乙醇残留量方法学研究(测定结果)
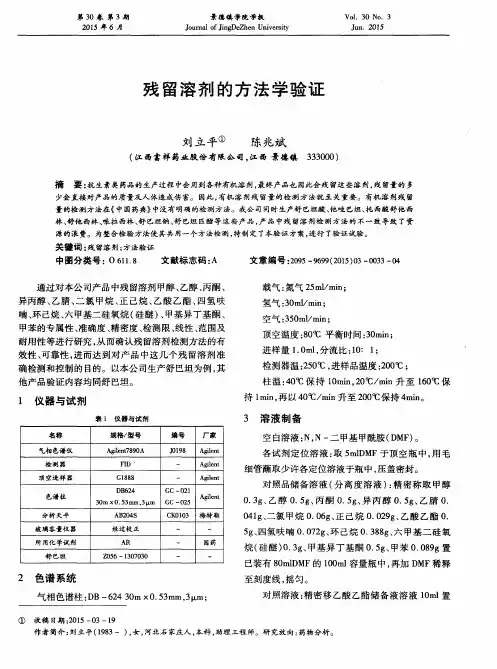
- 格式:doc
- 大小:2.52 MB
- 文档页数:13

GC法测曲尼司特中乙醇残留溶剂量摘要目的:建立测定曲尼司特中乙醇残留溶剂量的气相色谱方法。
方法:气相色谱条件:毛细管柱OV-1701(0.32mm×30m×1μm);氢火焰离子化检测器(FID);进样口温度210℃,检测器温度220℃;柱温初始温度为50℃,保持8分钟,以20℃·min-1升温至200℃,保持5分钟。
载气为氮气。
内标法,进样量为1μL。
结论:该方法可用于检测曲尼司特中乙醇残留溶剂量。
关键词GC法;曲尼司特;乙醇;气相色谱法0 引言曲尼司特原料药在生产过程中加入了有机溶剂乙醇,按《中国药典》2010年版二部附录ⅧP残留溶剂测定法要求,需建立良好的检测方法对乙醇残留量进行控制。
本文用气相内标法,以异丙醇为内标物,采用梯度升温法检测乙醇残留量。
方法灵敏度高、重现性好、为曲尼司特中乙醇残留量检测提供了可靠的方法。
1 仪器与试剂岛津GC-2014C;FID;OV-1701色谱柱;乙醇为色谱纯;N,N-二甲基甲酰胺和异丙醇均为分析纯。
2 溶液制备内标溶液:取异丙醇50mg,用N,N-二甲基甲酰胺稀释成1mg/mL溶液。
对照溶液:取乙醇31.5 mg,用内标溶液稀释至25mL,摇匀,取2mL~10mL 量瓶,用内标溶液稀释成250μg/ mL溶液。
样品溶液:取样品0.5g至10mL量瓶,用N,N—二甲基甲酰胺稀释至刻度,摇匀。
乙醇定位溶液:取乙醇31.5mg,用N,N—二甲基甲酰胺稀释至25mL,摇匀,取2mL至10mL量瓶,用N,N—二甲基甲酰胺稀释成250μg/ mL溶液。
3 色谱条件程序升温,起始温度为50℃,维持8分钟,再以每分钟20℃的速率升温至200℃,维持5分钟,进样口温度210℃,检测器温度220℃。
直接进样1μl。
FID 检测器,内标法。
色谱柱:OV-1701(0.32mm×30m×1μm)4 方法学试验4.1 专属性试验将溶剂及上述内标溶液、对照溶液、乙醇定位溶液、样品溶液、在上述色谱条件下进样,色谱图如下:由溶剂DMF图谱可知,溶剂对试验无干扰;由其他图谱可知,样品中无异丙醇峰,乙醇与异丙醇峰分离完全,故选异丙醇为内标物。



药品中有机溶剂残留量及其测定天津药学2001年6月第13卷第3期3讨论本文所收集的9项研究已证实氟西汀对强迫症有显着疗效.与氯米帕明比较没有显着性差异.本研究则是运用Mete分析方法进一步综台和加强了上述论文的观点,更进一步证实飘西汀对强迫症疗效确切,与目前被认可的氯米帕明没有较大的差异,差异的效应也比较小在1O种不良反应的比较中,氟西汀组出现失眠和恶心的机率为氯米帕明的4倍上,2组差异显着.而氯米帕明组的排尿困难,便秘,锥外反应,口干及其他不良反应明显多于氟西汀组,病因分析也提示,它们是飘西汀的几倍乃至几十倍导致这种差异的原因可能与药物结构特征和剂量有关.在临床使用上,可根据氟西汀与氯米帕明的不同特点,针对病人的体质恰当地选择以减少其不良反应和联用其它药物.参考文献1向虎.杜悔英,邦崇芬,等国产氟西打治疗强迫症的对照研究临床精神医学杂志.1997,7(1):342苏中华,王健一韩鹏,等.氟西汀治疗强迫症的双盲对照研究临床精神医学杂志,1997,7(1):363王健,王希凤,张吉营,等.氟西汀与氯米帕明治疗强迫症的双盲对照研究中国神经精神疾病杂志,1997,23(4):2344陈挂兵,蔡则环盐醴氟西汀治疗强迫症临来开放性对照研究.中原精神医学学刊,1998,4(4):2165刘绍梅,孙良是.氟西汀与氯米帕明治疗强迫症对照研究上海精神医学,1998,新10(3):1736王树阳,牛倥鸿,张英杰,等.奥西汀与氯丙眯嘛治疗强迫症的对照研究.实用精神医学,1999.6(2):887胡华,张安t张永芳,等.氯丙咪嚷及氟西汀治疗强迫症疗效与5一羟色胺吉量的动态观察,中国神经精神疾病杂志,19g9.25(2)658崔承英,各广臣t泰金兰,等氟西汀与氯丙咪啼治疗强迫症的对照研究.四川精神卫生,2000,13(1);529王健军氟酉订与氯米帕明治疗强迫症的对照研究医药导报, 2000,19f4):32610盘卫东数理精神医学(三)Meta分析lI精神医学,1996,9(】)62(收稿日期2061041) ComparativeStudyollFluxetineandClomiprBmineinTreatmentof ObsessiveCompulsiveDisorder:Mela—analysisTangGanyi.FanHede(ShenzhenLiuhunHospltal,Shenzhen 5180021;WenYuguan(GuangzhouMenlalHospital,Guangzhou 510370);DuZeguangfShenzhenKangningHospitalShenzhen51SO01)ABSTRACIObjectire:Tostudythedifferenceso1f[uoxetine(Flu) andclomipramine(CIo)intherapeuticeffectsandsideeffectsMeth ads?Meta—ana[ysiswascarriedfor9pubtishedpapersoncompara—tirestudyofFluandClo,Results:ThesignificantdifferenceofFlu groupirtpre—andpost—tre&tmeratwasfound(Z1—2964,P< 0?O1),anditstherapenficeffectsize…(dl=5.73)?in dicatingthe therapeuticeffectwasgreat.NodifferencewasfoundbetweenStoup FluandCloaftertreatmefit(Z2=123,P<0.n5),anddifferenceef—fieacpwasnotsignificant(d2—0.2j)Theincidenceofinsomniaand nauseationingroupFlu~-presignKican[pdifferentfromStoupClo (x2=14.58,P<0吼?x2=1232,P<O01),anditseffectsizewas verygreat(d=O.86,d=0.790),theiroddratic(OR1~-ereB.41and4.26,respectivelyTheothersideeffects~-ereloweringroupFlu thanthatingroupClo(x2=18.1O~7313,P<OO1)Theeffectsize ofothersideeffectsingroupClom1.0l~373withhighlevel ConclusionsTherea托differencesbetweenFluandCloinsideef.1eers,buthotintherapeuticeffectsKEYWORDSfluoxetine,c[omipramine,obsessivecompu[swediaor der,Meta,——ana[ysh药品中有机溶剂残留量及其测定王卫(天津市药品检验所天津300070)中图分类号:R9271文献标识码:A文章鳙号:1006—5687(20,31)03—0026--,35药品中的残留溶剂是指在原料药或赋形剂的生产中,以及在制剂制备过程中产生或使用的有机挥发性化合物,他们在工艺中不能完全除尽.由于残留溶剂没有疗效,故所有残留溶剂均应尽可能除去,以符台产品规范.GMP或其他基本的质量要求.近年来,药品中残存有机溶剂的毒性和致癌作用日益引起各方面的关注.为了保护患者免受药品中残留有机溶剂的伤害,对药品在生产过程中引八的有机溶剂残留量需进行测定1ICH中关于有机藩剂残冒■的规定ICH(TnternatlonalConferenceonHarmonizarionofY echnicalRequirementsforRegistrationofPharmaceuticals forHumanUse:人用药品注册技术规范国际协调会)是由日美欧三方的管理当局(MHW,厚生省,FDA,CPMP)以及制药团体(JPMA:日本制药协会,PHRMA,EFPIA)其6个单位组成的.目的是为将最新研制出的新药尽快应用于全球的医疗临床机构,消除由于各国间药品注册技术规范不同而形成的贸易壁垒ICH为了协调统一各国对药品残留溶剂的限量,从l994年起就开始着手编撰关于有机溶剂残留量的指导原则,日.美,欧盟三方组织了多次会议,目前已进入具体实施阶段.根据残留有机溶剂对人体可能造成危害的程度,ICH关于有机溶剂残留量的指导原则将有机溶剂分为3类第1类:避免使用的溶剂——对人体有致癌性,或有较大致癌嫌疑及对环境造成危害的溶剂=这类溶剂共有5种.由于它们不可接受的毒性,应当避免在原料药,辅料以及制剂的生天津药学2001年6月第l3卷第3期产过程中使用.然而为了生产一种有特殊疗效的药品,不可避免要使用这类溶剂时.除非经过其他论证.其用量应限制在表1所示的范围内表1药品中残留的第一类溶耕第2类:限制使用的溶剂一一可能引起神经中毒或畸变等不可逆毒性,或怀疑具有其他严重的但可逆的毒性.因其固有的毒性.应限制使用.共26种(见表2).为限制药品中残留溶剂的水平并避免与其他术语相混淆.在ICH美于有机溶剂残留量的指导原则中,采用了一个新的术语”允许每日暴露量(permitteddabexposure.PDF)计算公式为:PDF—N()EL(最合适的动物试验中的最大安全量)/F1×F2xF3×F4×F5在公式中假定人的体重为50.修正因子F1~F5是用束将动物试验的数据适用于人的固于.表2药品中残留的第2类溶剂上袁中.有机溶剂在药品中的限量,有两种方法计算.其中方法1是:原料,辅料或制剂中残留溶剂的浓度限量.以百万分之几(10)来表示,计算公式为:浓度(10一)一l000xPDE/剂量此公式计算药品假设的剂量为10g/日.其中PDErag/日计.剂量以g/日计.日剂量不超过10g时.不需再计算日剂量超过10g时,应考虑采用方法2计算,即不需要考虑药品中每种成分都要符台方法1中给的限量值.而是将药品各组分中存在的某些残留溶剂的量相加,每日所接受的该残留溶剂的总量应低于PDE的量.第三类:具有低毒性的溶剂~这类溶剂包括人们认为在药物中一般量存在肘对人体无害的溶剂,但该类溶剂中许多尚未进行长期毒性或致癌研究急性毒性或短期毒性试验表明.这类溶剂几乎无毒,无遗传毒性.每日允许暴露量为50mg/日(按方法1计算,为5000×10或0.5)或更高些.它们包括:乙酸,丙酮,苯甲醚,l一丁醇,2一丁醇,乙酸乙脂,甲基特丁基醚.异丙苯,二甲基亚砜,乙醇,乙醚,甲酸乙酯,甲酸,庚烷,乙酸异丁酯,乙胺异丁酯,乙酸甲酯,3一甲基l丁醇,甲基乙基酮,甲基异丁基酮,2一甲基一l,丙醇,戊烷,l一度醇, l一丙醇,2丙醇.乙酸丙酯及四氢呋喃,共28种.除了上述3类残留溶剂外.ICH在关于有机溶剂残留量的指导原则中,还提到了第4类溶剂,这类溶剂还没有适当的毒性数据来得出其PDE值.需要生产厂家提供使用这类溶剂的安全数据.原辅料及制剂中的残留溶剂均包括在这一指导原则的范围内,但不包括尚在临床研究阶段的新药及已上市的药品指导原则适用于所有剂型及给药途径.在某些情况下如短期(<30天)服用或局部给药:允许残留溶剂的限量略高,但必须提出使用该限量的正当理由.而且要具体问题具体处理.ICH建议使用一些低毒性的溶剂并对某些残留溶剂提出可接受毒性水平保证用药安全.对原辅料以及制剂生产中所用的大多数有机溶剂的安全指数作出了评价,并提出了限量和分析方法.2各国药典中收裁有机溶剂残留■检童的情况药品中残留溶剂检查常用的方法是气相色谱法中国药典l995年版,英国药典1998年版(BP1998)咀及美国药典第23版(USPXXIII,1995),分别开始在附录中收载了有机溶剂残留量检’日本药局方截止到第19版尚未收载有机溶剂残留量检查.2-1在中国药典2000年版附录的有机溶剂残留量项下.28天津药学2001年6月第l3卷第3期对某些溶剂的残留量限度,按照ICH的标准进行了修订(见表3).目前,中国药典要求必须控制的玻留有机溶剂包括苯,氯仿,二氧六环,二氯甲烷,吡畦,甲苯及环氧乙烷如生产过程中涉及其他需要检查的有害有机溶剂,则应在各品种项下另作规定.有机溶剂残留量的测定方法为气相色谱法,氢火焰离子化检测器(FID)采用填充柱,以二乙烯苯一己基乙烯苯型高分子多孔小球(0,25~018mm)为固定相.进样方式为溶液直接进样或顶空进样.以内标法测定时,内标物与待测物的2个色谱峰的分离度应大于1.5,每个标准溶被进样5 次,所得待测物与内标物峰面积之比的相对标准差应不大于5{若以外标法测定时,所得待测物峰面积的相对标准差不大于10裹3中国药典1995年版与中国药典2000年版有机溶剂残叠■项下收藏溶剂的隈魔比较()22D)≤15.2.3美国药典第24板中有机挥发性杂质(ChemicalTests 467,OrganicV olatileImpurities)项下收载了5种必须控制的有机溶剂:苯,氯仿,1,4-二氧六环,二氯甲烷和三氯乙烯采用气相色谱法测定,氢火焰离子化检测器(FID),毛细管柱,进样方式为溶液直接进样或顶空进样.按外标法进行计算,各成分峰的分离度应≥1,连续进样时.各成分色谱峰面积的相对标准差(RSD)≤15.2.4各国药典中收载有机溶剂残留量检查的比较(表4).裹4各国药典中收藏有机溶剂残冒■检查的比较中国药典2000年版,USPXXI’~及BPZO00中,对有机溶剂残留量的限量均与ICH一致.与BP1998收载的7种残留有机溶剂相比,BP2000中收载了TcH中规定的第1,2,3类溶剂3残留溶剂检聋的分析方法验证有机溶剂豉留量的测定.应属于杂质的定量测定根据中国药典2000年版附录XIXA”药品质量标准分析方法验证”中的要求,在有机溶剂残留量测定的方法学研究过程中,应对杂质测定的准确度,重复性,专署性,定量限,线性,范围和耐用性进行考察.4有机溶剂残留量的测定在苟糟分析中的应用目前t随着科学技术的进步,国内外陆续有大量的新药上市.为保证用药安全,各国对新药在研究过程中的质量控制均有严格的规定,其中规定药物原料或制剂中有机溶剂残留量必须进行检查国内关于有机溶剂残留量分析的报道日益增多,多采用气相色谱法畏『定有机溶剂残留量,按进样方式的不同,可分为溶液直接进样法及顶空进样法}按所使用色谱柱的类型,可分为填充柱测定及石英毛细管柱测定4?l溶液直接进样为通常采用的方法,适用于受热不易分解的样品.此{去的绝对进样量大,对于测定残留量的有机溶剂,天津药学2001年6月第13卷第3期29可弥补检测器灵敏度的不足,操作简便易行.另一方面,由于进样量太,使得溶剂峰响应很大(除水,氧仿等以外),增加了分析时间一般应采取程序升温的方法,将溶剂快速除去42嘎空进样法一般用于测定低沸点(沸点在i00C以下)的残留溶剂,其优点是可以使其气化后进样,气化的残留有机溶剂的浓度与溶液中相比有很太增加.进样室在较低温度下操作,可避免组分分解顶空进样可相对地减少用于溶解样品的沸点较高的溶剂(如二甲亚砜,二甲基甲酰胺)的进样量,缩短分析时间.但对溶剂的纯度要求较高,尤其不能含有低沸点的杂质,否则会严重干扰测定.顶空进样可减少样品本身可能对色谱系统带来的干扰或严重圬染.采用自动的顶空进样器进样.内标法计算,样品的重现性较好目前,采用嘎空进样法测定有机溶剂残留量的报道日渐增多沙棘油是从沙棘(HippophaerhamnoidesL.)果实或种子中经正已烷提取得到的植物油,其中混有很多不易挥发的植物油.为了测定残留的正已烷,同时避免了色谱柱的污染. 高海等建立了顶空进样法测定沙棘油中正已烷的含量,色谱柱采用Chromasorb担体,以0.25TCP为固定液,方法的回收率为100.2,正已烷的浓度在0.04~O.2mg/ml范围内线性关系良好,最低检出限为1蒋文强等采用嘎空进样法测定了佐匹克隆中异丙醚的残留量,采用外标法,最低捡出限为0.16,g,方法重现性好,适用于药厂生产中的质量控制.高立勤等川采用GDX一401柱,以二甲基亚砜为溶剂,测定了辛伐他汀中残留溶剂甲苯和环已烷的含量,方法的线性和回收率良好.另据报道,窦爱兰”等采用顶空进样法测定了甲硝唑原料中环氧乙烷的残留量.王伟Ⅲ等采用顶空进样法测定了伊索拉啶中乙醇的残留量.胡连生等采用顶空气相色谱法测定了子仁霜中丙酮的残留量.扬绍英等t采用顶空进样法测定了心痛停片中二氧甲烷残留量.4.3填充柱包括玻璃柱及不错钢柱.填充柱的成本低,不易损坏.但柱教低.一般理论扳数在1×10.左右.采用填充柱测定有机溶剂残留量是较常用的方法.莫匹罗星(mopiroxin).又名假单孢菌酸A,是由荧光假单孢菌发酵而获得.在其精制工艺中需使用多种有机溶剂,为控制成品中的有机溶弃吐残留量,袁文玮等tto:建立了采用填充柱同时测定莫匹罗星中5种有机溶剂残留量的气相色谱方法.以甲氧基乙醇为溶剂,色谱柱为20PEG一20M,各组分均有良好的线性及回收率.头孢哌酮为广谱抗生素,目前生产工艺上多采用有机溶剂结晶法生产,为保证用药安全,降低毒副作用,韩国玲等”采用GDX一102柱,建立了头抱哌酮针剂中残留溶剂异丙醇的测定方法,用无水乙醇提取样品.以正丙醇为内标,异丙醇的浓度在3.I9×10~1.28×10~g/ml范围内.线性关系良好,r一0.9998,方法的回收率为94.4=另据报道,王卫等-采用GDX一4Ol柱测定了西沙必利中残留甲醇的含量,以二甲基甲酰胺为溶剂.线性范围是19.9~3187~g/ml回归方程:y5383×10+7.542×10一,r09998.方法简单,实用,结果准确.狱平平l_采用GDX一102柱测定盐酸氟丁打中异丙醇残留量,以水为溶剂,叔丁醇为内标,异丙醇在12~300 g范围内线性关系良好,回收率为102.0霍秀敏等划采用GDX-4Ol柱.以二甲基亚砜为溶剂,测定了卡维地蓿中甲醇,异丙醇及甲苯的含量,方法的城性及回收率良好,44石英毛细管柱的柱效很高,理论板教一般在I×10左右.但成本也较高,易损坏.鉴于毛细管柱优良的分离效能,因此在有机溶剂残留量的分析中有着不可替代的优势.王卫采用毛细管柱测定了盐酸多尼倍齐中醋酸乙酯和甲苯的含量,以水为溶剂,甲基异丁酮为内标,以lOO二甲基聚硅氧烷为固定相一柱温95C.实现了各组分的基线分离,乙酸乙酯在lo~100~g/m|浓度范围内,甲苯在I.78~I78~g/m]浓度范围内绒性关系良好,方法的回收率均为IO1.j绦新元等采用毛细管柱测定了吡喹酮中的乙醇,丙酮,二氧甲烷,醋酸乙酯和苯共5种有机溶剂残留量.以NN二甲基乙酰睦为溶剂,采用CP—Si]5CB石英毛细管柱.方法的线性及回收率良好.5部分药物中有机溶剂残留■的定(见裹5)襄5部分药物中有机溶剂残留■的谢定天津药学2001年6月第13卷第3期6小结5I1CH中规定的3类溶剂并非详尽无遗+其他可能使月I的溶剂还有待日后补充列八.第1,2类溶剂的建议限度或溶剂的分类会随着获得新的安全性资料而调整.原科或制剂中仅存在低毒性的第3类藩剂也可采用测定干燥失重的方法控制(例如规定该品种的干燥失重<05)62在中国药典2000年版附录中有机溶剂硅留量项下还未收载毛细管柱测定方法,如采用石英毛细管柱测定有机溶剂残留量+其色谱分辨率会明显优于填充柱.英国药典2000年版中,残留溶剂项下采用石英毛细管柱及2个色谱系统.已可将多达2O种有机溶剂进行有效分离.在中国药典2000年版修订时应考虑在附录的有机溶剂残留量项下增加采用毛细管柱测定方法.63因所测定的为溶剂在药品中的残留量.浓度均为10级.故要求测定仪器具有很高的灵敏度,在此灵敏度下对用于溶解样品所使用的溶剂.也提出很高的纯度要求.国产的分析纯(A.R)或优级纯(GR)试剂.在用于测定有机溶剂残留量时.均有不同程度的杂质干扰现在国内尚无专用于气相色谱测定有机溶剂残留量的溶剂.希望国内试剂生产厂家,能提供用于测定有机溶剂残留量的气相色谱专用的高纯度试剂6.4原料或制剂中的有机溶剂残留量+多使用气相色谱法及FID检测器测定,因仪器的灵敏度所限,对于ICH规定的毒性大限度低的第1类溶剂(如苯≤2×10)很难达到准确定量.氯化物的毒性一般都较大,在第1类溶剂中除苯以外均为氯化物.其所允许使用的限量也较低,又由于氯化物的FID检测器上的响应也低,因此用FID检测器来测定氯化nisation1997.Jul 16~18,BrusselsStep4draftpage24中国药典1995年版.附录ⅧP.有机溶剂残留量测定法英国药典1998版(BP1998f.AppendixYII,ResidualSolvems美国药典第23版(USPXXlll,1995)ChemicalTests467~rganieV oLatiLelmpurites中国药典2000年版.附录ⅧP有机溶剂残留量测定法英国药典2000版(BP2000AppendicesⅧIResidua【Solvents美国药典第24版(USPXXIII,1998)ChemicalTests467,”OrganicV otadtelmpurkes高海.孙文基静态顶空进样法测定沙棘油中溶剂残留量中草药.1992,03(5):243蒋文强.李迎秋佐匹克隆中溶剂残留量的气相色谱测定.中国医药工业杂志.1999.30(6):272高立勤.气相色谱法测定辛伐他汀中有机溶剂残留量现代药物分析论坛香港:世界医药出版杜.1999.12:168窦爱兰,郝丽晓.用顶空色谱法测定甲硝唑中环氧己烷的残留量.山西医科大学,1999,30(】):32王伟,韩光智.顶空法测定伊索拉啶中乙醇的残留量山东医药工业,1994.13(4):14扬绍英.陈志华气相色谱法检测药物中的残留溶剂天津药学.1996.8(4):71胡莲声,朱丹.用顶空气相色谱内标法测定子仁霜中藩剂残留量现代仪器.1999.(6):30袁文玮,王延明气相色谱法测定奠匹罗星中的几种硅留藩剂药物分析杂志.1995.1j(21:46郭国砖.姜春刚气相色谱法测定头孢娠酮粉针剂中溶煤残留量.中国药科大学.1998,29t2):112王卫.高立勤气相色谱法测定西沙必利中有机溶剂残留量天津药学,1999,11(3):55驮平平.气相色谱法分析盐酸氟丁订中异丙醇残留景药钎分析杂志,1995.15(增刊):154晷秀敏,王卫,袁文玮.等.气相色谱法测定卡维地祥}中有机藩剂残留量.2000中国药学会学术年会论文集(下册)王卫.气相色谱法测定盐酸多尼倍齐中有机溶剂残留量20bC中国药学会学术朵具有镇痛作用较强,持续时间较长,依赖性潜力较低的特点.不能替代阿片类毒品起戒毒作用:曲马朵1次用量≥100mg的剂量及多次给药时.不仅有常见的,也可能出现严重的不良反应.结论:曲马朵可用于缓解临床各种中度至敞重度疼痛.应加强管理,防止滥用美蕾词曲马朵临床应用药物不良反应中圈分类号:R97I文献标识码:A文章编号:1006—5687(2001)03—0030--03 34_0618g01234—67890l2。

乙醇及其代谢物在人体内的代谢动力
学研究
题目:乙醇及其代谢物在人体内的代谢动力学研究
摘要:本文主要研究了乙醇及其代谢物在人体内的代谢动力学。
通过对志愿者进行乙醇摄入后的血液和尿液样本分析,我们发现乙醇在体内的消除呈现一定的规律。
一、引言
乙醇是一种广泛使用的饮料,了解其在人体内的代谢过程对于评估乙醇相关的健康风险和药物相互作用具有重要意义。
二、材料与方法
招募健康志愿者,进行乙醇口服灌胃,分别在不同时间点采集血液和尿液样本。
利用高效液相色谱-质谱联用技术(HPLC-MS/MS)分析样本中的乙醇及其代谢物浓度。
三、结果与讨论
1. 乙醇的代谢动力学
乙醇在人体内的消除半衰期约为[具体数值]小时,代谢速度与个体的肝功能密切相关。
2. 代谢物的产生
乙醇在肝脏中被代谢为乙醛,然后进一步转化为乙酸。
我们检测到了这些代谢物在血液和尿液中的存在,并观察到它们的浓度随时间的变化趋势。
3. 性别和年龄的影响
我们发现性别和年龄对乙醇及其代谢物的代谢动力学参数存在一定的影响。
四、结论
本研究提供了关于乙醇及其代谢物在人体内代谢动力学的详细信息。
这些结果对于理解乙醇的生物学效应、评估饮酒相关的风险以及指导临床实践具有重要意义。
未来的研究将进一步探讨乙醇代谢的个体差异和环境因素的影响。
以上是一个关于乙醇及其代谢物在人体内的代谢动力学研究的简要概述,你可以根据具体的研究内容和需求进行修改和扩展。

药审中心发补意见汇总(化学药原料药部分)说明:本汇总是根据许多课题的补充意见进行的统计,完全遵循“补充意见”原文,未作任何改动。
1、合成工艺(1)原料·为关键起始原料,请提供合法来源证明文件、质量控制方法,分析可能引入的杂质对终产品的影响。
·请提供起始原料β-胸苷的购货发票、质量标准、出厂检验报告及研制单位的检验报告。
·提供4’-特丁基-4-氯丙苯基甲酮的来源证明文件及质控标准。
·原料和试剂的规格与来源。
(2)合成路线选择·详细说明所采用工艺路线的依据,补充各步化学反应式及详细的制备工艺流程图。
·补充有关文献资料以便对工艺进行评价。
·请提供详细的工艺流程图。
(3)对中间体的要求·请提供中间体的质控指标;说明主要中间体质控方法。
·由于3-(三氯锗基)丙酸为合成过程中的关键中间体,故请说明3-(三氯锗基)丙酸的精制方法,并提供三批样品的熔点数据,以确定3-(三氯锗基)的熔点。
·提供中间体质控的GC图谱;说明二苯溴代甲烷(含量>85%)、4-二苯甲氧基-1-乙氧羰基哌啶(含量>70%)未进行精制的原因;说明2(5)、3(1)、3(2)步反应终点如何控制。
(4)成品精制·提供详细的精制方法及依据。
·由于本品为一聚合物,故请提供资料以说明如何控制聚合反应以得到所需要产品。
(5)三废处理·原料药合成工艺中请提供三废处理的方法。
·说明三废处理的标准及三废处理的初步方案。
2、结构确证·由于本品为八元环的聚合物,故应提供有关资料以说明本品的聚合度,确证八元环的结构。
·原料药结构确证:建议补做国外对照品的有关图谱,并将测试结果与试制品的图谱进行比较,以充分证明本品的结构。
·关于结构确证项:(1)提供供试品精制的详细工艺;(2)补充元素分析的三批测试数据;(3)补在酸、碱条件下的UV图;(4)本品含一个结晶水的证据不足,建议补单晶的X-射线衍射谱;(5)对本部分资料进行修订及复核,说明所用仪器的型号。


气相色谱法测定酒剂及蒸馏酒中乙醇量肖顺经;曾庆艳;倪兆武;曾波【摘要】目的探讨酒剂和蒸馏酒中乙醇量的测定方法.方法采用气相色谱法.结果乙醇体积分数在2~7μL/mL范围内与峰面积线性关系良好,正丙醇理论板数大于1000,乙醇和正丙醇两峰的分离度大于4.结论气相色谱法便捷、精确、重现性好,是酒剂、酊剂及蒸馏酒中乙醇量的理想测定方法.【期刊名称】《中国药业》【年(卷),期】2010(019)018【总页数】1页(P40)【关键词】酒剂;蒸馏酒;气相色谱法;乙醇;含量测定【作者】肖顺经;曾庆艳;倪兆武;曾波【作者单位】云南省昭通市食品药品检验所,云南,昭通,657000;云南省昭通市食品药品检验所,云南,昭通,657000;云南省昭通市食品药品检验所,云南,昭通,657000;云南省昭通市食品药品检验所,云南,昭通,657000【正文语种】中文【中图分类】R284.1;R286.0酒剂、蒸馏酒及配制酒中乙醇量的测定方法各不相同,药典中酒剂的乙醇量测定方法为气相色谱法或蒸馏法(相对密度法)[1],《食品卫生检验方法》中蒸馏酒及配制酒的乙醇量测定方法为蒸馏法(比重计法)[2]。
相比之下,气相色谱法更方便、快捷、精确。
笔者参照《蒸馏酒及配制酒卫生标准的分析方法》,在药典中气相色谱法和乙醇量测定法的基础上,采用气相色谱法测定了酒剂及蒸馏酒中乙醇量,报道如下。
1 仪器与试药GC-14Bpf型气相色谱仪(日本岛津)。
白酒(市售,酒精度50%);藿香正气水(云南腾冲制药厂,批号为071110,080111,080208,080501;云南云河药业有限公司,批号为070401;云南天利药业有限责任公司,批号为20080202);无水乙醇(分析纯,成都市联合化工试剂研究所),正丙醇(分析纯,北京化学试剂公司),纯净水。
2 方法与结果2.1 色谱条件色谱柱:GDX-102填充柱(2 m);柱温:140℃;FID检测器温度:190℃;进样口:不分流,温度170℃;载气:氮气,流量100 mL/min;燃气:氢气,流量60 mL/min;助燃气:空气,流量500 mL/min;进样量:0.5 μL 。

高效液相色谱法测定药物残留量的方法学研究随着食品安全问题的日益引起人们的关注,药物残留量的检测也变得越来越重要。
高效液相色谱法(HPLC)作为一种先进的分析技术,已成为检测药物残留量的主要手段之一。
本文将从HPLC的基本原理、样品处理、色谱柱选择、检测条件等方面,阐述HPLC测定药物残留量的方法学研究。
1. HPLC的基本原理HPLC是一种基于液相流动速率控制物质分离的技术。
它是将样品溶解在溶剂中,通过高压泵将溶剂和样品混合后,通过流动相与固定相相互作用的机理,完成样品中成分的分离和测定。
由于HPLC所使用的流体动力学压力较高,使得分离过程更加快速高效,所以被称为高效液相色谱。
2. 样品处理在HPLC测定药物残留量的实验中,样品的预处理是非常重要的环节。
样品处理的目的是去除干扰物质和杂质,并将待测物质有效溶解在适当的溶剂中。
样品的处理方法有很多种,如固相萃取法、液液萃取法等。
其中,固相萃取法是一种非常常见的样品处理方法,特别适用于样品质量较差、复杂的样品,这种方法不仅效率高,而且可以有效降低成本。
3. 色谱柱选择在选择色谱柱时,需要考虑样品的性质、待测物质分子量以及它与固定相的性质等因素。
常用的色谱柱有C18、C8、C4、OBD、NH2、CN等,其中C18最为常用。
C18是一种疏水性的固定相,适用于一般常见的药物成分的分离,同时还具有很好的选择性和稳定性。
4. 检测条件HPLC检测药物残留量时,需要进行一系列的检测条件设置。
这包括流动相的组成、流速、柱温等因素。
流动相的组成是指样品中的溶剂组合。
通常情况下,可以采用乙腈-水的混合组成流动相,加入一定量的缓冲液以保持流动相的恒定pH 值,从而控制某些化合物的离子状态。
流速则是指流动相在色谱柱中的流速,根据需要可以在一定范围内进行调整。
柱温对于样品的分离也是非常关键的一个参数,它可以影响某些化合物的极性,从而影响改变它在某种固定相上的保留度。
5. 结语综上所述,HPLC是一种有效检测药物残留量的手段。

残留溶剂检测的方法学验证用“残留溶剂”作关键词搜索了一下,结果有好几百项内容,细细看来发现有许多内容都“不了了之”,最后都没有得到解答,或者是零零星星的一些意见,没有全面综合一点的。
因此请大家一起动动脑筋,希望深入讨论以下内容:1、需要验证的具体内容?2、每项内容验证的具体方法和要求?3、个别特殊问题和特殊经验?网友eagleast:1、专属性。
应该是各种测定的成分之间都达到基线分离(或者说分离度达到1.5以上),并且溶剂系统、内标物质之间应该没有相互干扰的现象。
这个应该没有什么难处吧?具体实施和一般的含量测定没有什么不同。
对于残留溶剂的专属性,一般的理解起来是比较容易,毕竟一个样品中的残留溶剂的种类是有限的。
不太象中药、生物样品等等的HPLC分析,会在色谱图中产生过多的干扰。
但是也有例外的,后面再谈。
原料药在生产过程中用到过的溶剂都会有残留的可能,它们有的来自投入的反应的原料,有的是原料在最后精制过程中用到的重结晶步骤中的溶剂,有的是物料在喷雾干燥过程中用到的有机溶剂。
这些一般都需要进行控制,选定后进行相关的检测。
是不是在原料生产过程中用过的有机溶剂都需要进行控制呢?当然不是。
例如,许多原料在最后的精制过程中都有重结晶步骤,为了提高产品的纯度,有时候会用大量的水、乙醇等等溶剂将样品进行洗涤,除去样品中残留的其他溶剂、杂质等等。
如果我们先前使用的有机溶剂在最后用的洗涤的溶剂中有比较强的溶解性,其实,是能够保证最后的产品中含有量在ICH指导原则限度以内的。
这时候,我们可以用经过验证的生产工艺条件下生产出来的原料多批次(3~10批次)进行检查,如果都远远低于ICH的限度,我们就可以有充足的理由在最后的质量标准中不对该溶剂进行控制了。
那么,会有哪些因素会导致干扰检测成分的现象发生呢?a、上面提到的在制造过程中使用到的溶剂。
b、样品在分析过程中的降解产生的。
对于这点,尤其需要注意。
有的样品在分析条件下会降解产生一些成分,而这些成分如果在分析条件下有相应,那么,就十分可能出现干扰测定的情况。
气相色谱法测定泊马度胺中5种残留溶剂徐茂红; 戴根来; 周美云【期刊名称】《《食品工业科技》》【年(卷),期】2019(040)019【总页数】6页(P256-260,285)【关键词】泊马度胺; 残留溶剂; 气相色谱【作者】徐茂红; 戴根来; 周美云【作者单位】皖西卫生职业学院安徽六安237005; 合肥久诺医药科技有限公司安徽合肥230088【正文语种】中文【中图分类】TS207.3泊马度胺(Pomalidomide),化学名为3-氨基-N-(2,6-二氧代-3-哌啶基)邻苯二甲酰亚胺,是美国Celgene公司研发的第三代免疫调节剂,于2013年2月首次在美国获准上市[1]。
其具有抗多发性骨髓瘤肿瘤细胞的关键功能,适用于既往接受过至少两种药物(包括来那度胺、硼替佐米)且最近治疗进行中或治疗完成60 d内疾病进展的多发性骨髓瘤患者[2]。
泊马度胺的生产工艺复杂,生产过程使用多种有机溶剂,如甲醇、乙醇、乙酸乙酯、二氧六环等[3-6]。
根据人用药品注册技术规范国际协调会(ICH)要求,必须对残留溶剂进行检测,以保障用药安全。
经文献调研,关于泊马度胺原料中残留溶剂的测定方法虽有报道,但文献中研究的残留溶剂种类较少,且未包含本研究中的溶剂,各国药典也均未收载。
在泊马度胺合成过程中使用的有机溶剂多达十余种,采用同一种方法检测所有溶剂很难实现。
本文在预试验的基础上,采用气相色谱法一次性测定甲醇、乙醇、乙酸乙酯、二氧六环、乙酸5种溶剂残留量,依据《中华人民共和国药典》(2015年版)对原料药中残留溶剂测定的要求[7],进行了系统的方法学验证,以期为泊马度胺的生产工艺改进和质量标准制定提供技术支持。
1 材料与方法1.1 材料与仪器泊马度胺样品(3批批号分别为130301、130302、130303) 合肥久诺医药科技有限公司;二甲基亚砜、甲醇、乙醇、乙酸乙酯、二氧六环、乙酸均为分析纯,购自国药集团化学试剂有限公司。
药物残留结果分析报告范文1. 研究目的和背景药物残留分析是一项重要的实验室测试,用于检测食品、环境和生物组织中是否存在药物残留物,并确定其浓度。
药物残留物对人类健康和生态环境构成潜在的风险。
因此,药物残留分析报告的编写对于食品安全和环境保护至关重要。
本报告旨在分析并讨论所得到的药物残留结果,以提供给相关利益相关者参考,用以制定相应的食品安全和环境保护政策。
2. 实验方法在本次实验中,选取了食品样品进行药物残留测试。
首先,收集了不同来源的食品样品,包括蔬菜、禽肉、水产品等。
然后,采用液相色谱-质谱联用仪(LC-MS/MS)进行药物残留物分析。
运用LC-MS/MS技术,可以对样品中的药物残留物进行定性和定量分析。
该技术具有高分辨率、高准确性和高灵敏度等特点,能够准确检测药物残留物的存在及其浓度。
3. 实验结果根据实验数据,我们得到了以下药物残留结果:样品编号食品类型药物残留物检测浓度(μg/kg)1 蔬菜农药A 502 蔬菜农药B 303 禽肉抗生素A 254 禽肉抗生素B 405 水产品兽药A 60根据上表所示的结果,可以看出样品中存在不同的药物残留物,并且检测浓度在每个样品之间有所不同。
这提示我们需要对食品供应链进行监管,确保食品的安全性和质量。
4. 结果分析4.1 蔬菜类食品药物残留物分析根据实验结果,蔬菜样品1中检测到农药A的残留物,浓度为50μg/kg。
农药A是一种广泛使用的农药,对人体健康具有一定的风险。
而样品2中检测到的农药B残留物浓度为30μg/kg,同样需要引起关注。
农药残留对人体健康有潜在的危害,因此需要加强食品供应链的监管,确保食品的安全性。
建议农药使用的合理规范和严格执行农药残留限量标准。
4.2 禽肉类食品药物残留物分析禽肉样品3中检测到抗生素A的残留物,浓度为25μg/kg;样品4中检测到抗生素B残留物的浓度为40μg/kg。
这表明禽肉在生产过程中可能使用了抗生素,这对人体健康构成潜在风险。
溶剂残留方法学验证溶剂残留检查多采用气相色谱法。
一般认为残留溶剂测定属限度检查,按相关指导原则只需提供专属性、检测限、定量限及进样精密度等方法学研究资料,故未进行线性、准确度和耐用性方面研究.残留量检查究竟是属于定量还是限度检查的范畴,《化学药物残留溶剂研究的技术指导原则》中未明确规定。
考虑到药物合成情况比较复杂,根据所使用的有机溶剂种类、数量等的不同,残留溶剂研究需要进行的程度、目标也可能不同,对方法学的要求也可能随之不同.当检测结果明显低于规定限度时,通常按限度检查要求,无需得出准确含量;当检测结果明显高于规定限度(尤其当检测结果为限度边缘时),或需残留溶剂量进行含量纯度折算时,需按定量检查要求进行方法学研究。
一、系统适用性试验取对照品溶液,进样,记录色谱图.柱效:以被测物的色谱峰计,填充柱法的理论板数应大于1000,毛细管色谱柱的理论板数应大于5000。
分离度:色谱图中被测物色谱峰与其相邻色谱峰的分离度应大于1.5。
重复性:以内标法测定时,对照品溶液连续进样5次,所得被测物与内标物峰面积之比的相对标准偏差(RSD)应不大于5%;以外标法测定时,所得被测物峰面积的相对标准偏差(RSD)应不大于10%。
二、专属性1、对各种残留溶剂定位和进行混合溶剂的分离度试验,并附代表性图谱. 取各种残留溶剂对照品溶液,分别单独进样,记录色谱图。
取混合溶液,进样,记录色谱图。
2、排除供试品中的未知杂质或其挥发性热降解产物对残留溶剂的测定产生的干扰。
①如果未知杂质或其挥发性热降解产物与被测物的保留值相同(共出峰),通常采用在另一种极性相反的色谱柱系统中对相同样品进行测定,比较不同色谱系统的测定结果的方法。
如二者结果一致,则可以排除测定中有共出峰的干扰;如二者结果不一致,则表明测定中有共出峰的干扰.②热降解产物与被测物的结构相同(如甲氧基热裂解产生甲醇),通常要通过测定已知不含该溶剂的对照样品来加以判断.三、检测限和定量限检测限为信噪比3:1时相应浓度或注入仪器的量。
盐酸利托君残留溶剂的测定摘要:目的建立测定盐酸利托君中有机溶剂残留量的方法。
方法采用溶液直接进样气相色谱法,载气为氮气, fid 检测器,色谱柱为dm-1701 毛细管柱。
结果乙醇、乙醚、吡啶的线性范围分别为0.10352mg.m l- 1~1.5528mg.m l- 1 (r= 0.999)、0.09936mg.m l- 1~1.4904mg.m l- 1 (r= 0.999)、4.976μg.m l- 1~74.64μg.m l- 1 (r= 0.9999);平均回收率分别为98.2%、98.0%和98.4%;rsd 分别为2.7%、3.9%和3.1%;结论所用方法简单、快捷、灵敏,可用于盐酸利托君中有机溶剂残留量的控制。
关键词:盐酸利托君有机溶剂残留量毛细管气相色谱法盐酸利托君是美国fda唯一批准和推荐使用的预防早产的药物,全世界已有80多个国家注册使用,其原料药生产过程中使用了乙醇、乙醚和吡啶等有机溶剂。
根据相关要求对其残留溶剂进行了检测,采用气相色谱法测定了以上溶剂的残留量。
1 仪器与试药天美gc7900气相色谱仪hw2000色谱工作站,无水乙醇、乙醚和吡啶为分析纯。
盐酸利托君原料(某制药公司,批号: 090224 、090226、090228);2 方法与结果2.1 色谱条件与系统适用性试验色谱柱:dm-1701(30m*φ0.32mm*0.25μm);进样口:180℃;氢焰检测器:200℃;柱温:55℃保持2min后以10℃/min升温至80℃,再以30℃/min升温至180℃,保持3min。
理论板数按乙醚峰计算应不低于5000。
各组分分离度应符合要求。
在上述色谱条件下,空白溶剂对本品三残留溶剂测定无干扰,各被测组分与相邻色谱峰分离度符合要求;理论板数按乙醚峰计为80000以上,不低于5000,符合要求。
2.2 样品溶液的制备分别精密称取样品约1.0g,置10ml量瓶中,加dmf溶解并稀释至刻度,摇匀,即得供试品溶液。
乙醇残余量
药用辅料中的残留溶剂系指在药用辅料生产过程中残留于成品中的有机溶剂。
由于残留溶剂有可能增加药用辅料的毒副作用,甚至会影响药物的稳定性,故所有的有机溶剂应尽可能地除去。
由于我公司蔗糖生产工艺中使用乙醇作为溶剂,为了保障药用辅料产品质量,故采用气相色谱仪(顶空法)对药用蔗糖中的乙醇残留量进行了测定。
1、仪器
GC-2014C气相色谱仪,FID检测器,岛津公司生产。
顶空进样器DK-3001A,北京中兴汇利科技发展有限公司生产。
2、试药
2.1乙醇
批号:20090108,级别:色谱级
厂家:天津市科密欧化学试剂可发中心
2.2水
生产使用纯化水
2.3正丙醇
批号:T20090521,级别:分析纯
厂家:国药集团化学试剂有限公司
2.4 样品
蔗糖(批号20111101、20111102、20111103),河南鲁尔康药业有限公司生产
3、色谱条件
3.1色谱柱:DB-624[6%氰丙苯基-94%二甲基聚硅氧烷]30m×0.53mm×3.0μm;安捷伦公司。
3.2检测条件:起始温度40℃,以每分钟5℃的速率升温至120℃,维持1分钟,顶空瓶平衡温度为90℃,平衡时间为30分钟。
进样体积1ml,载气为N2,FID检测器。
4、检测步骤
4.1溶液配制
4.1.1内标贮备液
精密量取正丙醇0.3125g于250ml容量瓶中,加水稀释至刻度,制成每1ml含正丙醇1.25mg的内标贮备液。
4.1.2对照品贮备溶液
精密称取乙醇250.8mg于100ml量瓶中,加水稀释至刻度,制成每1ml含乙醇2.508mg的对照品贮备溶液。
4.1.3对照品溶液
依次取0.1ml、0.2 ml、0.3 ml、0.4 mll对照品贮备溶液分别置4个100ml量瓶中,再分别加入1ml的内标贮备液,加水稀释至刻度,制成每1ml含乙醇2.51、5.02、7.52、10.0μg,含正丙醇12.5μg的系列浓度的对照品溶液。
4.1.4供试品溶液
精密称取样品1g置100ml容量瓶中,再精密量取1ml内标贮备液置容量瓶中,加水溶解,加水稀释至刻度,得供试品溶液。
4.2 测试
按照气相色谱测试方法,依次测定对照品、供试品记录结果;建立回归方程计算供试品中残余乙醇含量。
5、测定结果
5.1、对照品
5.2 供试品
6、结论
通过测定乙醇残余含量均低于0.5%,可以保证产品的质量安全。
测定图谱附后:
对照2:
对照3:
对照4:
供试20111101-01
供试20111101-02
供试20111102-01
供试20111102-02。