地中海贫血(全)
- 格式:ppt
- 大小:1.08 MB
- 文档页数:58


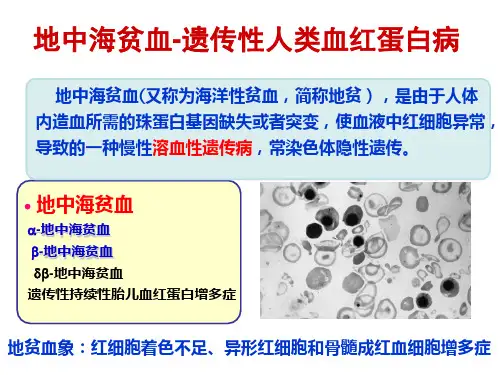
地中海贫血地中海贫血亦称为珠蛋白生成障碍性贫血或海洋性贫血(thalassemia),是一组遗传性溶血性贫血。
因珠蛋门基因缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少或不能合成,导致血红蛋白的组成成分发生改变,临床上主要表现为慢性溶血性贫血。
国外以地中海沿岸及东南亚各国多见,中国主要分布在长江以南各省,广东、广西、海南及四川等省(自治区)发病率较高,湖南、湖北及江两等省发病率稍低,北方各省少见。
胚胎期后组成珠蛋白的肽链有4种:α、β、γ、δ链,每种肽链各由相应的基因编码、。
因珠蛋白基缺失或点突变不同,肽链合成障碍各异,根据异常肽链类型将地中海贫血分为α、β、δβ、γ等类型,其中β和α地中海贫血较为常见。
β一地中海贫血:β一地中海贫血(简称β一地贫)是由于调控β珠蛋白的基因缺陷,导致β珠蛋白合成障碍的溶血性贫血。
1、发病遗传学人类β珠蛋白基因位于1 l号染色体短臂上(1 1p15.5),发生β一地贫的主要原因是基因的点突变,少数为基因缺失。
如果基因缺失或点突变导致β链的生成完全受到抑制,称为βo地贫;如果点突变仅使部分β链生成减少,则称为β+地贫。
β地贫基因变化复杂,已发现的点突变有200多种.1979年,中国发现第1个β一地贫基因点突变,迄今已累计发现相关基因点突变28种,最为常见的突变有6种:①β4l 42(‘TCTT),约占45%;②IVS一Ⅱ一654(c—T),约占24%;③1317(A—T),约占14%;④13—28 (A—T),约占9%;⑤[β7l一72(+A),约占2%;⑥[β26(G—A),约占2%。
根据酽或β+地贫基因的组合不同,临床表现为3型β一地贫:①重型:为βo基因的纯合子(βo/βo)、部分β+基因的纯合子(β+/β+)及部分βo和β+基因的双重杂合子(βo/β+);②中型:少数βo/βo,部分β+/β+,以及不典型β地贫杂合子、重型β地贫合并α或δβ地贫及某些变异型β地贫的纯合子等;③轻型:β+、βo、δβ地贫的杂合因子。

地中海贫血地中海贫血是由一组遗传性溶血性贫血。
全世界每年约有10万例重型地中海贫血的患儿诞生。
地中海贫血最常发生在意大利、希腊、中东、南亚及非洲地区。
1.各类地中海贫血有何不同?地中海贫血包括几种不同类型的贫血(红细胞缺乏)。
其中主要类型有两种:α地中海贫血和β地中海贫血,其分类依据是红细胞中携氧蛋白(即血红蛋白)的哪部分缺乏。
最重型的α地中海贫血,主要分布在南亚、中国、菲律宾地区,会导致胎儿或新生儿死亡。
而其他大多数α地中海贫血患儿的病情较轻,只有不同程度的贫血。
而β地中海贫血的表现,从特别严峻型到对健康无影响的不同程度。
重型地中海,被称为Cooley贫血,以1925年第一次发觉该症的医生名字命名的。
中间型地中海贫血,是一种轻度的Cooley贫血。
轻型地中海贫血,除血红蛋白特别外可能不会引起任何症状。
2.地中海贫血如何影响儿童?大多地中海贫血儿童诞生时看来是健康的,但生后一两年中开头变得苍白、倦怠、烦躁与食欲不振。
他们生长缓慢,还常消失皮肤黄疸。
假如不治疗,患儿肝、脾、心脏进行性增大。
骨骼变得细、脆,面部骨骼变形,地中海贫血的患儿看起来长得都很相像。
心衰和感染是未经治疗的地中海贫血患儿的主要死因。
3.有什么治疗方法?定期输血和使用抗生素能改善重型地中海贫血患儿的外观。
虽然中度地中海贫血患儿在并发症开头消失时会被要求输血,但一般是不要求输血的。
重型地中海贫血患儿定期输血(一般每3~4周一次)的目的在于维持其血红蛋白接近正常水平,以防止并发症消失。
这一治疗方式一般称为“超高量输血”,以促进患儿的生长发育与健康,同时还可预防心衰和骨骼变形。
不幸的是,反复输血会导致体内铁的蓄积,后者会损难过、肝及其他器官。
一种叫铁鳌合剂的药物被推举用以清除体内过量的铁,从而预防或延迟因超负荷铁所带来的问题。
这一药物通常会在患儿每天睡着的时候由一个便携式泵注入皮下。
经定期输血和铁鳌合剂治疗的重型地中海贫血患儿可存活20~30年,甚至更长。




是由于红细胞中⾎红蛋⽩的合成发⽣了障碍⽽致的溶⾎性贫⾎,该病在⼴东的发⽣率为5~6%。
地中海贫⾎主要分为两⼤类,由⾎红蛋⽩的α链合成减少⽽引起的贫⾎称为甲型地中海贫⾎,由⾎红蛋⽩β链合成减少⽽引起的贫⾎称为⼄型地中海贫⾎。
根据病情的轻重不同各⾃⼜分为轻型和重型。
重型甲型地中海贫⾎患者⼀般为死胎或出⽣后即死亡,死胎全⾝⽔肿、肚⼤、胎盘特别⼤,故⼜称为“⽔肿胎⼉综合征”。
⽽重型⼄型地中海贫⾎患者在出⽣时往往看不出异常,数⽉后则慢慢出现⾯⾊苍⽩,痴呆型⾯容,发育不良,并且贫⾎不断加重,常需间断的输⾎来维持⽣命。
就⽬前的医疗⽔平,即使竭尽全⼒治疗,病孩也难以活到成年,这给家庭和社会都造成了极其沉重的经济和精神负担。
与重型地中海贫⾎截然不同的是,轻型患者病情很轻,常没有症状或只有轻度贫⾎,他们的体⼒、智⼒、寿命均不受影响。
如果两个患同类的轻型地中海贫⾎的青年结婚,则有1/4的机会⽣出⼀个重型的患⼉,这就是为何⼴东这样的地中海贫⾎⾼发区不断有重型患⼉出⽣的原因。
⽬前对地中海贫⾎最有效的办法是进⾏产前诊断,以防⽌重症患⼉的出⽣。
[治法]:补脾肾,化瘀,利湿,理⽓
熟地20、黄芪30、党参18、⽩术15、云苓18、补⾻脂15、枸杞⼦15、⽞参18、⼥贞⼦15、⽩芍10、黄柏12、茵陈15、⿅⾓胶15(烊化)、⽩茅根20、旱莲草15
[加减]:
(1)伴有黄疽者,加⽥基黄15泽泻12
(2)腹内有结块者,加鸡⾎藤30莪术12鳖甲15(先煎)
(3)⼼阳虚弱者,加龙眼⾁15远志10。


地中海贫血基因诊断全套
(SEA型、3.7型、4.2型、β地中海贫血)项目简介
地中海贫血是全球广为流行的遗传性溶血性疾病,属于常染色体隐性遗传的人类血红蛋白病,全世界至少有3.45亿人携带地中海贫血的致病基因。
全球地中海贫血基因携带者频率高达2.62%,包括中国南方在内的东南亚地区,印度次大陆、地中海地区、中东、北非和太平洋地区都是该病的高发地区,尤其我国广西的携带率高达24%、广东高达11%。
由于携带者没有症状,然而婚配的下一代却有1/4的机会罹患严重溶血性贫血症状的重症地中海贫血,因而构成了严重的公共健康问题。
目前,对此类遗传病尚无有效的治疗方法,但通过产前诊断可避免重症地中海贫血患儿的出生,这是目前国际上公认的首选预防措施。
临床意义:
地中海贫血基因突变位点众多,本检测针对其中常见的3种α地贫基因缺失及17种常见的β地贫基因点突变进行检测,可覆盖95%的地贫基因突变。
1、用于婚前、产前筛查,指导产前诊断,避免重症胎儿的出生,是优生的重要措施
2、用于临床患者检测,进行α地中海贫血及β地中海贫血的诊断
适检情况
1、婚检、产前筛查:婚龄人群、孕妇及其丈夫
2、临床患者:血常规MCH、MCV偏低、有地贫临床表现
标本采集要求
1、填写地贫基因检测的专用申请单。
2、EDTA抗凝管取全血或新生儿脐带血2ml,2-8℃保存,当天送检
临床项目收费标准。

地中海贫血百科名片疾病分类病因及发病机制1临床表现β地中海贫血1α地中海贫血1诊断及鉴别诊断诊断1鉴别诊断1疾病治疗一般治疗1输血和去铁治疗1铁鳌合剂疾病预防展开疾病分类地中海贫血按照受累的氨基酸链来分类,组成珠蛋白的肽链有4种,即α、β、γ、δ链,分别由其相应的基因编码,这些基因的缺失或点突变可造成各种肽链的合成障碍,致使血红蛋白的组分改变。
通常将地中海贫血分为α、β、δβ和δ等4种类型,其中以β和α地中海贫血较为常见。
也可按照一个或两个基因缺损来分为轻型或重型地中海贫血。
病因及发病机制本病是由于珠蛋白基因的缺失或点突变所致。
1.β地中海贫血人类β珠蛋白基因簇位于11p15.5。
β地中海贫血(简称β地贫)的发生主要是由于基因的点突变,少数为基因缺失。
基因缺失和有些点突变可致β链的生成完全受抑制,称为β0地贫;有些点突变使β链的生成部分受抑制,则称为β+地贫。
β地贫基因突变较多,迄今已发现的突变点达100多种,国内已发现28种。
其中常见的突变有6种:①β41-42(-TCTT ),约占45%;② IVS-Ⅱ654 ( C → T ),约占24%;③β17 ( A →T );约占14%;④TATA盒- 28 ( A →T ),约占9%;⑤β71-72(+A ), 约占2%;⑥β26( G → A ),即HbE26,约占2%。
重型β地贫是β0或β+地贫的纯合子或β0 与β+地贫双重杂合子,因β链生成完全或几乎完全受到抑制,以致含有β链的HbA 合成减少或消失,而多余的α链则与γ链结合而成为HbF( a2 γ2),使HbF明显增加。
由于HbF的氧亲合力高,致患者组织缺氧。
过剩的α链沉积于幼红细胞和红细胞中,形成α链包涵体附着于红细胞膜上而使其变僵硬,在骨髓内大多被破坏而导致“无效造血”。
部分含有包涵体的红细胞虽能成熟并被释放至外周血,但当它们通过微循环时就容易被破坏;这种包涵体还影响红细胞膜的通透性,从而导致红细胞的寿命缩短。
地中海贫血地中海贫血又称海洋性贫血,是一组遗传性疾病,是人群中最常见的不完全显性的慢性溶血性贫血病。
其发病机制是合成血红蛋白的珠蛋白链减少或缺失导致血红蛋白结构异常,这种含有异常血红蛋白的红细胞变形性降低,寿命缩短,可以提前被人体的肝脾等破坏,导致贫血甚至发育等异常,这种疾病也就是医学上讲的溶血性贫血。
地中海贫血症属于一种可防难治的遗传性疾病,如果能在婚前就清楚了解自己的遗传背景,并且在产前做好地贫筛查和诊断,就可以有效把下一代患重型地贫的机会减至最低。
轻型地贫携带者同正常人婚配,其后代有 50%的机会成为轻型地贫携带者。
静止型地贫与轻型地贫婚配,有 1 /4机会生出地中海贫血患儿。
如果如果夫妻为同型地贫基因携带者,每次怀孕,胎儿有1/4的机会为正常,1/2的机会为基因携带者,另1/4的机会为重型地中海型贫血患者,而如果夫妻双方携带的是不同型的地贫基因,或者只有一方携带地贫基因,所生的孩子不会得地中海贫血。
地中海贫血患者在刚出生的时候也没有什么很明显的症状,表现的跟大部分新生儿一样正常,根本不能发现轻度地中海贫血症状。
但过了婴儿期后就会出现贫血、浑身疲乏无力、浑身水肿、肝和脾肿大以及出现轻度的黄疸。
随着年龄的增长,会出现眼睛距离变宽、鼻梁变扁等面容方面的改变,还会出现呼吸道感染,在服用一些药物会出现急性溶血加重贫血的症状,甚至导致溶血危象,有生命危险。
重度地中海贫血的患儿可能会出现死胎的现象,或者在出生后马上死亡。
也有一些患者是由中度地中海贫血导致成重度的,只是它的症状比中度的更加严重,直至导致死亡,一般都不可能活到成年。
地中海贫血基因是引起地中海贫血的直接原因,地中海贫血是一组遗传性肽链合成障碍导致的血红蛋白异常性疾病。
地中海贫血病人父母往往是轻型地贫病人,也可以说病理基因携带者,父母各自有一个病理基因,如果父母各自的病理基因同时遗传给子女,即子女有两个病理基因。
在中国,a-地贫基因携带率为2.64%。
地中海贫血的诊断标准地中海贫血(MediterraneanAnemia)是一种常染色体隐性遗传性血液疾病,其主要特征是造成血红蛋白水平低。
地中海贫血是一种罕见的遗传病,发病率约为1:500,约占全球血缺乏症的2%,在中国大陆的发病率尚不明确。
地中海贫血是由于某些染色体突变所引起的,多表现在欧洲和地中海地区的人群中。
诊断地中海贫血的标准可由血液检查和遗传学检查确定。
血液检查是确定地中海贫血的基本标准,包括血常规检查,血细胞检查及血液生化检查等内容。
血常规检查包括白细胞计数,红细胞计数,血红蛋白含量及血小板计数等参数检测,血小板计数过低是提示地中海贫血的明显标志。
另外血液细胞检查可发现血小板大小及形状的异常,如细胞核大小和细胞核形状发生变化,血小板增生,或者出现多核细胞等。
另外,遗传学检查也是确定地中海贫血的重要手段。
遗传学检查包括家族史收集、非编码RNA检查、血清学检查及DNA检查等,其中家族史收集是遗传学检查的基础,可从与患者相关的家庭成员中发现有相同病史的亲属,从而确定家族史。
非编码RNA检查可确定染色体的突变是否和患者的症状有关,血清学检查可观察血液中抗体的存在,有助于确定患者是否发生过免疫反应,DNA检查是确定地中海贫血的最重要检查手段,它可以为地中海贫血的病因研究奠定基础。
通过遗传学检查及血液学检查的结果,可以有效确定地中海贫血的诊断标准。
如果患者的血液检查结果显示血小板计数过低,并且在遗传学检查中发现有染色体突变,那么就能够确定患者患有地中海贫血。
地中海贫血是一种常染色体隐性遗传性血液疾病,必须对患者进行全面的检查,才能够确诊患病。
地中海贫血是一种罕见的遗传性血液疾病,在欧洲和地中海地区的人群中更加普遍,常以血小板计数过低的血液检查及染色体突变的遗传学检查结果来确诊。
如果患者血液检查及遗传学检查结果均满足地中海贫血的诊断标准,那么就能够确诊患有地中海贫血。
地中海贫血一经发现,需要及早进行治疗,以改善患者的生活质量。