O%(O by difference).A 5%Ru/TiO 2catalyst was prepared using the metal ox-ide,that is,titanium (IV)oxide (Alfa Aesar,Catalyst sup-port,Anatase 1/800pellet)with a pore volume of 0.38cc/g.The oxide was reduced in particle size to smaller than 500m m,dried overnight (12h),and cooled to room temperature before the metal impregnation step.The metal was added by incipient wetness impregnation with an aqueous solution of ruthenium (III)chloride hydrate (Aldrich,99.98%trace met-als basis).The ruthenium precursor salt was first dissolved in deionized water in the appropriate amount to obtain 5wt %of ruthenium metal of the total mass of the catalyst.The pre-pared precursor solution was added drop-by-drop on the tita-nia with mixing using a glass mortar and pestle.After the impregnation,the catalyst was dried at 120o C for 12h,and oxidized in flowing air at 400o C for 4h starting with a heat-ing rate of 10C/min,and finally cooled to room temperature.The catalyst was pelletized and subsequently crushed and sieved to particles with a size ranging from 450to 850m mto minimize the pressure drop when packed into the catalyst bed coupled with the pyrolysis reactor during operation.The detailed synthesis and characterization of the catalyst has been described in model compound studies of Boonyasuwat et al.38and Pham et al.39The catalyst was then reduced in situ as described below.Bench-scale fast pyrolysis reactor with directly coupled catalyst bedA continuous bench-scale fast pyrolysis reactor with a flexi-ble capacity of 40–200g/h biomass was designed,fabricated,and used in this work.This system can allow the addition of selective catalytic stages to upgrade the hot pyrolysis vapor in situ .This system is schematically shown in Figure 3.The pyrolysis reactor was made of a stainless steel tube with an ID of 0.930in.and length of 12in.The pyrolysis re-actor was externally heated with a one-zone electric furnace,where the actual pyrolysis temperature was monitored by a Type K thermocouple inserted in the center of the reactor tube.The pyrolysis temperature used for these experiments was 500 C.The twin-screw feeder model KT-20(from KTRON America Pitman,NJ)was controlled by a KTRON digidrive.The dried biomass,preloaded in the feeder hopper,was conveyed by the twin-screw auger into the pyrolysis reactor hot zone through a 1/200transport tube.A carrier gas,0.5SLPM He,was introduced in the upper part of the transport tube to facilitate the particles’transfer.During the pyrolysis experiment,a flow of H 2preheated to 500 C was fed from the bottom of the pyrolysis reactor tube at a rate of 0.7SLPM,which together with the top-fed carrier gas provides an overall gas residence time inside the pyrolysis reactor of 2s.The reactor pressure was just above atmos-pheric,with a hydrogen partial pressure of about 0.58atm.The exiting pyrolysis gas–vapor mixture first passed through the cyclone,and then instantly entered the direct-coupled fixed bed catalytic reactor.This was made of a stainlesssteelFigure 2.Strategy for upgrading phenolics from bio-oil.The pathway proposed here is based on phenolic model compound studies using guaiacol over the bifunctional 5%Ru/TiO 2catalyst.2278DOI 10.1002/aicPublished on behalf of the AIChEJuly 2013Vol.59,No.7AIChE Journaltube with an I.D of 0.930in.and a length of 7in.The cata-lytic bed was heated and maintained at 400 C by external heating tapes,where 4–6g of 5%Ru/TiO 2was packed in the center and sandwiched by two layers of ceramic wool at both ends.Silica beads were used as a bed for noncatalytic “blank”experiments.The catalyst was reduced in situ at 400 C for 2h before biomass feed was initiated.After the catalyst bed,the upgraded vapor mixture was then condensed by a train of two condensing traps cooled by ice water (trap 1)and liquid nitrogen (trap 2),respectively,where liquid products were accumulated for further analysis.This arrangement allowed online measurement of the gas composition after trap 1due to condensation of CO 2in trap 2during the experiment.An electrostatic precipitator was initially used,but it col-lected essentially nothing after trap 2and was ultimately removed.A wet test meter was used to measure the flow rate for the postcondensation gas mixture,before its release to the vent.It should be noted that in this system the activity of the catalyst cannot be monitored continuously in situ to deter-mine the deactivation at a certain point of operating time (time on stream).The approach used here to assess this is operation as a semibatch process,that is,the biomass is con-tinuously fed into the pyrolysis reactor,while the produced vapor is condensed and accumulated in the two condensers.After operation feeding 20–30g of biomass,feed was sus-pended and the traps replaced,upon which biomass feed operation was resumed.During this interval,the gas flows and system temperatures were maintained.The designation of each catalytic run number,for example,1st,2nd,and so on refers to the accumulated products from feeding about 20–30g of biomass.The typical biomass feed rate used in this work is about 40g/h.The catalyst amount used in this work for oak and switchgrass pyrolysis was 4and 6g,respectively,thus corresponding to a catalyst/feed ratio of about 0.1and 0.15h 21,somewhat analogous to a cat/oilratio.This mode of operation then shows,for these “run”increments,the continuing extent of catalytic activity as additional biomass is fed to the system for successive “runs.”These increment sizes allowed sufficient collection of liquid product to complete material balance,composition,and prop-erty analyses.The reactor operation in this work is entrained flow,wherein the particles are carried into the pyrolysis reactor by the transport gas as well as gravity and mix with the main carrier moving upwards from the bottom.The feed rate and the flow rates were chosen so that the char formed was not blown out of the reactor during pyrolysis operation.Under this set of conditions,the cyclone did not accumulate solids during pyrolysis operation.Gas composition analysisDuring the pyrolysis operation,the noncondensable gaseswere quantitatively analyzed by a CARLE VR Series 400AGC equipped with a thermal conductivity detector that was cali-brated with gas mixture standards.The gas was sampled af-ter trap 1about every 15min via a 30mL syringe and injected immediately into the GC.GC-FID,GCMSGC/MS analysis was performed on a Shimadzu GCMS-QP2010S for qualitatively identifying different species in liq-uid bio-oil samples using the NIST database.The column used was a DB-1701(60m,0.25mm ID,0.25m m)with a program set at 45o C for 4min and ramped to 280o C at 3o C/min and held for 20min.The injector temperature was kept at 250o C with the helium flow rate maintained at 1mL/min.41Bio-oil samples for this analysis were prepared as 10%solutions in methanol prior to injection.Combined with the GC-MS analysis,an HP 6890A GC equipped with a flame ionization detector (FID)was used to determine the quantitative compositions of the liquid samples.The same GC column and the same GC conditions as described in the GC-MS analysis earlier were used so that compounds identified by GC-MS would elute at the same retention time and order from the GC-FID as well.Addition-ally,known compounds were used to validate the MS identifi-cations and retention times for most of the quantified components.About 0.1m l of neat liquid product sample was injected for these analyses.When there were separate aqueous and organic phases,they were analyzed separately and then summed together to get overall compositions after multiplying by their respective mass fractions (wt %of per biomass feed).ViscosityViscosity was measured by Brookfield DV-G 1Pro pro-grammable viscometer.A water bath was connected to the viscometer sample cup,with the temperature set at 40 C.Approximately 0.5ml of sample was placed at the center of the sample cup for each measurement.Each measurement was repeated for each sample and calibration checks were made.Multiple shear rates were measured,however only those at 120s 21are reported.Gel permeation chromatography (GPC)Molecular weights of bio-oil samples were obtained by a Waters GPC system using a Waters styragel HR column with THF as solvent and a flow rate of 1ml/m using a Waters 515pumping system with a 717autosampler.AFigure 3.Reactor diagram of bench-scale fast pyroly-sis system.The catalyst bed follows the pyrolysis reactor,directly coupled after the cyclone.The ESP was removed as no product was found to be collected after the train of ice water and liquid nitrogen traps.T1and T2refer to the thermocouples that monitor the inside reactor and cata-lyst bed temperatures,respectively.AIChE JournalJuly 2013Vol.59,No.7Published on behalf of the AIChEDOI 10.1002/aic2279CH-30column heater maintained a30 C column tempera-ture.Samples were prepared with about a1wt%sample in HPLC grade THF and placed in the autosampler tray.For each measurement,the injection volume was10m l.Each sample was analyzed twice.Detection was by a Waters486 tunable absorbance detector with the wavelength set at254 nm.The UV response was positive over the retention time range for the model compound standards,including the light oxygenates.However,no response calibration was attempted. The GPC column was calibrated using a series of polysty-rene standard chemicals with a molecular weight range from 400to40,000and another seven smaller compounds includ-ing guaiacol,naphthalene,toluene,furfural,acetol,acetone, and acetic acid were added to extend the calibration’s lower end,because a major portion of the biomass liquid products have a molecular weight less than400Da.Water content and elemental analysisWater content analysis by weight percent of each sample was carried out by TitroLine Karl-Fischer analyzer(Titro-Line KF,SCHOTT Instruments)following the standard instrument procedure.Elemental analysis(for Carbon and Hydrogen,with oxygen content by difference)was per-formed on switchgrass and oak samples,pyrolysis bio-oils and chars,using a CE-440Elemental analyzer,following their procedures.ResultsCatalytic and noncatalytic pyrolysis of oak sawdustAs mentioned in the experimental section,the liquid prod-ucts were separated and collected by two traps in series, cooled by ice water(Trap1)followed by liquid nitrogen (Trap2).For the noncatalytic pyrolysis runs,liquid phases in both of the two traps appear as homogeneousfluids except that there was always some heavy part that would not pour or pipette,usually termed as“pyrolytic lignin”in the litera-ture,deposited at the bottom in thefirst trap.This material is completely soluble in THF.After catalytic treatment,the most apparent change is that there is phase separation in both of the traps.The aqueous phase formed in thefirst trap, which appears to be pale and clear is mainly water and forms the top layer in Trap1.The oil phase stays at the bot-tom,which appears much less viscous than the heavy depos-its in thefirst trap from the blank run.The liquid product collected in the second trap also separates into two phases; however,the oil phase is lighter and sits above the lower pale translucent aqueous phase.Table1summarizes the results comparing the blank and the“first”catalytic run using4g of5%Ru/TiO2.Previous tests showed no effect of the carrier gas composition on the blank pyrolysis run.In one typical blank run,the products yields,on a%of biomass basis,are7.0%char,24.3%gas, and60.2%total liquid.For the catalytic run,a larger amount of gaseous products,about33%,are produced at the expense of less total liquid yield,49.9%,which includes an overalloil phase(including those in both Trap1and Trap2) accounting for25.3wt%and an aqueous phase of24.6wt %.The combined analyses of the aqueous phases(from both traps)contains mostly water,with a content as high as80%, while the combined oil phases contains mainly oxygenated organic compounds with only9%water.The total water pro-duced,summed for all phases,is increased from13.2%in Table1.ProductPhaseMassBalancesofOakPyrolysisat5CwithandwithoutCatalyticVapor-upgradingonawt%ofBiomassfedBasisOakProductNoncatalytic,Oak4g5%Ru/TiO21stRun,Oakwt%C%O%H%H2O%H/C(dry)*O/C(dry)wt%C%O%H%H2O%H/C(dry)O/C(dry)Organicphase6.2(1.5)†57.8(1.5)6.9(1.9)72.4(4.2)13.2(1.3)1.6(.11).59(.4)25.3(1.6)36.9(1.7)13.6(1.1)31.5(2.8)2.3(.3)1.21(.1).21(.3)Aqueousphase24.6(1.8)4.5(.6)41.3(2.7)4.7(3.9)19.6(2.)1.92(.15).93(.5)Char7.13.1.93.17.13.1.93.1Gas24.3(1.4)23.1(1.3)28.2(1.6)1.(.6)33.(2.8)3.2(2.5)36.7(3.4)13.(1.)Total91.5(1.4)93.9(1.6)91.(2.3)85.5(4.5)13.2(1.3)89.9(1.5)84.6(2.)93.5(2.5)88.3(4.7)21.9(1.4)*“Dry”referstothebasiswithsubtractionofthewatercontentinthesample.†Dataarereportedastheaverageof2repeatrunsandthenumbersinparenthesesarethestandarddeviations.2280DOI10.1002/aic Published on behalf of the AIChE July2013Vol.59,No.7AIChE Journal。