Chemoffice_2008_使用教程
- 格式:pdf
- 大小:1.39 MB
- 文档页数:20

操作简易的化学画图软件----ChemWindow一、分子式、简式的书写:①按下键盘上的Caps Lock 键,点击键后再点击Label键,在文档中输入H2SO4 即可得到:;同理可写出有机分子式,在必要的时候加上括号:如输入CH3(CH2)3CH3时候可得到:②检查:运行“Other”菜单下的“Check Chemistry”和“Check Spelling”检查结构式,保证正确性。
例如:写一个错误的戊烷的分子式:,选中这个分子式,运行Check Chemistry(或按下F10键),弹出以下对话框:语句“The valence is not correct.”意思是:化合价不正确。
若分子式用于特殊场合,点击“Ignore”或“Ignore All”按钮可以忽略错误,即保持错误的状态。
二、离子式的书写:数字在英文字母之前或之后输入,系统默认为下标;但数字在“-”或“+”之后为上标;先输入数字,后输入“-”或“+”,则数字为下标。
如硫离子的书写:输入“SO42-”显示为“”,这显然不是我们需要的,正确的操作是先输入“SO4-2-”后再删除前一个“-”即可得到。
切换到中文输入法也可以输入中文,用按钮可设置上下标:如①在大写状态下,点击按钮,写出硫酸的分子式:②当光标停在下标“4”时,点击下标按钮,输入(浓)“Caption”工具是另外一种文字输入工具,但不能识别上下标,只要用于中文标题或者大写字母的输入。
三、其他1.高聚物先写出高聚物的单体,再单击中括号,在文档中拖放出一个中括号,适当缩放,在右下角的方框中填入“n”最后将单体放入:如聚氯乙烯的书写:2.电子式右击文档空白得到的菜单中,最下面一列是专门用于绘制电子式的:以Na2O2电子式的绘制为例:①选择,经过适当缩放后复制一份,将两个点并排,组合②用文字工具添加两个O,并将电子移到相应位置:③点击文字工具,选择“·”,输入两个“×”,并移动到相应的位置④点击,在文档中拖放出一个中括号,适当缩放,最后写出两个钠离子,如图:3.电子云和分子轨道的绘制方法同“电子式”的绘制:在菜单中找出合适的电子云模型,先画出雏形后,再拉伸放大。
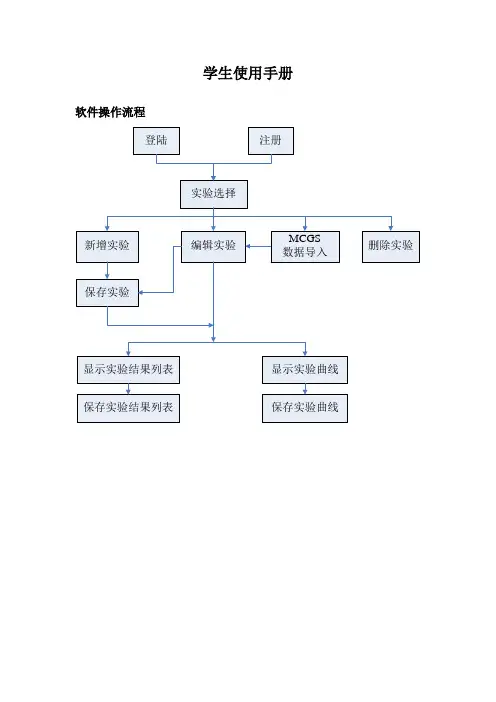
学生使用手册软件操作流程1.登陆与注册身份确认进入软件登陆画面,首先选择登陆身份,学生用户请选择“学生”注册第一次使用本软件的学生需要先进行注册,以便在系统中留下有关信息。
方法是点击[注册]按钮,弹出注册对话框,如图所示。
在对话框中输入学号等有关信息,其中“学号”、“密码”、“密码确认”是必填信息,其他为选填信息。
点击[确定],在确认无误后保存。
登陆已经注册的学生用户,在下次进入登陆画面的时候,可以直接输入学号和密码,并点击[登陆]按钮进入系统2.实验原始数据处理a)新增实验实验原始数据输入学生可以点击工具栏上新增实验按钮,或选择菜单【实验原始数据】→【新增实验】以清空“实验原始数据表”,然后即可在表上输入实验原始数据。
按钮[插入行]和[删除行]用于在输入时插入一个空白数据行和删除一个数据行。
登陆后首次进入主界面时,系统自动处于新增实验状态,可以直接输入实验数据。
实验保存实验数据输入完毕后必须进行保存:点击工具栏上实验保存按钮,或选择菜单【实验原始数据】→【保存实验】,弹出如图所示对话框。
从装置下拉式列表中选择实验所使用的装置,并点击[保存]按钮确定。
实验保存后就可以查看实验结果和曲线,具体操作见“实验结果显示与保存”说明。
b)编辑实验实验打开要对已有的实验进行编辑或查看实验结果和曲线需要先打开实验:点击工具栏上编辑实验按钮,或选择菜单【实验原始数据】→【编辑实验】,弹出如图所示对话框,从实验装置下拉列表中选择装置,然后在实验列表中选择所要打开的实验,点击[打开]按钮打开实验。
实验类型中,“基本型”表示用户手动输入数据的实验,“数字型”表示从MCGS导入数据的实验。
实验数据编辑修改实验打开后,可以直接在“实验原始数据表”上对实验数据进行修改。
要保存修改后的结果,请点击工具栏上实验保存按钮,或选择菜单【实验原始数据】→【保存实验】,确认之后保存。
另外,实验打开后可以对该实验的结果和曲线进行查看和保存,具体操作见“实验结果显示与保存”说明。

office中操作化学结构式1.引言在科学研究和学术写作中,经常需要插入化学结构式来更加清晰、准确地表达化学概念和实验结果。
M ic ro sof t Of fi ce的办公软件套件中,如W or d、Ex ce l和P o we rP oi nt,提供了丰富的功能和工具来操作和插入化学结构式,方便用户进行科研和学术交流。
本文将介绍如何在O f fi ce中使用这些功能,快速而准确地插入和编辑化学结构式。
2.使用Wor d插入化学结构式2.1借助化学插件为了在Wo rd中插入化学结构式,我们可以使用一些强大的化学插件,如C he mD ra w、Ch emO f fi ce和C he mD raw P ro fe ss io na l等。
这些插件提供了丰富的绘图工具和化学符号库,能够满足我们的各种化学结构绘制需求。
安装插件后,我们可以通过以下步骤插入化学结构式:1.打开Wo rd文档,定位到要插入化学结构式的位置;2.点击“插入”选项卡中的“化学式”按钮,选择需要的结构式类型;3.在绘图工具中绘制结构式,或者从符号库中选择现有的化学结构;4.点击“确定”按钮,插入化学结构式到W or d文档中。
2.2使用内置公式编辑器除了使用化学插件,我们还可以使用W ord内置的公式编辑器来插入化学结构式。
虽然功能相对简单,但对于一些简单的化学结构式绘制还是非常实用的。
以下是使用内置公式编辑器插入化学结构式的步骤:1.打开Wo rd文档,定位到要插入化学结构式的位置;2.点击“插入”选项卡中的“公式”按钮,进入公式编辑模式;3.在编辑器中输入化学结构式的代码,使用化学符号和数学运算符等;4.通过编辑器提供的工具和选项,调整结构式的风格和排版;5.点击“确定”按钮,插入化学结构式到W or d文档中。
2.3使用快捷键和符号插入对于一些简单的化学结构式,我们还可以通过Wo rd内置的快捷键和符号插入功能,快速地插入化学结构式。


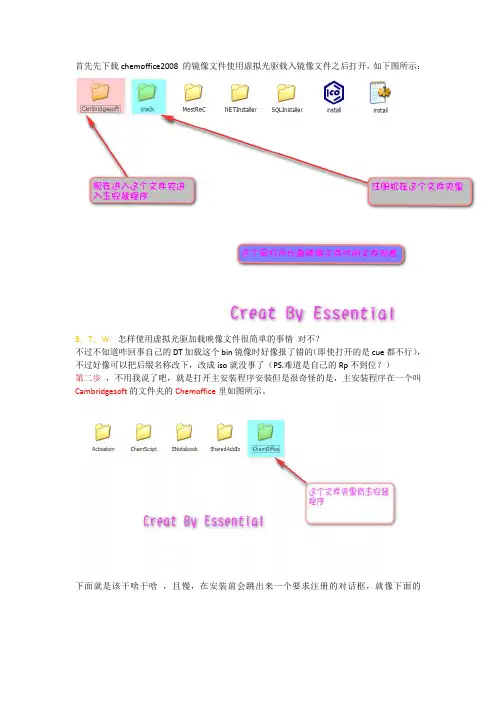
首先先下载chemoffice2008 的镜像文件使用虚拟光驱载入镜像文件之后打开,如下图所示:
B.T.W.怎样使用虚拟光驱加载映像文件很简单的事情对不?
不过不知道咋回事自己的DT加载这个bin镜像时好像报了错的(即使打开的是cue都不行),不过好像可以把后缀名称改下,改成iso就没事了(PS.难道是自己的Rp不到位?)
第二步,不用我说了吧,就是打开主安装程序安装但是很奇怪的是,主安装程序在一个叫Cambridgesoft的文件夹的Chemoffice里如图所示。
下面就是该干啥干啥,且慢,在安装前会跳出来一个要求注册的对话框,就像下面的
现在打开注册机(呵呵,注册机在那里?大家看第一幅图的说明)就像是这样
现在就按找第三幅的提示办~!Creat By Essential。

计算化学实验Chem3D软件指南Chem3D开始:点击桌面“Chem3D Ultra 10.0”图标Chem3D一览菜单和工具栏区组成区坐标区光谱区输出结果区构象区2D分子区3D绘图区Chem3D详解菜单和工具栏区文件打开输入打印设置编辑拷贝粘贴修改选择显示工具结构测量旋转饱和修正重叠计算界面结果密度面轨道电势面分子面电影旋转轨迹数据库窗口排列帮助说明例子Chem3D详解文件新分子模型样本分子打开文件中的分子结构保存Chem3D详解编辑撤消操作重复操作清除当前分子Chem3D详解视图分子显示方式显示取向工具栏分子组成ChemDraw直角坐标Z-矩阵测量列表参数表输出区构象区光谱显示区文件输入原子类型MM2类型Chem3D详解结构测量列表分子位置和取向设置Z-matrix饱和价键(加氢)自动调整分子结构重叠分子对接Chem3D详解计算模块计算结果管理停止计算二面角计算(构象搜索)EHMO计算电荷轨道MM2分子动力学模块分子性质计算模块GAMESS计算界面Gaussian计算界面Jaguar计算界面Mopac计算界面Chem3D详解计算模块二面角计算(构象搜索)扫描一个二面角扫描两个二面角二面角计算(构象搜索) 扫描一个二面角二面角计算(构象搜索) 扫描二个二面角构象过渡态Chem3D详解计算模块EHMO电荷分子面总电子密度面分子轨道Chem3D详解计算模块EHMO电荷Chem3D详解计算模块EHMO总电子密度面等值面图isoval=0.01Chem3D详解计算模块EHMO分子轨道等值面图isoval=0.01MM2分子动力学模块Chem3D详解计算模块运行MM2输入文件优化构型(能量极小化)分子动力学性质计算打印力场参数MM2分子动力学模块Chem3D详解计算模块力场参数M2 Constant Value QualityCubic stretch constant-2.000 4Quarticstretch constant2.333 4X-B,C,N,O-Y Stretch-Bend interaction force constant0.120 4 X-B,C,N,O-H Stretch-Bend interaction force constant0.090 4 Sexticbending constant (* 10**8)7.000 4Cutoff distance for charge/charge interactions35.000 4Cutoff distance for charge/dipole interactions25.000 4Cutoff distance for dipole/dipole interactions18.000 4Cutoff distance for vander Waalsinteractions10.000 4MM2 Atom RadiusEps WeightReduct Lone Pairs Quality 5 1.500 0.047 1.008 0.915 0 4 2 1.940 0.044 12.000 0.000 0 4 Bond KS Bond Length Dipole Quality2-5 4.600 1.100 0.000 42-2 9.600 1.337 0.000 4Angle KB XR2 XRH XH2 Quality 2-2-5 0.360 120.0 120.5 0.0 42-2-2 0.430 120.0 0.0 0.0 4Atoms Force Constant Quality2-2 0.050 42-5 0.050 4Torsional V1 V2 V3 Quality5-2-2-5 0.000 15.000 0.000 42-2-2-5 0.000 9.000 -1.060 42-2-2-2 -0.930 8.000 0.000 4PiAtom Electron Ionization Repulsion Quality 2 1 -11.160 11.134 4PiBond DForce DLength Quality2-2 4.600 0.166 43, 4次键伸缩常数键伸缩-弯曲作用参数截断距离LJ非键参数键伸缩参数键弯曲(角)参数键扭转参数π键(共轭)修正参数MM2分子动力学模块Chem3D详解计算模块优化构型(能量极小化)收敛要求: 最小均方根梯度(力)输出结果能量分解总能量MM2分子动力学模块Chem3D详解计算模块分子动力学模拟步长结构输出间隔轨迹步数升温速率目标温度参数一览MM2分子动力学模块Chem3D详解计算模块分子动力学模拟暂停/开始停止MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果时间(10-15秒)总能势能温度MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果分子平动能= 0分子转动能= 0非平衡平衡态MM2分子动力学模块Chem3D详解计算模块分子动力学模拟输出结果总能守恒!温度控制!MM2分子动力学模块Chem3D详解计算模块π键键级能量Chem3D详解计算模块分子性质计算模块Chem3D详解计算模块GAMESS优化构型优化过渡态分子性质频率分析红外光谱NMR光谱创建输入文件查看输出结果文件运行输入文件Chem3D详解计算模块GAMESS作业类型方法基组波函数类型基组极化函数基组扩散函数基组指数优化方法限制性部分优化坐标自旋多重度= 单电子数+1净电荷Chem3D详解计算模块GAMESS高级设置-1最大SCF循环次数优化最大步数收敛要求(力)温度(仅用于热力学计算)溶剂模型溶剂参数初始猜测方法分子对称性点群内存要求Chem3D详解计算模块GAMESS性质偶极电子密度静电势Lowdin电荷和布居分析Mulliken电荷和布居分析电子势能电子总能量电子动能Chem3D详解计算模块GAMESS计算方法激发态从头算理论密度泛函理论基态Chem3D详解计算模块GAMESS等电子密度面(0.001)HF/STO-3G IR光谱苯甲醇NMR光谱Mopac计算界面Chem3D详解计算模块优化平衡构型优化过渡态分子性质IR光谱Mopac计算界面Chem3D详解计算模块作业类型方法波函数优化方法溶剂模型限制性优化收敛要求坐标Mopac计算界面Chem3D详解计算模块电荷偶极静电势生成焓超精细耦合常数电离能电荷溶剂介电常数Chem3D详解分子面模块分子面探测半径显示模式颜色模式分辨率选择分子轨道等值面值溶剂面总电子密度面总自旋密度面静电势面分子轨道清除所有面Chem3D详解分子面模块SA面总电子密度面isoval=0.01总电子密度面/静电势isoval=0.01静电势isoval=1Connolly面HOMO轨道LUMO轨道甲醇Chem3D详解工具栏新分子打开保存拷贝剪切粘贴恢复重复印模型背景特效红蓝镜像坐标原子符号原子编号残基全屏电影播放选择平移旋转缩放移动选中键型清除饱和调整符输入擦写分子面探测半径显示配色分辨率分子轨道等值状态优化旋转停止Chem3D详解文件格式晶体结构文件Gaussian输入输出文件cif文件pdb文件(蛋白质)Chem3D详解晶体结构文件有机分子晶体: CCDC数据库, 草酸晶体Pbca空间群(61)OXALAC06.datChem3D详解晶体结构文件无机晶体: ICSD, AMCSDAmerican Mineralogist Crystal Structural Database http://rruff.geo.arizona. edu/AMS/amcsd.php不能正确处理周期性的体系!!Chem3D详解生物大分子pdb文件(蛋白质)RSCB PDB蛋白质晶体结构数据库/pdb/home/home.do1AAQChem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件需要把Gamess输出文件修改一下才能用Jmol5.0正确读出!将这些含“REDUCED MASSES”的行全部删除! 然后保存退出此处只留一个空行!Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件旋转分子缩放平移选择测量轨迹/频率Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件打开修改后的GAMESS输出文件*.out打开“extras”→“vibrate ...”Chem3D详解振动模式用JMOL显示GAMESS的频率分析输出文件选择频率观察“绕CO键的内转动”。

ChemDraw 的操作以下技巧是近期频繁使用ChemDraw中总结出来的,适用版本是11.0(即ChemOffice 2008).希望对搞化学的朋友有所帮助。
1. 把默认的页数设置为多页。
如果你经常在一个页面中要画很多分子式导致一页放不下,ChemDraw不会象Word一样能够自动给你增加页数,要手动设置:点击“File--Document Settings--Documents size"中设置为你所需要的页数,比如说 3 pages. 但如果这样设置,仅对当前文档有效。
要想以后新建的文件都是如此,办法是:新建一个空文档,不要输入任何内容,然后设置文档为3页(或任意你所要的页数),再点击“File--Save Style Sheet”,在出现的“另存为”对话框的左左下角点击“go to chemdraw item"按钮,转入”ChemDraw Item “文件夹,再选择文件类型为“CDS类型”,保存,覆盖掉原来这个目录下的New Document.cds。
再启动ChemDraw就可以了。
道理:ChemDraw每次以这个ChemDraw item目录下的New Document.cds为模板启动新建文档的。
当然,你可以作更多的格式修改,如法炮制即可。
2. 快速编辑结构图的几个办法2.1 当方块形的光标出现在键上时,按1可以自动变为单键,2自动变为双键,3自动变为叁键。
键盘与鼠标配合使用,挺方便!2.2 当方形块的光标出现是原子上时,按1,2,3分别修改为正丁基、仲丁基、叔丁基,按4可以修改为苯基Ph,等等。
按小写s(即没有同时按下Shift或启动大写锁定),原子变为S,按c原子变为显示出来的碳原子,按m出现Me即甲基的缩写,e出现Et乙基的缩写。
不需要双击进入编辑状态就可以修改,十分方便。
自己多试试就知道出现的是什么。
还可以根据你的需要进行修改,详见第三条"自定义快捷键"。

操作简易的化学画图软件----ChemWindowChemWindow是一个能绘出各种结构和形状的化学分子结构及化学图形的免费软件。
最新版本是 6.0。
ChemWindow为软件,可在相关网站下载,如在中国化学软件网() 及中国化学化工网() 上都能下载Chemwindow 6.0 标准版。
ChemWindow 6. 0 主要有4个功能模块组成:(1) 分子结构绘制,包括化学键绘制、各种环结构绘制、化学反应机理的表示、相应文字说明、电子云表示等功能;(2) 化学实验装置图绘制,包括各种接口的玻璃仪器的图形模板;(3) 化工工艺流程图绘制,具有各类化工设备的示意图形库;(4) 提供大量的各种复杂分子结构通用模板,直接提取应用。
所有功能模块采用丰富的工具栏选择,界面友好,使用便捷。
如图所示为Chemwindow 6.0的界面:新安装时界面上的一些工具是没有打开的,公欲善其事,必先利其器,因此可以根据自己的习惯定制不同的风格,方法如下:点击“view”菜单,选择其中的一项,选中的项目在菜单中用“√”显示。
但不必打开所以的选项,一来打开过多小窗口影响速度,二来按住带有红色三角形的按钮即可看到“view”菜单中的全部按钮;另外,在文档中的空白处任意右击也可以得到这些常用的按钮。
常用按钮的选择如图所示:View菜单右击得到的菜单首先来认识一些常用按钮:Standard Tools(标准工具):其中:用于选取绘图区中的某一范围;用于选取整体中的部分;为文字工具,用于书写分子式等,但有所区别;:键线工具,用于绘制单键,双键或其他键线在“Bond Tools”菜单或者右击即可看到::各类环状;按住键可以获得更多环状结构物质;如::按住这些工具均可得到和右击文档空白得到一样的菜单。
为编辑图形的按钮,功能分别为,将选中的图形:置于顶层、置于底层、组合、拆分、自由旋转、水平旋转、垂直旋转、对齐。
一、简单有机结构式的绘制:法一:利用“Other”菜单中的“Make Stick Structure”或“Make Labeled Structure”进行转换:如戊烷的键线式的绘制:①首先写出分子式:②用选择工具选中分子式,点击“Other”菜单中的“Make Stick Structure”,得到戊烷的键线式:③选中键线式,点击“Other”菜单中的“Make Labeled Structure”则又可以得到分子式。

使用 ChemDraw 小小技巧 1. 点击 File 选择 Apply Document Settings from 中的 ACS Document 1996,这样你所 画的分子式,就是美国化学会杂志的格式,这种格式的优点是图较小,特别合适在 Word 中使用,推荐使用。
点击 File 选择 Preferences 弹出对话框,点击 Open/Save 项,将 Open files 的 Default Style Sheet 设置为 ACS Document 1996,其通常在如下 目录:C:\Documents and Settings\All Users\Application Data\CambridgeSoft\ChemOffice2008\ChemDraw\ChemDraw Items\ACS Document 1996.cds,这样当你打开 Chemdraw 时,缺省的设置就是 ACS Document 1996。
要 做幻灯片的话,可选择 Slides。
2. 按 Ctrl+L 组合键,可以固定结构式的键长,也就是所画线段的长度,这样就可 避免有的键长有的键短的现象,将鼠标放在一根键的末端或结点上,当有蓝色框出 现时(没出现则画出的键不跟此点相连)单击,软件会自动按键角 120 度生成键长 固定的一根键,再单击一次也是一样,这样出现的键角十分标准,请勿单击后拖 动鼠标来画另一根键,这样键长和键角都不标准,结构式跟东施一样十分难看。
如再按一次 Ctrl+L,键长不再固定,因此按这种组合键可以反复在这两种模式切 换。
3. 点击 View 选上 Show Main Toolbar 会出现工具模块,使用这个模块点击上面的 按钮,然后在主窗口单击,你就可以直接画六边形、苯环等,按住时拖动鼠标还可 旋转这些图形;也有一个按钮有离子符号“+”、“-”以及自由基符号“·”,点 击后再单击主窗口中结构式上的碳原子或者其他原子,就可使这些原子带上正负号 等;还有一个按钮,点击后有许多画好的结构模块供选择,如氨基酸、多环化合物 等,特别方便。
第一节简单的结构画法1.1 基本功能1.画原子, 化学键(单键, 双键, 三键, 共价键, 等等), 和标签.2.翻转分子结构式.3.选择, 旋转和扩大与缩小结构图4.将结构式输出至文件夹, 文件及打印机.5.清理屏幕.在结构模式下进行以下操作:1.2 使用常用画图工具1.常用画图工具是默认的。
画直鏈和分支结构,画周期表中指定的原子。
2.在画图模式(Draw Mode)下,从原子工具条选择碳原子。
3.单击空白处,画出CH4。
4.单击CH4增加一CH3集团,以标准键长连接。
双击同一个碳原子,画出CH3CH3CH3。
5.单击Set Bond Vertically, 将从上一步得到的结构旋转至CH3CH3CH3。
2 6.单击结构工具栏中常用画图工具。
7.单击最右边的碳原子,画出CH 3CH 33。
8.重复以上步骤,画出一些结构CH 3CH 3CH 3,CH 3CH 3CH 3和CH 3C H 3CH 3。
(1) 双键和三键1.在上面的第7步中的最后一个单键上再次单击获得双键:C H 3CH 2CH 32.再次在双键上单击获得三键:CH 3CHCH 33.再次在三键上点击将重新得到单键.(2) 逐个去除原子1.点击Delete 键. 在以上的分子结构上进行逐一去除原子的练习. (3) 恢复键(Undo)1.单击Undo 键 。
这会将ChemSketch 的界面恢复到以前的设置.注意: 点击 Undo 键将会击活重做(Redo)键. Undo 键可以连续点击50次.2.多次单击恢复(Undo)键,直到将其恢复至以下结构:CH 3CH 3CH 33(4) 改变原子1.单击元素周期表( Periodic Table)键,打开周期表( Periodic Table of Elements):2.单击元素周期表中的键3.单击最左边的碳(Carbon), 用氟原子替代碳原子:CH 3FCH 3注意: 当你选择了元素周期表中的某个元素时, 相应的元素键将自动加到原子工具条中。
化学绘图软件—Chemwindow一、ChemWindow软件的特点及功能该软件具有快速便捷的操作工具、用来画分子图形的各种常用操作均用图标示于工具箱中,不必记忆任何操作口令,亦不必采用下拉式菜单(虽然也配有此功能)。
如对分子图形的组合、水平、垂直翻转及任意角度旋转等操作直接从工具箱中用鼠标点相应图标即可完成。
该软件还提供了大量画分子式或分子图形所需的各种“散装元件”,如各种类型的化学键、分子母环(从三元到八元环,包括六元环的船式和椅式构型)、化学分子轨道、圆电荷、球(椭球)等。
在模板工具中存储有大量常用的杂环、稠环供选择。
使用户在使用过程中变得象小孩子玩积木那样简单,在3.0版中具有丰富的图形屏幕显示,对图形不同部分可用不同的颜色进行显示。
该软件另一大特点就是强大的分子图形编辑功能。
对分子图形可进行组合,分块处埋,即可将许多物体结合成一个物体进行处理,或将一个物体分解成许多部分,使得在编辑时可根据需要用不同的方式来编辑。
对整体分于图形既可进行放大、缩小、旋转等操作也能对局部进行精确微调。
其还具有一般绘图软件所不具备的化学分子式的上下标标记功能及每个操作对象都有“敲击箱”(hit box),这对于图形的各种操作处理,特别是局部处理提供了很大的灵活性。
需要画分子结构式的三维平面图时,该仪件具有将图形中重叠部分进行断开处理的功能,使之呈现出立体感。
但是,ChemWindow 不具有OLE(对象链接与嵌入)的界面支持,使得它与其它软件之间的共享受到限制,尤其是在其它软件中(如Word,PowerPoint等)需对ChemWindow图形进行修改时,更显得繁琐。
二、ChemWindow 的使用方法和技巧ChemWindow 的操作绝大部分由鼠标便可完成,基本使用方法借助Help亦可较快学会。
下面介绍作者在使用过程中的一些点滴经验和体会1.绘制ChemWindow图形的几点技巧在绘制复杂分子结构式时,利用复制、水平或垂直翻转等操作可绘出对称协调的分子结构式图形。
C hemoffice2008使用教程ChembioOffice简介ChembioOffice是由CambridgeSoft开发的综合性科学应用软件包。
该软件包是为广大从事化学、生物研究领域的科研人员个人使用而设计开发的产品。
同时,这个产品又可以共享解决方案,给研究机构的所有科技工作者带来效益。
利用ChemBioOffice你可以方便的进行化学生物结构绘图、分子模型及仿真、可以将化合物名称直接转为结构图,省去绘图的麻烦;也可以对已知结构的化合物命名,给出正确的化合物名。
目前最新的版本为Chemoffice 2010。
Chemoffice2008包括以下几个部分:1.ChemDraw Ultra11.0:化学结构绘图。
2.Chem3D Ultra11.0:分子模型及仿真。
3.ChemFinder Ultra11.0:化学信息搜寻整合系统4.E-Notebook11.0:整理化学信息、文件和数据,并从中取得所要的结果。
Chemoffice安装方法:1.运行setup.exe2.输入证书时输入:Name:lZ0email:lz0@Serial:011-256104-71993.然后点击"Activate Later"4.复制crack.reg文件到C:\Program Files\CambridgeSoft\ChemOffice2008\Common\DLLs 双击确定即可ChemDraw的使用方法:1.绘制结构式以水杨酸(salicylic acid)为例步骤:1.打开ChemDraw2.单击垂直工具栏右下角的图标,鼠标变成苯环的样子,在绘图区单击鼠标出现一个苯环3.单击图标,将鼠标移至苯环的一个角上,出现深色的正方形连接点4.自连接点横向拉出一根实线单键,松开鼠标,自单键终点向右下方再次拉出一根单键,与前一根单键夹角约为109°,其余类推。
5.选中垂直工具栏中的文本工具,分别将鼠标移至应该出现羟基或氧原子的位置,待出现连接点后,点击键盘上的O键。
即可得如图所示图形:2.根据化合物名称得到结构式【Structure】/【Convert Name to Structure】,弹出对话框在输入框中输入化合物的英文名,点击OK即可出现结构式即可得如图所示图形:ChemDraw可以根据结构式预测分子的1H和13C核磁共振化学位移。
选中此结构,【Structure】/【Predict1H-NMR-Shifts】,出现该化合物的1H核磁共振化学位移值及图谱,如下图:ChemNMR1H Estimation11.06.98salicylic acidEstimation quality is indicated by color:good,medium,rough10812624PPMProtocol of the H-1NMR Prediction:Node Shift Base+ment(ppm rel.to TMS)OH 5.35 5.00aromatic C-OH0.35general correctionsOH11.011.00carboxylic acidCH 6.987.261-benzene-0.531-O0.211-C(=O)O0.04general correctionsCH8.047.261-benzene-0.171-O0.871-C(=O)O0.08general correctionsCH7.627.261-benzene-0.171-O0.341-C(=O)O0.19general correctionsCH7.517.261-benzene-0.441-O同理可得C13谱图。
ChemDraw可以对化合物结构进行分析计算。
选中化合物结构式,【View】/【Show Analysis Window】,弹出分析窗口,包含该化合物的分子简式、摩尔质量、同位素分布图,元素分析组成比例等数据。
【View】/【Show Chemical Properties Window】,弹出化学性质窗口,包含该化合物的沸点、熔点、临界温度、临界压力、临街体积、Gibbs自由能、LogP、MR、Herry’s Law、生成热、ClogP、CMR等。
可得:Chemical Formula:C7H6O3Exact Mass:138.03Molecular Weight:138.12m/z:138.03(100.0%),139.04(7.8%)Elemental Analysis:C,60.87;H,4.38;O,34.75ChemDraw提供了一张使用起来十分方便的元素周期表。
【View】/【Show Periodic Table Window】即可打开元素周期表窗口。
单击周期表上的元素符号,就可以得到该元素的物理性质。
单击表中的按钮,可以打开或关闭周期表下方的物理性质详细列表。
绘制实验装置ChemDraw可以用磨口玻璃仪器接插件迅速搭建化学反应装置。
以简单的蒸馏装置为例:【View】/【Other ToolBars】/【Clipware,Part1】或【Clipware,Part2】打开玻璃仪器模板窗口(或从垂直工具栏中打开)Chem3D的使用Chem3D是ChemOffice的组成部分,它能很好地同ChemDraw一起协同工作,ChemDraw 上画出的二维结构式可以正确地自动转换为三维结构。
Chem3D Ultra版还包括了一个很好的半经验量子化学计算程序MOPAC,还可以与著名的从头计算程序Gaussian连接,作为它的输入、输出界面,能够以三维的方式显示量子化学计算结果,如分子轨道、电荷密度分布等。
打开上述文件可得如图所示的界面:1)利用键工具建立模型单击工具栏上的单键按钮将鼠标移动至模型窗口,按住鼠标左键拖出一条直线,放开鼠标即成乙烷的立体模型将鼠标移至C(1)原子上,向外拖出一条直线,放开鼠标即成丙烷立体模型将鼠标移至C(2)原子上,向外拖出一条直线,放开鼠标即成丁烷立体模型2)利用文本工具建立模型单击工具栏文本工具按钮将鼠标移至模型窗口,单击鼠标出现文本输入框,在输入框中输入“C4H10”,按回车键,Chem3D自动将输入的分子式变成丁烷3D模型若化合物带有支链,可将支链用括号括起来。
(注意:CH必须为大写!)回车得如下界面:3)使用子结构建立模型Chem3D提供了子结构库,我们可以选择其中的子结构,然后将他们拼装起来,组成复杂结构。
【View】/【Parameter Table】/【Substructure】菜单命令,弹出【Substructure】窗口如图:单击【Phenyl】(苯基)的【Model】选中之复制粘贴即可。
单击工具栏上的复制按钮,复制该结构在3D模型窗口中,单击粘贴按钮,将子结构粘贴至窗口,使用轨迹球按钮使其处于正面结构,【Structure】/【Reflect Model】/【Invert Through Origin】将图形置于与原来呈镜面对称的位置再次单击粘贴按钮,在窗口中就复制了两个苯环单击工具栏中的虚键按钮,将两个苯环连接起来,即得所要建立的3D模型。
4)使用模板建立模型【File】/【Sample Files】/【Nano】/【Buckminsterfullerene-C60】命令,出现C60的3D 模型。
还可以在此基础上修改模型,例如可以接上一些官能团。
ChemFinderChemFinder是一个智能型的快速化学搜索引擎,所提供的ChemInfo是目前世界上最丰富的数据库之一,包含ChemACX、ChemINDEX、ChemRXN、ChemMSDX等,并不断有新的数据库加入。
Chem Finder可以从本机或网上搜寻Word、Excel、Power point、ChemDraw、ISIS格式的分子结构文件,还可以与Excel结合。
可连接的关系式数据库包括Oracle及Access,输入的格式包括ChemDraw、MDL ISIS SD及RD文件等。
1)根据结构式检索执行【开始】/【程序】/【ChemOffice】/【Chem Finder Ultra11.0】命令,启动ChemFinder 首先打开的式【ChemFinder】对话框,包含三个选项卡,单击【取消】在左侧Expelor窗口中最下端选择【Favorites】,出现“Favorites”文件夹,点击该文件夹双击选中“CS_DEMO.CFX”数据库,即打开该数据库在【Structure】输入框中已经有了一个苯环结构,这是该数据库的一个结构,【Formula】显示其分子式为“C6H6”,【MolWeight】显示其分子量为“78.1118”,【Molname】显示的是其英文文件名称“Benzene”,【Synonyms】显示苯的所有别名。
单击【Enter Query】清空各窗口双击【Structure】窗口,出现ChemDraw绘制分子式的工具栏【Tools】。
用ChemDraw的【Tools】工具绘制环丁烷,单击【Find】按钮查找。
2)根据分子式检索还可以直接输入分子式紧缩相关资料。
Chem Finder具有模糊检索功能,不必输入准确的分子式。
接前一步操作,单击【Enter Query】清空各窗口单击【Formula】窗口,输“C9H8O4”,按回车键单击【Enter Query】清空各窗口,单击【Formula】窗口,输入“C5-6N2”,按回车键,执行【View】/【Switch Views】命令,显示窗口变成列表,每执行一次,显示形式不同。
3)根据化学名称检索可以直接输入化学名(英文名)检索相关资料。
Chem Finder具有模糊检索功能,检索化学名时可用通配符表示不清楚的字符,我们也不必为分子的名称怎么拼写而发愁了。
以查找尼古丁的分子式为例:接前一步操作,单击【Enter Query】清空各窗口,单击【Molname】窗口,输入“﹡nico ﹡”,按回车键,单击工具栏上的【Next Record】,可以显示出尼古丁分子来4)根据相对分子质量检索可以直接输入相对分子质量来检索相关分子。
相对分子质量也可以模糊检索,只要大致确定一个范围就可以了。
接前一步操作,单击【Enter Query 】清空各窗口,单击【MolWeight 】窗口,输入“160-170”,按回车键。