微生物16S测序数据的正确打开方式
- 格式:doc
- 大小:31.00 KB
- 文档页数:10

微生物分子生态技术16S在微生物学领域,微生物分子生态技术16S以其广泛的应用范围和独特的研究价值备受。
该技术通过对其目标DNA进行特异性扩增和测序,有助于我们对微生物群落的结构和功能进行深入了解。
本文将详细介绍微生物分子生态技术16S的概念、工作原理和实验流程,以及如何分析和解读实验结果。
微生物分子生态技术16S是一种以细菌的16S rRNA基因作为靶标,通过PCR扩增和测序等技术对细菌进行分类和鉴定的方法。
16S rRNA 基因是细菌染色体上编码rRNA的基因,具有高度的保守性和特异性,可用于区分不同种类的细菌。
通过比较16S rRNA基因的序列差异,我们可以对细菌进行精确的分类和鉴定。
微生物分子生态技术16S的工作原理是:首先提取样品中的总DNA,然后利用引物特异性扩增16S rRNA基因的部分序列。
扩增后的产物经过测序仪进行高通量测序,得到原始序列数据。
通过对这些数据的生物信息学分析,可以获得样品中细菌的种类、丰度等信息。
该技术适用于研究微生物群落的结构和功能,以及生态环境和人体健康等方面的应用。
实验流程包括样本采集、DNA提取、PCR扩增和测序等环节。
在样本采集阶段,需根据研究目标选择合适的样品类型和采集方法。
在DNA 提取阶段,需要选择适当的试剂盒,以最大限度地提取出样品中的总DNA。
在PCR扩增阶段,需设计针对16S rRNA基因的特异性引物,并优化扩增条件以获得最佳的扩增效果。
在测序阶段,需使用高通量测序技术在短时间内获取大量的序列数据。
对于实验结果的分析与解读,主要包括物种组成、多样性指数和关联分析等方面。
通过对比不同样品的序列数据,可以获得样品中的优势菌群和稀有菌群。
利用多样性指数分析样品的物种丰富程度和分布情况,有助于我们了解生态系统的稳定性和健康状况。
通过关联分析探究不同细菌之间的相互关系,有助于我们深入了解微生物群落的生态功能。
在生态环境方面,微生物分子生态技术16S可用于研究土壤、水体等环境中的微生物群落结构与功能。

菌群16s测序解读-回复16S测序是一种常见的菌群分析技术,通过对细菌16S rRNA基因进行测序和比对,可以了解菌群的组成和多样性。
本文将以中括号内的内容为主题,一步一步回答相关问题,带你了解16S测序解读的过程。
1. [什么是16S测序?]16S测序是一种分析微生物菌群组成的技术,其基础是对细菌的16S rRNA基因进行测序。
16S rRNA是一种高度保守且广泛存在于细菌中的基因,在不同菌种之间具有一定的差异,可以用于鉴定和分类细菌。
通过对16S rRNA基因进行PCR扩增和测序,可以获取菌群的遗传信息。
2. [为什么要进行16S测序?]菌群的组成和多样性对生态系统的功能和健康有重要影响,因此了解菌群的结构和组成对于研究微生物生态学、人体健康等具有重要意义。
传统的菌群研究方法往往需要进行菌落计数、培养和鉴定等步骤,耗时且无法覆盖所有细菌。
而16S测序作为一种高通量的方法,可以快速、准确地获取大量细菌的遗传信息,为菌群的分析和比较提供了有效手段。
3. [16S测序的具体步骤是什么?]16S测序的具体步骤包括:样品收集和处理、DNA提取、16S rRNA基因扩增、文库构建、高通量测序和数据分析。
首先,收集样品并进行表面消毒等预处理,以避免外源细菌的污染。
然后,提取样品中的总DNA,其中包括目标菌群的DNA。
接着,利用特异引物对16S rRNA基因进行PCR 扩增,获得目标菌群的16S rRNA基因片段。
将扩增得到的片段纳入文库,然后进行高通量测序,获得大量的16S rRNA序列数据。
最后,对测序数据进行质控、序列拼接和比对,然后进行菌群组成和多样性分析。
4. [如何解读16S测序数据?]解读16S测序数据需要进行一系列的数据分析和生物信息学处理。
首先,进行质控筛选,去除低质量的序列,并对序列进行去嵌合。
然后,对序列进行拼接,使其成为完整的16S rRNA基因片段。
随后,利用生物信息学工具将序列比对到已知菌种的数据库中,进行分类和注释。

16s扩增子测序流程引言:16s扩增子测序是一种常用的微生物学研究方法,通过对16s rRNA 基因进行扩增和测序,可以获取微生物群落的组成信息。
本文将详细介绍16s扩增子测序的流程,包括样品收集、DNA提取、扩增子设计、PCR扩增、测序和数据分析等环节。
一、样品收集:在进行16s扩增子测序之前,需要准备好待测样品。
样品可以来自环境样品、动物体内、人体内等各种来源。
在收集样品时,需要注意避免污染和样品损伤,保证样品的质量和完整性。
二、DNA提取:样品收集后,需要提取样品中的总DNA。
DNA提取的方法有很多种,常用的有CTAB法、酚/氯仿法、商用DNA提取试剂盒等。
提取的DNA应具有高纯度和高浓度,以保证后续的PCR扩增和测序的质量。
三、扩增子设计:扩增子是指用于扩增16s rRNA基因的引物。
根据不同的研究目的和物种组成,可以设计不同的扩增子。
常用的扩增子包括V1-V9区域的引物。
在设计扩增子时,需要考虑引物的特异性和覆盖范围,以确保扩增到目标基因片段。
四、PCR扩增:PCR扩增是16s扩增子测序的关键步骤。
在PCR扩增中,需要将样品中的16s rRNA基因片段扩增到目标长度。
PCR反应体系包括模板DNA、引物、dNTPs、聚合酶和缓冲液等。
PCR扩增的条件包括温度、时间和循环数等,需要根据具体的扩增子设计和样品特点进行优化。
五、测序:PCR扩增后的产物需要进行测序。
测序技术有很多种,包括传统的Sanger测序和高通量测序技术。
根据实验室条件和研究目的,选择合适的测序平台和方法进行测序。
测序的结果将得到样品中16s rRNA基因片段的序列信息。
六、数据分析:测序后的数据需要进行生物信息学分析。
首先,对测序得到的序列进行质量控制和去噪处理,去除低质量和低复杂度的序列。
然后,将序列比对到参考数据库(如Greengenes、SILVA等),进行分类学注释和物种分析。
最后,可以使用多样性分析方法(如Alpha多样性和Beta多样性分析)对微生物群落进行描述和比较。

16s rrna报告解读摘要:1.16s rRNA简介2.16s rRNA报告解读方法3.报告结果分析与应用4.报告的局限性与未来发展方向正文:随着分子生物学技术的发展,16s rRNA报告已成为微生物学、生态学等领域的重要研究手段。
16s rRNA是细菌核糖体的一部分,具有种属特异性,可通过高通量测序技术对微生物群落进行定性和定量分析。
本文将介绍16s rRNA报告的解读方法、结果分析与应用,以及报告的局限性与未来发展方向。
一、16s rRNA简介16s rRNA存在于细菌细胞中,负责蛋白质生物合成。
由于不同细菌物种的16s rRNA序列存在差异,可通过测定该序列对微生物进行分类和鉴定。
目前,16s rRNA测序已成为微生物多样性研究的主要手段。
二、16s rRNA报告解读方法1.序列分析:将测序得到的原始数据进行质控、比对、组装等处理,得到序列。
然后,通过序列之间的相似性分析,对微生物物种进行分类和鉴定。
2.生物信息学分析:基于序列数据,进行物种多样性、群落结构、代谢途径等分析。
常用的生物信息学工具包括MetaPhlAn、Kraken等。
3.统计分析:对生物信息学结果进行统计,评估样本间的差异性和相似性,为实验结果提供依据。
三、报告结果分析与应用1.物种多样性分析:通过统计不同物种的序列数量,评估样本中的微生物多样性。
2.群落结构分析:分析不同物种在样本中的相对丰度,揭示微生物群落的组成和变化。
3.功能基因分析:基于代谢途径和基因功能,分析微生物群落的代谢活性。
4.应用:16s rRNA报告可用于医学、环境、农业等领域的微生物学研究,为疾病诊断、环境保护、农业生产等提供科学依据。
四、报告的局限性与未来发展方向1.局限性:16s rRNA报告无法区分共生和病原微生物,且受样本制备和测序深度等因素影响。
2.未来发展方向:发展更高效的测序技术、生物信息学方法,以及整合多组学数据进行综合分析,提高报告的准确性和应用价值。

16S扩增子测序分析流程1.样品采集:从研究对象的生态环境中采集样品,如土壤、水样、动植物组织等。
2.DNA提取:从样品中提取总DNA。
目前常用的提取方法有商业试剂盒法和酚氯仿法等。
3.扩增16SrRNA基因:使用引物对样品中的16SrRNA基因进行PCR扩增。
常用的引物对包括V3-V4、V4等。
与此同时,可以通过引入带有索引序列的引物,将不同样品进行区分,以进行多样品同时测序。
4.文库构建:将PCR扩增产物纯化、定量后,进行文库构建。
可以使用商业试剂盒进行文库构建。
5. 测序:采用高通量测序平台(如Illumina MiSeq)对文库进行测序。
常采用双端测序,可同时测序16S rRNA的两个可变区域。
测序结果通常以FASTQ格式存储。
6.数据预处理:对测序数据进行初步处理,包括去除低质量序列、去除引物序列、去除低质量碱基等。
7. 序列拼接:对样品的两个测序reads进行拼接,形成完整的16S rRNA序列。
拼接工具有PANDASeq、FLASH等。
8.降噪:通过降低测序错误和测序深度变异对测序数据进行降噪,常用的方法包括UNOISE2、DADA2等。
9.代表序列挑选:从降噪后的序列中选择代表序列。
可以使用聚类算法如CD-HIT、UPARSE等。
10. 物种注释:将代表序列与参考数据库(如Greengenes、Silva)进行比对,进行物种注释。
11. 多样性分析:根据物种注释结果,进行多样性分析,如Alpha多样性(如丰富度和均匀度)和Beta多样性(如Unifrac距离)。
12.统计分析:使用统计学方法对不同样品的多样性指数和物种丰度等进行比较和统计。
13.功能注释:对代表序列进行功能注释,可以利用专门的功能注释数据库如PICRUSt等。
14.结果可视化:将分析结果进行可视化,如绘制条形图、热图、PCoA图等。
15.结果解读和讨论:根据分析结果进行解读和讨论,提出结论并撰写论文或报告。
需要注意的是,16S扩增子测序分析流程中的每个步骤都有许多选择和可调整的参数,因此具体的实验流程和分析方法可能会因不同研究目的和需求而有所不同。

16S rDNA鉴定细菌的方法细菌16S rDNA鉴定主要分为7个部分:1.提取细菌基因组DNA,2.设计/选择引物进行PCR扩增,电泳检测纯度与大小。
3.琼脂糖凝胶电泳分离4.胶回收目的片段5.目的片段测序。
6.BLAST比对获取相似片段。
7.构建系统进化树试剂:1、培养基:通常选择组分简单且细菌生长良好的培养基(培养基组分过于复杂会影响DNA 的提取效果,也可以在裂解细菌前用TE缓冲液对菌体进行洗涤。
)。
2、1M Tris-HCl (pH7.4, 7.6, 8.0)(1L):121.1g Tris,加浓盐酸约(70ml, 60ml, 42ml),高温高盐灭菌后,室温保存。
冷却到室温后调pH,每升高1℃,pH大约下降0.03个单位。
3、0.5M EDTA(pH8.0)(1L):186.1g Na2EDTA•2H2O,用NaOH调pH至8.0(约20g),高温高压灭菌,室温保存。
4、10×TE Buffer(pH7.4,7.6,8.0)(1L):组分:100 mM Tris-HCl,10 mM EDTA。
1M Tris-HCl (pH7.4,7.6,8.0)取100ml,0.5M EDTA(pH8.0)取20ml。
高温高压灭菌,室温保存。
1×TE Buffer用10×TE Buffer稀释10倍即可。
5、10%SDS(W/V):称10g,68℃加热溶解,用浓盐酸调pH至7.2。
室温保存。
用之前在65℃溶解。
配置时要戴口罩。
6、5M NaCl:称292.2gNaCl,高温高压灭菌,4℃保存。
7、CTAB/NaCl(10%CTAB,0.7M NaCl):溶解4.1g NaCl,加10g CTAB(十六烷基三甲基溴化铵),加热搅拌。
用之前在65℃溶解。
8、氯仿/异戊醇:按氯仿:异戊醇=24:1(V/V)的比例加入异戊醇。
9、酚/氯仿/异戊醇(25:24:1):按苯酚与氯仿/异戊醇=1:1的比例混合Tris-HCl平衡苯酚与氯仿/异戊醇。

16SrDNA鉴定菌株的标准操作规程1.适用范围本标准规定了通过特定引物对细菌的16SrDNA片段进行PCR扩增,然后对扩增片段进行序列分析比对,快速获得细菌种属信息的操作规程。
本标准适用于未知细菌的快速种属分析,以及为细菌的生化鉴定提供指导信息。
2.方法和原理16SrDNA鉴定是指用利用细菌16SrDNA序列测序的方法对细菌进行种属鉴定。
包括细菌基因组DNA提取、16SrDNA特异引物PCR扩增、扩增产物纯化、DNA测序、序列比对等步骤。
是一种快速获得细菌种属信息的方法。
细菌rRNA(核糖体RNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。
16S rDNA是细菌染色体上编码16S rRNA相对应的DNA序列。
16S rDNA由于大小适中,约1.5Kb左右,既能体现不同菌属之间的差异,又能利用测序技术较容易地得到其序列,故被细菌学家和分类学家接受。
在 16S rRNA 分子中,可变区序列因细菌不同而异,恒定区序列基本保守,所以可利用恒定区序列设计引物,将16S rDNA片段扩增出来,利用可变区序列的差异来对不同菌属、菌种的细菌进行分类鉴定。
16SrDNA序列的前500bp序列变化较大,包含有丰富的细菌种属的特异性信息,所以对于绝大多数菌株来说,只需要第一对引物测前500bp序列即可鉴别出细菌的菌属。
针对科学论文发表或是前500bp无法鉴别的情况,需要进行16SrDNA的全序列扩增和测序,得到较为全面的16SrDNA的序列信息。
由于测序仪一次反应最多只能测出700bp的有效序列,为了结果的可靠性,通常将16SrDNA全长序列分成3部分进行测序。
3.设备和材料3.1器材移液器(1000μL、200μL 、100μL、10μL);涡旋振荡器;Eppendorf MixMate;离心机;水浴锅;电泳仪;制冰机;低温冰箱;PCR仪:Veriti 96 Well Thermal Cycler;凝胶成像仪:VersaDoc MP 4000;基因分析仪:AB3500、AB31303.2试剂DNA快速提取试剂:PrepMan Ultra;琼脂糖;PCR试剂:Taq酶,10×Taq Buffer(Mg2+),dNTPs,ddH2O等;ExoSAP-IT;测序试剂:BigDye Terminator,5×Sequencing Buffer;BigDye XTerminator Purification Kit;3.3耗材移液器吸头:1000μL、200μL、10μL;离心管:1.5mL、200μL;Micro Amp TM Optical 96-Well Reaction Plate;页脚内容2Micro Amp TM Optical Adhesive Film ;3.4 引物16SrDNA 名称 序列 扩增长度第1部分 正向引物27F 5'-AGA GTT TGA TCC TGG CTC AG-3'500 bp 左右反向引物519R 5'- GWA TTA CCG CGG CKG CTG -3'第2部分正向引物357F5'- CTC CTA CGG GAG GCA GCA G-3' 750bp 左右反向引物1115R 5'-AGG GTT GCG CTC GTT GC-3' 第3部分正向引物926F5'- AAA CTY AAA KGA ATT GAC GG-3'560 bp 左右反向引物1492R 5'-TAC GGC TAC CTT GTT ACG ACT T-3'其中M=C:A, Y=C:T. K=G:T, R=A:G, S=G:C. W=A:T; all 1:14. 操作流程5.实验方法5.1核酸提取:挑取单菌落,然后置于装有100μLPrepMan Ultra的离心管中,涡旋震荡混匀30s左右,然后100℃水浴10min后,以离心机最大转速离心3min,取10μL上清液与490μL ddH2O(即稀释50倍),混匀作为下步PCR的模板DNA。

16srrna鉴定菌株的标准操作规程1. 准备工作:清洗实验室器具、准备培养基和试剂、保持操作环境的无菌状态。
2. 提取细菌样品:从菌落、液体培养基或环境样品中选择一个菌落较纯净的菌株。
使用无菌操作工具,在无菌条件下,用菌液或菌落均匀涂抹于无菌平板上。
3. 培养菌株:将无菌平板培养基转移到适合该菌株生长的培养条件下。
通常情况下,大多数菌株可以在37℃下培养24小时后获得足够的菌量。
4. 提取菌株的16S rRNA基因:使用合适的菌株提取方法提取菌株的总DNA,并使用PCR方法扩增16S rRNA基因。
PCR 反应条件可根据实验室的标准方法或相关文献进行设置。
5. 准备电泳样品:将扩增的16S rRNA基因产物与DNA分子量标记物一同混合,通过琼脂糖凝胶电泳进行分离和检测。
6. 电泳分析:将准备好的电泳样品注入琼脂糖凝胶孔中,进行电泳分析。
根据相对位置和迁移速度,确定16S rRNA基因的大小。
7. 分离目标DNA带:使用无菌操作工具,在琼脂糖凝胶上切割目标DNA带。
8. 提取目标DNA带:使用合适的目标DNA提取方法,从琼脂糖凝胶上提取目标DNA带。
9. DNA序列测定:将提取的目标DNA带进行测序,可以委托测序机构进行测序,也可以使用实验室的测序设备进行测序。
10. 序列分析:使用生物信息学工具对测序结果进行分析,包括序列比对、物种鉴定和进化分析等。
11. 物种鉴定结果的确认:将测序结果与数据库中的已知序列进行比对,确定菌株的物种鉴定结果。
12. 数据和结果的解释:根据测序结果和数据库比对结果进行数据分析和结果解释。
上述是针对16S rRNA基因进行鉴定的一般操作流程,具体操作规程可能因实验室的要求和设备的不同而有所差异。
实施过程中应始终保持无菌操作,确保结果的准确性和可靠性。

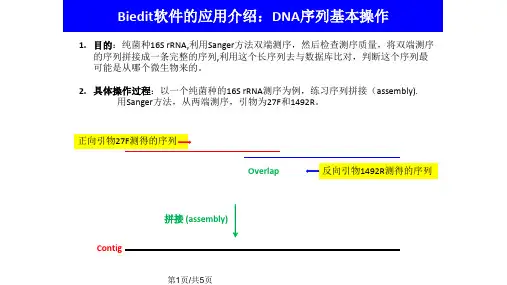
16SrDNA测序分析流程
宏基因组测序是指对微生物群体进行高通量测序,分析特定环境中微生物群体基因组成及功能、微生物群体的多样性与丰度,进而分析微生物与环境、微生物与宿主之间的关系,发现具有特定功能的基因。
宏基因组测序无需分离纯培养微生物,较大扩展了微生物资源的利用,为环境微生物群落的研究提供了有效工具。
宏基因组深度测序可以揭示或估计环境中真实的物种多样性和遗传多样性,挖掘具有应用价值的基因资源,应用于开发新的微生物活性物质。
宏基因组研究分两个方向:扩增子测序和全基因组测序。
扩增子测序,涉及特定序列位点的PCR扩增,通常是16S/18S rDNA。
宏基因组的物种分类,一般用OUT(operational taxonomic unit),即可操作单元来表示。
通常原生生物使用16S rDNA来衡量,真核生物的OUT使用18S rDNA来衡量。
主要应用在以下几个方面:通过宏基因组测序的方法,将肠道微生物与疾病进行关联分析以揭示疾病与健康个体间的微生物差异;鉴定特定环境中的特定微生物发现耐受菌种及相关基因。
可以研究物种的分类,研究与特定环境相关的代谢通路,以及通过不同样品的比较研究微生物群落内部、微生物与环境、微生物与宿主之间的关系。
扩增子序列(16rDNA)分析流程:
结果示例:
图1 不同样本OUT(operat ional taxonomic units)丰度的热图展示
图2 实验组和对照组在属水平上生物种类分布的柱状图和饼状图
图3 系统发生树。

菌群16s测序解读菌群16s测序,是一种广泛应用于微生物群落研究的技术。
通过对16s rRNA基因进行测序,并对其序列进行分析,可以对群落中存在的细菌进行分类和鉴定。
这项技术能够帮助我们了解细菌的多样性、丰度和功能,对于研究环境微生物群落结构、人体菌群与健康之间的关系等具有重要意义。
在进行菌群16s测序时,首先需要从样品中提取细菌的DNA,并进行PCR扩增。
常用的扩增方法是利用16s rRNA的保守序列设计引物,将目标序列扩增出来。
扩增后的DNA片段进一步纯化和测序,最终得到的测序数据即为16s rRNA的序列信息。
在得到16s测序数据后,需要对数据进行处理和解读。
首先是序列质量控制,去除低质量的序列。
接下来,对序列进行去噪声处理,去除PCR扩增产生的噪声和测序错误。
然后,利用聚类算法对序列进行分割,将相似的序列分为同一个OTU(操作分类单元)。
最后,通过比对数据库,将OTU与已知菌种进行分类和鉴定。
通过菌群16s测序,我们可以获得菌群的多样性指数。
多样性指数反映了菌群的物种丰富度和均一性。
常用的多样性指数包括Shannon指数、Simpson指数等。
这些指数能够告诉我们菌群内的物种多样性和优势菌种。
除了菌群的多样性,16s测序还可以帮助我们了解菌群的功能。
通过比对菌群16s测序数据和已知的功能基因数据库,我们可以推断菌群在代谢、信号传导、抗性等方面的功能。
这对于研究菌群与宿主的相互作用机制、菌群在环境中的重要功能等具有重要意义。
菌群16s测序的应用十分广泛。
在环境科学方面,可以通过菌群16s测序了解不同环境样品中细菌的组成和功能,帮助了解细菌在环境生物地球化学循环中的作用。
在医学研究中,菌群16s测序可以用于研究肠道菌群与人体健康之间的关系,探索微生物在疾病发生发展中的作用。
在食品安全检测中,通过菌群16s测序可以追踪食品中可能存在的致病菌,提供相关的病原菌检测和风险评估。
尽管菌群16s测序在微生物群落研究中具有广泛应用,但也存在一些限制。
16s rdna测序原理16s rDNA测序是一种用于研究微生物群落结构和多样性的重要技术。
它通过对细菌和古菌16s rDNA基因的测序,可以揭示微生物的分类和系统发育关系,帮助科学家更好地了解微生物在自然界中的分布和功能。
下面,我们将详细介绍16s rDNA测序的原理及其在微生物学研究中的应用。
首先,我们需要了解16s rDNA是什么。
16s rDNA是细菌和古菌细胞中的一个重要基因,它编码了16s rRNA,是细菌和古菌的核糖体小亚基的组成部分。
由于16s rDNA在不同的细菌和古菌中存在一定的差异,因此可以作为分类和鉴定微生物的分子标记。
通过对16s rDNA序列的测定和比对,可以揭示微生物的遗传关系和系统发育地位。
在进行16s rDNA测序时,首先需要从样品中提取微生物的总DNA。
接下来,利用特异性引物对16s rDNA基因进行PCR扩增,得到16s rDNA的扩增产物。
然后,对扩增产物进行测序,获取16s rDNA的序列信息。
最后,利用生物信息学方法对测得的16s rDNA序列进行分析和比对,从而揭示微生物的分类和系统发育关系。
在微生物学研究中,16s rDNA测序被广泛应用于微生物群落结构和多样性的研究。
通过对不同环境样品中微生物16s rDNA序列的测定和分析,可以揭示微生物在不同环境中的分布和多样性,帮助科学家更好地了解微生物在自然界中的生态功能。
此外,16s rDNA测序还可以用于鉴定和分类未培养的微生物,为微生物资源的开发和利用提供重要的信息。
总之,16s rDNA测序是一种重要的技术手段,可以帮助科学家揭示微生物的分类和系统发育关系,了解微生物在自然界中的分布和功能。
随着测序技术的不断发展和进步,相信16s rDNA测序在微生物学研究中会发挥越来越重要的作用,为我们认识微生物世界提供更多的信息和启示。
16s测序方法16s测序方法是一种常用的高通量测序技术,可以用于对DNA或RNA样本的全基因组或转录组进行测序。
本文将介绍16s测序方法的原理、应用及其在生物医学领域中的作用。
一、16s测序方法的原理16s测序方法是通过对16s rRNA基因进行测序来研究微生物的遗传多样性。
16s rRNA基因存在于细菌和古细菌的基因组中,具有高度保守的区域和可变的区域。
通过PCR扩增16s rRNA基因,然后对扩增产物进行测序,可以得到样本中存在的不同微生物的16s rRNA序列。
1. 研究微生物多样性:通过对不同环境样本中的16s rRNA进行测序,可以了解微生物的多样性、组成和分布情况,包括土壤、水体、肠道等各种环境。
2. 比较微生物群落结构:通过对不同样本的16s rRNA进行测序,可以比较它们的微生物群落结构,了解其相似性和差异性,从而揭示微生物在不同环境中的生态功能。
3. 诊断和监测微生物感染:通过对临床样本(如血液、尿液、粪便)中的16s rRNA进行测序,可以快速准确地鉴定微生物感染的种类和数量,对临床诊断和治疗具有重要意义。
4. 研究微生物与宿主的相互作用:通过对宿主体内微生物的16s rRNA进行测序,可以了解微生物与宿主的相互作用关系,从而揭示微生物对宿主健康的影响机制。
三、16s测序方法在生物医学领域中的作用1. 揭示微生物与人类健康的关系:通过对人体不同部位的16s rRNA进行测序,可以了解微生物与人类健康的关系,包括肠道微生物与肠道疾病、皮肤微生物与皮肤疾病等。
2. 个体化医学研究:通过对个体的16s rRNA进行测序,可以了解个体微生物组的特点,从而为个体化医学提供基础数据,包括个体化诊断、个体化治疗等。
3. 新药研发和药物安全性评价:通过对微生物的16s rRNA进行测序,可以了解微生物对药物的代谢和耐药性,为新药研发和药物安全性评价提供参考依据。
16s测序方法是一种重要的高通量测序技术,可以用于研究微生物的遗传多样性、微生物群落结构、微生物感染的诊断和监测,以及微生物与宿主的相互作用关系。
细菌16SrDNA测序鉴定一、细菌总DNA提取二、16SrDNA的扩增1、以单菌落或菌悬液为模板的16SrDNA扩增体系25ul体系Buffer(含有Mg2+):2.5 uldNTP:2.5 ul:3,-primer:0.5ul5,-primer:0.5ul模板:1个单菌落(或1 ul在LB培养液中37℃,12h的菌悬液)吐温20:2ulTaq酶:0.3水:16.7(或15.7ul)细菌16S rDNA的通用引物为Pf-AGAGTTTGATCCTGGCTCAG和Rf-TACCTTGTTACCACTT许敬亮博士论文66页:扩增采用通用的引物,其正向序列为:5'-AGAGTTTGATCCTGGCTCAG-3',反向互补序列为: 5'-TTCAGCATTGTTCCAT-3'。
黄婷婷博士论文70页:5’端:5'-AGAGTTTGATCCTGGCTCAG-3'(Escherichia coli bases 8 to 27),3’端:5'-TACCTTGTTACGACTT-3'(Escherichia coli bases 1507 to 1492)。
2、以总DNA为模板的16SrDNA扩增体系25ul体系Buffer(含有Mg2+):2.5 ul (购买)dNTP:2.5 ul:3,-primer:0.5ul5,-primer:0.5ul模板:1 ulTaq酶:0.3(或0.2ul)双蒸水:17.7(或17.8ul)3、PCR反应条件94℃,5min;95℃,变性30s;50℃~52℃(可根据扩增情况进行适当调节),退火30s;72℃,延伸1min;30个循环;72℃,延伸15min;10℃(or4℃),保温10min(or nh)。
4、检测扩增效果取1~3 ul扩增产物,适量load buffer,以DL2000为Marker,上0.75%琼脂糖凝胶跑电泳(电压挑低些80~100,胶长些,利于各扩增产物分离及回收切胶)注:扩增条件摸索时每个模板只扩1管即可三、16SrDNA的回收扩增出的每一管16SrDNA经0.75%琼脂糖凝胶电泳检测后,将效果好的各管16SrDNA合并使达到60~100ul样量,100ul16SrDNA+10ul溴酚蓝跑电泳回收胶(电泳液需换成新鲜的,DL2000为Marker),EB染色后在紫外灯下切下16SrDNA 条带放入1.5ml离心管中(离心管事先称重),按DNA凝胶回收试剂盒操作说明回收16SrDNA,取回收16SrDNA 2ul(Dl2000为Marker)跑电泳观察浓度(亮度)与纯度(条带数与位置)。
根据16S预测微生物群落功能最全攻略最近,越来越多的证据表明:自然环境(如海洋、土壤等)中的微生物群落功能(functional)组成而非物种(taxonomic)组成与环境因子密切相关;换言之,相似环境中的微生物群落功能更相似,而行使功能的微生物物种组成可能差异较大(Gibbons et al. 2017; Louca et al. 2016; Nelson et al. 2016)。
这说明,除了揭示环境中有哪些微生物之外,揭示微生物群落功能轮廓尤为重要。
目前,微生物生态研究中常用的揭示微生物群落功能的方法有宏基因组测序、宏转录组测序、宏蛋白组测序、宏代谢组分析等。
这些方法优点突出,能较准确、真实地反映不同层面的微生物群落功能特征。
但其价格较高,一般实验室难以承受大批量样本的实验;另外,数据量巨大,数据处理也是难点。
有没有较经济、又适合大样本的方法呢?有!那就是基于marker基因扩增子高通量测序的功能预测。
上次卢瑟菌给大家介绍了根据真菌ITS序列预测真菌群落功能的工具——FUNGuild (点击了解更多)今天,卢瑟菌就和大家介绍基于原核16SrDNA高通量测序结果对微生物群落功能(function)或表型(phenotype)进行预测的四种方法——PICRUSt、Tax4Fun、FAPROTAX及BugBase。
1PICRUSt简介PICRUSt全称为Phylogenetic Investigationof Communities by Reconstruction of Unobserved States,由Langille等人于2013年开发,文章发表在Nature Biotechnology上(Langille et al. 2013)。
它是最早被开发的基于16S rRNA基因序列预测微生物群落功能的工具,包括在线版(/galaxy/root?tool_id=PICRUSt_normalize)和基于MacOS X或Linux系统的下载安装版(/picrust/install.html#install)。
菌群16s测序解读-回复什么是菌群16s测序?菌群16s测序是一种利用高通量测序技术对微生物群体的16s rRNA基因片段进行测序的方法。
16s rRNA基因是细菌和古菌特有的基因,其序列在不同的菌种之间存在一定的差异性,因此可以作为菌类的指纹序列。
通过对16s rRNA基因的测序及分析,可以了解菌群的物种组成、丰度及群落结构等信息。
这种测序方法被广泛应用于微生物学研究、环境监测、生态学等领域。
菌群16s测序的步骤:1. 样品采集和DNA提取:样品可以是土壤、水体、消化道内容物等,根据研究目的不同选择不同的采样方法。
提取的DNA作为测序的模板。
2. PCR扩增:根据16s rRNA基因序列的保守区域设计引物对DNA进行PCR扩增。
由于16s rRNA基因片段较短,可以通过引物设计选择特定的靶标区域,如V3-V4区域。
3. 片段纯化:扩增后的DNA片段通过凝胶电泳或其他方法分离纯化,去除杂质和引物。
4. 样品索引:为了进行多个样品的混合测序,需要为每个样品设计独特的DNA序列索引。
索引序列在PCR扩增时与待测序的片段一起扩增。
5. 测序:将纯化的DNA片段进行高通量测序,常使用Illumina MiSeq 或Ion Torrent平台进行。
6. 数据处理与分析:得到测序数据后,需要进行数据处理和分析。
首先对测序数据进行质控和过滤,去除低质量的序列和嵌合体。
然后使用专业的生物信息学软件对16s rRNA基因片段进行分类、物种注释和群落分析。
7. 结果解读与应用:通过菌群16s测序可以获取菌群的物种组成和丰度分布,可以进一步探究微生物群体在不同环境中的分布规律、生态功能和相互作用等问题。
这些研究结果对生态学、环境科学、农业、医学等领域都具有重要的意义。
菌群16s测序的优势:1. 高通量:菌群16s测序利用高通量测序平台,可以在短时间内获得大量的序列数据。
2. 物种注释准确性高:16s rRNA基因在不同菌类中具有高度保守性,通过对该基因片段进行测序,可以准确地对菌群进行分类和物种注释。
微生物16S测序数据的正确打开方式16S rRNA基因测序(也称16S rDNA测序)是最常用的菌群多样性分析的手段。
对于新手,如果收到一份不讲“人话”的16S测序分析报告,很快就会被各种生态学术语、各种指数、各种分析方法弄晕。
7个问题串起16S测序的核心结果怎么办?用你的研究逻辑来梳理16S测序数据(图1)。
简单地说,做16S测序是为了鉴定样本中的微生物(细菌)群组成,找微生物群与疾病或表型的相关性。
详细地说,1)首先想了解在不同组样本中各有哪些微生物存在和丰富度(对应于菌群鉴定和α多样性分析);2)接着想看不同样本组间微生物群组成是否存在差异(对应于β多样性分析);3)如果是,那么就有必要找出引起不同组样本微生物群差异的关键菌。
如果不是,那说明微生物群比如肠道菌群与疾病或表型可能并不相关(基于已有的研究,这种可能性比较小);4)找到了关键菌,在临床上,很自然会想到,这些(个)关键菌是否可以作为Biomarker(对应于疾病诊断模型构建),比如用于区分糖尿病前期患者与健康组的标志物;5)以及这些(个)菌是否与临床指标具有相关性(对应于菌群与临床指标的相关性分析);也会进一步想到,既然不同组的微生物群落存在差异,又与疾病具有相关性,6)那么这些菌群是如何影响宿主的,可能参与了哪些代谢途径(对应于菌群基因功能预测);7)这些预测到的菌群功能是否与疾病有关,通常是肯定的。
最后把这些结果整合起来分析,可以初步得出菌群组成的变化是如何与疾病或表型相关的。
顺着上述7个生物学问题来看16S测序结果,你会轻松拨开迷雾,直达核心结果。
图1 7个问题串起16S测序的核心结果6张图就够发菌群与疾病相关性文章编者对2019发表的数十篇以16S测序为主的肠道菌群与疾病关系研究文章(IF 5至10分)的内容进行了分析和归纳,发现大部分文章的Results部分都是由图1所列的核心结果组成。
以联川生物医学16S测序报告为例,具体讲解16S测序文章中的核心结果及其对应的图表。
微生物16S测序数据的正确打开方式16S rRNA基因测序(也称16S rDNA测序)是最常用的菌群多样性分析的手段。
对于新手,如果收到一份不讲“人话”的16S测序分析报告,很快就会被各种生态学术语、各种指数、各种分析方法弄晕。
7个问题串起16S测序的核心结果怎么办?用你的研究逻辑来梳理16S测序数据(图1)。
简单地说,做16S测序是为了鉴定样本中的微生物(细菌)群组成,找微生物群与疾病或表型的相关性。
详细地说,1)首先想了解在不同组样本中各有哪些微生物存在和丰富度(对应于菌群鉴定和α多样性分析);2)接着想看不同样本组间微生物群组成是否存在差异(对应于β多样性分析);3)如果是,那么就有必要找出引起不同组样本微生物群差异的关键菌。
如果不是,那说明微生物群比如肠道菌群与疾病或表型可能并不相关(基于已有的研究,这种可能性比较小);4)找到了关键菌,在临床上,很自然会想到,这些(个)关键菌是否可以作为Biomarker(对应于疾病诊断模型构建),比如用于区分糖尿病前期患者与健康组的标志物;5)以及这些(个)菌是否与临床指标具有相关性(对应于菌群与临床指标的相关性分析);也会进一步想到,既然不同组的微生物群落存在差异,又与疾病具有相关性,6)那么这些菌群是如何影响宿主的,可能参与了哪些代谢途径(对应于菌群基因功能预测);7)这些预测到的菌群功能是否与疾病有关,通常是肯定的。
最后把这些结果整合起来分析,可以初步得出菌群组成的变化是如何与疾病或表型相关的。
顺着上述7个生物学问题来看16S测序结果,你会轻松拨开迷雾,直达核心结果。
图1 7个问题串起16S测序的核心结果6张图就够发菌群与疾病相关性文章编者对2019发表的数十篇以16S测序为主的肠道菌群与疾病关系研究文章(IF 5至10分)的内容进行了分析和归纳,发现大部分文章的Results部分都是由图1所列的核心结果组成。
以联川生物医学16S测序报告为例,具体讲解16S测序文章中的核心结果及其对应的图表。
(1)菌群鉴定与物种分布采用最新的QIIME 2分析流程,并使用更严谨的DADA2算法对扩增子数据进行去噪,相当于以100%的相似性聚类(取代传统的OTU聚类),仅对低质量序列进行去除和校正等,获取扩增子序列变异,然后去冗余,即得到feature(特征)数据。
将feature数据和16S数据库(如SILVA、NT-16S)进行序列比对,可以对样本中检测到的细菌从界(Kingdom)、门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)多个分类学层级进行物种鉴定和注释。
然后根据各个分类层级上的物种相对丰度以物种分布堆叠图(图2A-C,以门水平为例)来直观展示。
图2A 物种分布堆叠图图2B 物种聚类堆叠图图2C 物种相对丰度Heatmap在联川医学16S测序报告中,会提供上述三种主流的物种分布堆叠图,你可以选择其一使用。
在图2A、B中,不同颜色的柱子对应不同的物种,柱子的长短代表该物种所占比例的大小。
图2B中左侧采用Bray-Curtis距离法分析样本间菌群组成的相似性并进行聚类。
图2C中展示了不同细菌物种在不同样本中的相对丰度情况,颜色越红,丰度越高,颜色越蓝,丰度越低。
(2)菌群α多样性α多样性是度量单个样本内有多少种微生物物种,以及每个物种所占比例的指标。
在报告中,采用5种常用指数来度量α多样性:Observed species和Chao1反映样本中物种丰富度,但不考虑每个物种的占比情况(均匀度);Shannon 和Simpson反映物种的丰富度和均匀度;Good’s Coverage 反映样本的测序深度。
使用Wilcoxon秩和检验对上述各个指数的样本数据进行分析,筛选出各样本组比较中显著差异的α多样性指数并绘制小提琴图(图3)。
图3 小提琴图表示α多样性指数小提琴图集合了箱形图和密度图的特征。
上图以Good’s Coverage为例,左上角给出了差异分析使用的检验方法和计算得到的p值。
当p<0.01,表示差异极显著;当p<0.05,表示差异显著;当p>0.05,则表示无显著性差异。
(3)菌群β多样性β多样性是度量不同样本间菌群组成的相似度大小的指标,即关注各样本间的菌群组成差异。
α多样性关注样本自身的菌群丰富度和均匀度,而β多样性关注样本间的菌群组成与分布的差异。
只有当样本(组)间菌群组成存在差异,才有可能进一步探讨菌群失调与疾病的关系。
在报告中,采用主流的PCA、PCoA、NMDS、ANOSIM、Adonis、UPGMA等多种分析方法来考察和区分样本间的菌群组成差异(图4,以PCoA为例)。
图4 PCoA分析结果图上图中每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
左图是基于Unweighted UniFrac的PCoA分析结果,右图是基于Weighted UniFrac的PCoA分析结果。
在这个例子中,采用Weighted UniFrac的PCoA分析更能把不同组的样本区分开来,且p值<0.01,具有显著统计学差异。
需要说明的是,PCoA分析本身是没有p值计算的,p值来自于ANOSIM 分析的结果。
在绘图时,把p值加入了PCoA图中。
由于每个项目的实验设计和样本菌群组成差异巨大,无法预先知道哪种β多样性分析方法是将样本间菌群差异区分开的最优方法。
因此,在报告中提供了多种β多样性分析方法和产生的图片,在撰写文章时,你只需要从中选出最能解释生物学问题的图片用在文章中即可(通常展示一个或者两个β多样性分析结果)。
(4)显著差异菌群分析通过β多样性分析,可以确定不同组间的微生物群落是存在差异的,接着需要进一步找出哪些菌(群)引起了组间的群落差异。
只有找出核心菌(群),才能明确下一步的研究方向。
在报告中,使用目前在文献中高频出现的方法——LEfSe(Linear discriminant analysis Effect Size),来做菌群差异分析,寻找生物标志物(Biomarker)。
该方法综合了统计学上的差异分析和该差异物种对分组结果的影响力得分值,同时强调了统计学意义和生物相关性。
LEfSe分析结果图,通常包括进化分支图(图5A)和LDA值分布柱状图(图5B)。
需要说明的是,联川不仅提供LEfSe筛选差异菌群,还提供其他多种方法,如随机森林分析(图9)、秩和检验等。
图5A LDA值分布柱状图上图主要展示了LDA score大于预设值的显著差异物种(less_strict设为2;more_strict 设为4),即具有统计学差异的Biomarker;柱状图的颜色代表各自的组别,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
图5B 进化分支图上图中,小圆圈: 图中由内至外辐射的圆圈代表了由界(单个圆圈)至属(或种)的分类级别。
不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈直径大小与相对丰度大小呈正比。
颜色:无显著差异的物种统一上黄色,差异显著的物种Biomarker跟随组别进行上色,红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应。
(5)菌群标志物预测能力评估受试者工作特征(ROC)曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。
在报告中,通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定哪种菌(群)具有最佳的诊断价值(图6)。
图6 菌群标志物ROC曲线分析上图以灵敏度为纵坐标,特异度为横坐标绘制曲线。
ROC 曲线越靠近左上角,试验的准确性就越高。
若AUC值为1.0,反映出对两个群组的完美区分,且不存在预测误差。
若AUC 值在1.0和0.5之间,在AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC 在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。
AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。
AUC<0.5不符合真实情况,在实际中极少出现。
(6)菌群基因功能预测因为菌群功能预测软件PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)的出现,研究者能进一步基于16S测序数据预测菌群可能参与的代谢通路(尽管并没有测定菌群基因信息),以便能初步讨论菌群组成变化与疾病或表型是如何关联在一起的。
在联川报告中,使用最新的PICRUSt 2,相比上一版,用于预测的参考基因组数据库已扩展超过10倍,可以获得包括COG,EC,KO,PFAM,TIGRFAM等数据库对菌群的基因功能注释结果。
然后,再使用STAMP软件进行差异分析,得到在不同样本组中显著差异的菌群基因功能(图7,以pathway结果为例)。
如果要系统研究菌群携带的基因及其功能,则应该做宏基因组测序。
图7 PICRUSt 2预测菌群基因功能上图中比较了不同组菌群的KEGG pathway,并筛选出具有显著性组间差异的 pathway。
左边柱状图代表某代谢通路的丰度分别占两组样本中所有代谢通路的百分比,右边为corrected p值。
至此,一篇医学微生物组16S测序文章的主要结果和图表就基本齐备了。
当然,完整的医学16S测序报告还包括更多内容(图8),而且16S测序数据还有许多扩展性以及个性化的分析图表(图9),联川会根据研究者的具体需求来提供。
图8 联川生物医学16S测序报告内容图9 16S测序数据个性化分析图表(部分)值得参考的菌群与疾病关系的研究思路甲状腺癌患者的肠道菌群和代谢谱的变化(IF4.982)发表期刊:International Journal of Cancer影响因子:4.982发表时间:2019研究内容:肠道菌群、代谢谱与甲状腺癌之间的关系样本数量:16S测序:30例甲状腺癌(TC)vs 35例健康对照(HCs);代谢组:15例TC vs 15例HCs样本类型:粪便样本实验方法:16S rRNA基因测序+非靶向代谢组检测实验设计:所有受试者均为汉族,出生于中国东北地区,饮食结构相似研究思路:采用目前主流的肠道菌群与疾病关系的研究策略,联合肠道菌群16S测序和粪便代谢组(极具性价比的检测组合),并结合临床指标来一起讨论,给出甲状腺癌患者的肠道菌群特征和代谢谱,潜在的疾病标志物,以及肠道菌群影响肿瘤发生发展的潜在途径。