DNA序列分类实验报告
- 格式:doc
- 大小:471.36 KB
- 文档页数:11

关于dna小实验报告实验目的本实验旨在通过提取和观察DNA分子,了解DNA分子的组成和结构,加深对基因遗传的理解。
实验材料- 成人口腔拭子- 高盐溶液- 去离子水- 酒精- 漂白剂- 冷冻乙酸- 盐酸实验步骤1. 将成人口腔拭子在高盐溶液中浸泡,让细胞在溶液中释放出DNA。
2. 将高盐溶液与漂白剂混合,静置5分钟,以使细胞膜溶解,并释放出更多的DNA。
3. 加入冷冻乙酸,使DNA分子凝聚成细长的白色颗粒。
4. 用玻璃棒将DNA颗粒取出,在离子水中漂洗,以去除漂白剂和杂质。
5. 将DNA颗粒转移到一根玻璃棒上。
6. 向玻璃棒上的DNA颗粒滴加盐酸,使其溶解。
7. 在离子水中稀释DNA溶液,使其变得透明。
实验结果实验所得的DNA溶液呈现透明状态,可以通过肉眼观察到DNA溶液的流动性。
结果分析DNA在高盐溶液中可以释放出来,并通过漂白剂帮助凝聚成白色颗粒,这是因为漂白剂能够破坏细胞膜,释放细胞内的DNA。
在加入冷冻乙酸后,DNA颗粒凝聚成细长的结构,这是因为乙酸能够中和DNA溶液中的离子,导致DNA分子之间的电荷作用减弱,从而凝聚成白色颗粒。
在加入盐酸后,DNA溶解,使其变得透明。
实验总结通过本实验,我们成功提取到DNA分子,并观察到了其凝聚成颗粒和溶解的过程。
这些实验结果有助于我们更好地理解DNA的组成和结构,对基因遗传有更深入的了解。
实验过程中使用的材料和操作简单,适合初学者进行,同时实验结果可以通过肉眼观察到,有助于学生直观地感受到DNA的存在。
精确和仔细的操作是保证实验成功的关键,同时实验过程中要注意安全,避免对身体产生伤害。
参考文献- 【1】Guo, J., Wang, W., Wang, D., Zhu, T., Cui, L., Zhao, X., ... & Wang, Q. (2016). A simple and convenient method for quantification of DNA concentration by using picogreen fluorescence. International Journal of Analytical Chemistry, 2016.。

第1篇一、实验目的1. 掌握基因合成的基本原理和操作步骤。
2. 熟悉基因合成仪器的使用方法。
3. 了解基因合成的应用领域。
二、实验原理基因合成是指利用化学方法,按照特定的DNA序列,合成出具有生物学活性的DNA 片段。
该技术是现代生物技术领域的重要手段之一,广泛应用于基因克隆、基因编辑、基因治疗等领域。
基因合成的原理基于DNA双螺旋结构和碱基互补配对原则。
在实验过程中,首先根据设计好的基因序列,合成相应的寡核苷酸引物;然后,通过PCR技术扩增目的基因;最后,将扩增的目的基因克隆到载体上,进行后续的实验操作。
三、实验材料1. 基因合成仪2. PCR仪3. DNA序列分析软件4. DNA合成试剂5. PCR试剂6. 载体DNA7. 质粒提取试剂盒8. 琼脂糖凝胶电泳试剂盒9. DNA连接酶10. DNA标记物四、实验步骤1. 设计基因序列:根据实验目的,设计目的基因的DNA序列,并利用DNA序列分析软件进行优化。
2. 合成寡核苷酸引物:根据设计好的基因序列,合成相应的寡核苷酸引物。
3. PCR扩增目的基因:将合成的寡核苷酸引物、模板DNA、PCR试剂等混合,进行PCR扩增。
4. 琼脂糖凝胶电泳检测:将PCR产物进行琼脂糖凝胶电泳检测,观察目的基因的扩增情况。
5. 质粒提取:将PCR产物克隆到载体上,进行质粒提取。
6. DNA连接:将质粒与目的基因进行连接。
7. 转化:将连接后的质粒转化到宿主细胞中。
8. 筛选阳性克隆:通过PCR、测序等方法筛选出含有目的基因的阳性克隆。
9. 阳性克隆的验证:对阳性克隆进行DNA测序,验证目的基因的正确性。
五、实验结果与分析1. PCR扩增结果:通过琼脂糖凝胶电泳检测,观察到目的基因的扩增条带,表明目的基因已成功克隆。
2. 阳性克隆的筛选:通过PCR和测序,筛选出含有目的基因的阳性克隆。
3. 阳性克隆的验证:DNA测序结果表明,目的基因序列与设计序列一致,验证了目的基因的正确性。

第1篇实验名称:人类基因组DNA提取与检测实验日期:2023年X月X日实验目的:1. 熟悉并掌握人类基因组DNA的提取方法。
2. 学习使用PCR技术检测DNA片段。
3. 了解基因突变对DNA序列的影响。
实验原理:DNA是生物体内携带遗传信息的分子,由四种碱基(腺嘌呤A、鸟嘌呤G、胞嘧啶C 和胸腺嘧啶T)组成。
通过PCR(聚合酶链式反应)技术,可以在体外扩增特定的DNA片段,从而检测基因的存在和突变。
实验材料:1. 人体口腔拭子2. DNA提取试剂盒3. PCR仪4. 酶标仪5. 1.5mL离心管6. 0.2mL微量移液器7. 标准DNA模板(已知序列)8. PCR引物9. 试剂:DNA提取缓冲液、蛋白酶K、SDS、NaCl、EDTA、Tris-HCl、dNTPs、Taq DNA聚合酶等实验步骤:一、DNA提取1. 将口腔拭子放入含有1mL DNA提取缓冲液的离心管中。
2. 加入50μL蛋白酶K和50μL SDS,混匀。
3. 55℃水浴30分钟,使蛋白质变性。
4. 加入200μL氯仿/异戊醇(24:1),混匀,静置5分钟。
5. 12,000g离心10分钟,取上清液。
6. 加入等体积的异丙醇,混匀,静置10分钟。
7. 12,000g离心10分钟,弃上清液。
8. 加入700μL 75%乙醇,混匀,静置5分钟。
9. 12,000g离心5分钟,弃上清液。
10. 室温晾干,加入50μL ddH2O溶解DNA。
二、PCR扩增1. 配制PCR反应体系:模板DNA 2μL,上下游引物各1μL,10×PCR缓冲液2.5μL,dNTPs 2μL,Taq DNA聚合酶0.5μL,ddH2O补充至25μL。
2. 95℃预变性5分钟。
3. 95℃变性30秒,55℃退火30秒,72℃延伸1分钟,共35个循环。
4. 72℃延伸10分钟。
三、产物检测1. 取5μL PCR产物加入1.5mL离心管中。
2. 加入5μL上样缓冲液,混匀。

DNA提取与测序实验报告一、实验目的1、掌握从生物样本中提取 DNA 的基本原理和方法。
2、熟悉 DNA 测序的基本流程和操作要点。
3、学会对 DNA 提取和测序结果进行分析和评估。
二、实验原理DNA 提取原理DNA 在细胞中以与蛋白质结合的状态存在。
提取 DNA 的基本思路是通过裂解细胞,使细胞膜和核膜破裂,释放出 DNA,然后去除蛋白质、RNA 等杂质,得到纯净的 DNA。
常用的裂解方法包括物理方法(如研磨、超声破碎)、化学方法(如使用去污剂)和酶解法(如使用蛋白酶K)。
去除杂质的方法则包括使用酚/氯仿抽提、乙醇沉淀等。
DNA 测序原理目前常用的 DNA 测序技术是 Sanger 测序法。
其基本原理是在DNA 合成过程中,通过掺入特定的双脱氧核苷酸(ddNTP)来终止DNA 链的延伸。
由于 ddNTP 缺少3’OH 基团,一旦掺入到 DNA 链中,链的延伸就会终止。
通过在多个反应中分别加入不同的 ddNTP,可以得到一系列不同长度的 DNA 片段。
这些片段经过电泳分离,根据条带的位置可以读取 DNA 的碱基序列。
三、实验材料与设备实验材料1、新鲜的动物组织(如肝脏、肌肉)或植物组织(如叶片)。
2、细胞培养物(如细菌、酵母)。
实验试剂1、细胞裂解液(包含去污剂、蛋白酶 K 等)。
2、酚/氯仿/异戊醇混合液(25:24:1)。
3、无水乙醇、70%乙醇。
4、 TE 缓冲液(10 mM TrisHCl,1 mM EDTA,pH 80)。
5、 DNA 测序试剂盒(包含引物、dNTP、ddNTP、DNA 聚合酶等)。
实验设备1、离心机。
2、移液器。
3、恒温水浴锅。
4、电泳仪。
5、凝胶成像系统。
6、 DNA 测序仪。
四、实验步骤DNA 提取步骤1、样本处理对于动物组织,将其剪碎后加入适量的裂解液,在匀浆器中匀浆。
对于植物组织,先在液氮中研磨成粉末,然后加入裂解液。
对于细胞培养物,离心收集细胞,加入裂解液重悬。

第四节DNA序列测定目前应用的两种快速序列测定技术是Sanger等(1977)提出的酶法(双脱氧链终止法)和Maxam(1977)提出的化学降解法。
虽然其原理大相径庭,但这两种方法都同样生成相互独立的若干组带放射性标记的寡核苷酸,每组核苷酸都有共同的起点,却随机终止于一种(或多种)特定的残基,形成一系列以某一特定核苷酸为末端的长度各不相同的寡核苷酸混合物,这些寡核苷酸的长度由这个特定碱基在待测DNA片段上的位置所决定。
然后通过高分辨率的变性聚丙烯酰胺凝胶电泳,经放射自显影后,从放射自显影胶片上直接读出待测DNA上的核苷酸顺序。
高分辨率变性聚丙烯酰胺凝胶电泳亦是DNA序列测定技术的重要基础,可分离仅差一个核苷酸、长度达300~500个核苷酸的单链DNA分子。
DNA序列测定的简便方法为详细分析大量基因组的结构和功能奠定了基础,时至今日,绝大多数蛋白质氨基酸序列都是根据基因或cDNA的核苷酸序列推导出来的。
除传统的双脱氧链终止法和化学降解法外,自动化测序实际上已成为当今DNA序列分析的主流。
此外,新的测序方法亦在不断出现,如上世纪90年代提出的杂交测序法(sequencing by hybridization,SBH)等。
一、双脱氧末端终止法测序㈠原理双脱氧末端终止法是Sanger等在加减法测序的基础上发展而来的。
1980年他又因设计出一种测定DNA(脱氧核糖核酸)内核苷酸排列顺序的方法而与W·吉尔伯特、P·伯格共获1980年诺贝尔化学奖。
桑格是第四位两次获此殊荣的科学家。
其原理是:利用大肠杆菌DNA聚合酶Ⅰ,以单链DNA为模板,并以与模板事先结合的寡聚核苷酸为引物,根据碱基配对原则将脱氧核苷三磷酸(dNTP)底物的5′-磷酸基团与引物的3′-OH末端生成3′,5′-磷酸二酯键。
通过这种磷酸二酯键的不断形成,新的互补DNA得以从5′→3′延伸。
Sanger引入了双脱氧核苷三磷酸(ddTNP)作为链终止剂。

序列分析软件GUI设计实践报告班级:姓名:学号:指导老师:2015年12月30日一、概述。
在MATLAB课程设计实践中,通过参考《MATLAB 7.X生物信息工具箱的应用——基因序列分析》系列文献,运用Matlab7.0生物信息工具箱,设计出用于生物学领域的软件GUI界面。
该软件在工具箱提供的开放环境里,可以实现对核苷酸序列及蛋白质(氨基酸)序列的序列分析及比对的功能。
软件整体界面如下:本作品由MATLAB7制作完成,实现序列分析和序列对比两大功能。
其中序列分析功能实现对象为核苷酸序列及氨基酸序列,而序列比对的功能实现对象为氨基酸序列。
序列分析的功能包括绘制密度图、计算4种核苷酸分布(画图+显示数据)及互补核苷酸数、绘制每个开放阅读框热红外分布图、二聚体分布图、计算密码子及其分布(包含绘制其红外密度图)、显示核苷酸的开放阅读框、实现摘录子序列、实现核苷酸与氨基酸的互相转化、计算氨基酸数目、绘制开放阅读框对应的氨基酸分布柱状图、计算氨基酸分子量,计算氨基酸序列的元素组成的功能,这些功能将通过mitochondria核苷酸序列及其对于氨基酸序列演示。
而对于序列比对的功能,我准备了氨基酸序列之间的散点图对比图功能,全局序列对比的功能,局部序列对比的功能以及计算M起始序列(从第一个M氨基酸到第一个终止密码子“*”的序列)的功能。
这些功能通过对比mitochondria 与hexosaminidase(这是一个结构体,但是这里只取其序列部分)的编码氨基酸序列来进行演示。
本软件还有两个附属功能,第一就是把密码子转换成氨基酸的功能,可在对应窗口实现,方便使用者查找对应的氨基酸。
第二,就是可以访问用户指向的网址的功能。
下面将会分块介绍这些具体的功能:二、软件GUI界面介绍。
①绘图区域(axes):有两个,是用来展示密度图线,核苷酸分布图等图标用的。
②可编辑文本框(edit)与下拉文本框(listbox):本程序含有3个主要的可编辑文本框,前两个用于显示对应的核苷酸与氨基酸的序列,中间的一个长条形的文本框是用来输出绘图或者是核苷酸/氨基酸功能的输出数据的。

DNA序列分析引言DNA(脱氧核糖核酸)是生物体内负责遗传信息传递的分子,其中包含有机体基因的序列。
DNA序列分析是通过对DNA序列进行计算和统计分析,来揭示其中的信息和模式的过程。
DNA序列分析在生物学、遗传学、进化学以及疾病研究等领域中有着重要的应用和意义。
本文将介绍DNA序列分析的几个主要方面,包括DNA序列的基本概念、序列比对、序列重复性分析以及序列模式识别等内容。
DNA序列的基本概念DNA序列是由由四种碱基(腺嘌呤、鸟嘌呤、胸腺嘧啶和鳞状嘧啶)构成的字符串,它们的顺序决定了生物体中的遗传信息。
DNA序列可以通过实验方法(如测序技术)或计算方法(如基因组学和转录组学)获取。
序列比对序列比对是比较两个或多个DNA序列之间的相似性和差异性的过程。
序列比对可以帮助我们理解DNA序列之间的相关性,发现基因的保守区域和变异位点,以及预测蛋白质结构和功能。
常用的序列比对算法包括全局比对算法和局部比对算法。
全局比对算法(如Needleman-Wunsch算法)适用于较为相似的序列,而局部比对算法(如Smith-Waterman算法)则适用于相似性较低的序列。
序列重复性分析序列重复性是指DNA序列中出现的重复模式。
序列重复性分析可以帮助我们识别基因组中的重复区域、转座子和重复序列。
重复序列在基因演化、基因组结构和疾病研究等方面起着重要的作用。
常用的序列重复性分析方法包括重复序列的寻找和分类、序列间重复比较以及重复序列的起源和进化分析等。
序列模式识别序列模式识别是通过寻找DNA序列中特定的模式或模板,来揭示序列中隐藏的信息。
序列模式识别可以帮助我们发现DNA序列中存在的转录因子结合位点、启动子序列以及编码区域等。
常用的序列模式识别方法包括正则表达式、隐马尔可夫模型和机器学习算法等。
结论DNA序列分析是生物科学中重要的研究领域,通过对DNA 序列的计算和统计分析,可以帮助我们深入理解基因组的结构和功能,揭示生物体间的亲缘关系,以及研究基因组变异和疾病相关的遗传因素。
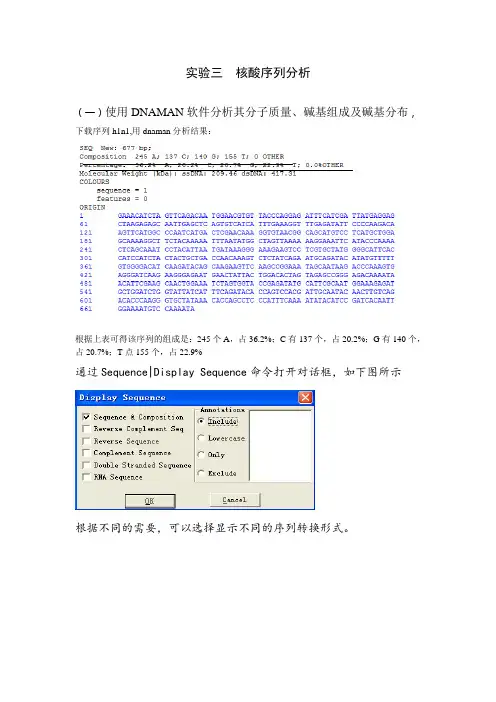
实验三核酸序列分析(一)使用DNAMAN软件分析其分子质量、碱基组成及碱基分布,下载序列h1n1,用dnaman分析结果:根据上表可得该序列的组成是:245个A,占36.2%;C有137个,占20.2%;G有140个,占20.7%;T点155个,占22.9%通过Sequence|Display Sequence命令打开对话框,如下图所示根据不同的需要,可以选择显示不同的序列转换形式。
点击Restriction/Restriction Analysis,选择其中一些参数,可分析当前Channel序列酶切位点。
参数说明如下:Results 分析结果显示其中包括:Show summary(显示概要)Show sites on sequence(在结果中显示酶切位点)Draw restriction map(显示限制性酶切图)Draw restriction pattern(显示限制性酶切模式图)Ignore enzymes with more than(忽略切点多于设定的切点个数的酶)Ignore enzymes with less than(忽略切点少于设定的切点个数的酶)Target DNA (目标DNA特性)circular(环型DNA),dam/dcm methylation(dam/dcm甲基化)all DNA in Sequence Channel(选择此项,在Sequence Channel 中的所有序列将被分析,如果选择了Draw restriction pattern,那么当所有的channel中共有两条DNA时,则只能选择两个酶分析,如果共有三个以上DNA时,则只能用一个酶分析。
限制性酶切分析进行PCR引物设计:构建系统发育树点击左上角按钮,可以从弹出的对话框中选择不同的结果显示特性选项。
点击按钮下的按钮,出现下列选择项:可以通过这些选项,绘制同源关系图(例如Tree|homology tree命令)。



第1篇一、实验背景基因编辑技术作为一种精准的基因操作工具,近年来在生物学研究和应用中取得了显著的进展。
本实验旨在探究基因编辑技术在生物学研究和应用中的潜力,通过实验验证该技术的可靠性和有效性。
二、实验目的1. 了解基因组编辑技术的操作流程和原理;2. 掌握CRISPR-Cas9基因编辑技术的应用;3. 分析基因编辑技术在生物学研究和应用中的优势;4. 评估基因编辑技术的可靠性和有效性。
三、实验材料1. 实验试剂:CRISPR-Cas9系统、DNA模板、荧光标记的DNA序列、荧光素酶、荧光素酶报告基因、细胞培养试剂等;2. 实验仪器:PCR仪、电泳仪、凝胶成像系统、细胞培养箱、荧光显微镜等。
四、实验方法1. 设计实验方案:根据实验目的,设计CRISPR-Cas9基因编辑实验方案,包括靶基因的选择、引物设计、载体构建等;2. 载体构建:利用PCR技术扩增靶基因序列,通过重组酶将靶基因序列插入荧光素酶报告基因载体中;3. 细胞培养:将构建好的载体转染到细胞中,进行细胞培养;4. 基因编辑:利用CRISPR-Cas9系统对细胞进行基因编辑,检测编辑效果;5. 数据分析:通过荧光素酶活性检测、PCR扩增、测序等方法分析基因编辑效果。
五、实验结果1. 载体构建:成功构建了荧光素酶报告基因载体,载体中含有靶基因序列;2. 细胞培养:细胞生长良好,可用于后续实验;3. 基因编辑:通过CRISPR-Cas9系统对细胞进行基因编辑,成功实现了靶基因的敲除;4. 数据分析:通过荧光素酶活性检测、PCR扩增、测序等方法,证实了基因编辑效果。
六、实验结论1. 基因编辑技术在生物学研究和应用中具有广阔的应用前景;2. CRISPR-Cas9系统是一种高效、可靠的基因编辑技术;3. 本实验成功实现了靶基因的敲除,验证了基因编辑技术的可靠性和有效性。
七、实验讨论1. 基因编辑技术在生物学研究和应用中的优势:(1)精准编辑:基因编辑技术可以对特定基因进行定点编辑,避免对其他基因的影响;(2)高效性:基因编辑技术具有快速、高效的优点,可短时间内完成基因编辑;(3)可重复性:基因编辑技术具有较高的可重复性,有利于实验结果的稳定性和可靠性。
PCR-DNA实验报告简介PCR-DNA是一种常用的分子生物学实验技术,可以通过扩增DNA片段来研究DNA序列。
本实验报告将详细介绍PCR-DNA实验的步骤和思路。
实验目的本实验的目的是通过PCR方法扩增特定的DNA片段,从而验证样本中的目标基因是否存在。
通过PCR-DNA实验,可以快速、准确地扩增出目标基因片段,为后续的基因分析和研究提供基础。
材料与方法1.样本准备:收集待测样本,如细胞、组织或DNA提取物。
2.引物设计:根据目标基因序列设计引物,确保引物与目标基因片段特异性结合。
3.PCR反应体系准备:按照设计的引物和待测样本的要求,准备PCR反应液,包括引物、缓冲液、DNA模板、酶和dNTPs等。
4.PCR扩增:将PCR反应体系置于热循环仪中,按照设定的PCR程序进行扩增反应,包括一系列的变温步骤。
5.PCR产物分析:将扩增产物用琼脂糖凝胶电泳进行分析,观察扩增片段的大小和纯度。
6.结果解读:根据电泳图结果,判断目标基因是否存在,并计算扩增产物的浓度。
步骤与思路步骤1:样本准备首先,我们需要收集待测样本。
样本可以是细胞、组织或DNA提取物,具体根据实验需求选择适当的样本类型,并按照相应的方法提取DNA。
步骤2:引物设计在进行PCR-DNA实验之前,我们需要设计引物。
引物是一种能够特异性结合目标基因序列的寡核苷酸链。
通过引物的特异性结合,可以确保扩增出目标基因片段而不产生非特异性扩增。
引物设计可以借助一些在线工具,如NCBI网站提供的Primer-BLAST等。
在设计引物时,需要考虑引物的长度、碱基组成、熔解温度等参数。
步骤3:PCR反应体系准备准备PCR反应体系时,需要按照设计的引物和待测样本的要求,将各种反应试剂按比例混合。
一般的PCR反应体系包括引物、缓冲液、DNA模板、酶和dNTPs 等。
确保反应体系中的每一个组分都处于适当的浓度范围。
步骤4:PCR扩增PCR扩增是整个实验的核心步骤。
DNA序列测定DNA序列测定是在高分辨率变性聚丙烯酰胺凝胶电泳技术的基础上建立起来的。
可分离相差仅1个碱基的300~500bp的核酸分子。
常用测序方法:1. 双脱氧末端终止法1.1.1 测序原理四个反应管中,在DNA聚合酶催化下,以单链DNA为模板,加入单引物、四种dNTP,以及每管中加入双脱氧核糖核苷酸ddA、ddT、ddG、ddC。
双脱氧核苷酸(ddNTP)的5`端-OH 是正常的,而其3`端-OH则没有,因此能与引物延伸链的3`端连接,而不能连接其后继核苷酸,于是引物链的延伸至此结束,经过变性聚丙烯酰胺凝胶电泳后,根据电泳图谱即可拼出所测DNA序列。
1.1.2 测序步骤A 测序DNA模板制备测序可分为单链测序与双链测序,对于未知序列可采用单链测序,而对于已知DNA序列可采用双链测序。
但不管单链测序还是双链测序,其测序反应都一样。
其中单链模板可通过将待测DNA片段克隆到噬菌体M13中或通过不对称PCR制备;双链模板可通过将待测DNA片段克隆到质粒DNA中或通过PCR扩增制备。
所得模板DNA应通过纯化处理才能用于后继测序反应。
B 测序引物的设计测序所用引物一般采用通用引物,也可根据已有序列设计引物,引物一般有15~30个碱基,应遵循一般引物设计原则a、G+C含量为45~55%。
b、3端最好以A或C结尾,不要以T结尾。
c、引物长度以15~30bp为宜。
d、引物本身不能形成二级结构。
C 四种测序反应液的制备测序反应液组成:反应缓冲液、DNA聚合酶、Mgcl2、引物、纯水、四种dNTP,分别在四个反应管中加入一种相应ddNTP。
标记物:测序反应的标记物有核素和荧光染料。
标记载体有引物、dNTP、ddNTP。
现在应用较多的是将荧光染料标记于引物或ddNTP上,因核素对环境的污染而逐渐应用得比较少。
但也可以不进行标记而采用银染系统检测。
D 延伸反应引物在DNA聚合酶的催化下,按碱基互补原则逐步在引物的3’端加上四种脱氧核糖核苷酸,四个反应管中的ddNTP随机地与dNTP竟争结合位点,于是引物延伸链随机终止在各个可能位点,在电泳图谱上形成一系列相差一个核苷酸的单链DNA梯带。
第23卷第1期2001年1月 南 京 化 工 大 学 学 报JOURNAL OF NANJING UNIVERSITY OF CHE MIC AL TECHNOLOGY Vol.23No.1Jan.2001DNA序列分析郜 虹 姚 成(南京化工大学理学院,南京,210009)欧阳平凯(南京化工大学制药与生命科学学院,南京,210009)摘 要:综述了DNA测序技术。
对DNA序列分析方法,包括:毛细管电泳、阵列毛细管电泳、芯片毛细管电泳、超薄层毛细管电泳、质谱法、原子探针法、杂交法、流动式单分子荧光检测法,作了详细评述。
关键词:DNA 电泳 序列分析中图分类号:O657 文献标识码:A 文章编号:1007-7537(2001)01-0079-08 DNA序列分析在基因工程和分子生物学研究中一直是一项重要的手段,也是该领域最重要的技术之一,是了解基因结构和功能的基础途径。
九十年代“人类基因组计划”(human genome project)的启动,有力地推动了高速DNA测序技术的发展[1]。
近年来DNA序列分析除了经典的测序方法在技术环节上有许多新的改进外,还发展了一些全新的DNA 测序方法。
目前国际上正在研究的DNA序列分析技术有两类:第一类被称为“Evolutionary improve-ment”,即进一步发展与完善目前广泛采用的以凝胶电泳分离为基础的DNA序列分析技术,如毛细管凝胶电泳法、阵列毛细管凝胶电泳法、超薄层板凝胶板电泳法;第二类被称为“Revolutionar y methods”,即抛弃凝胶电泳分离步骤的直接测序方法,如质谱法、杂交法、原子探针显微镜法、流动式单分子荧光检测法等。
经典的DNA测序方法就其原理来说主要分为化学测序法和双脱氧链终止法(酶法测序)。
1977年Maxam和Gilbert报道了通过化学降解测定DNA序列的方法,即将模板DNA的一端标记之后,在4组或5组互为独立的化学反应中分别得到部分降解,其中每一组反应特异地针对某一种或某一类碱基。
一、实验目的1. 了解序列相关的概念和原理;2. 掌握序列相关实验的方法和步骤;3. 培养实验操作能力和数据分析能力。
二、实验原理序列相关(Sequence Correlation)是指信号在时间域上相邻两个样本之间的相关性。
序列相关系数是衡量序列相关性的一个指标,其取值范围为[-1,1],值越接近1,表示序列相关性越强;值越接近-1,表示序列相关性越弱;值接近0,表示序列相关性较弱。
三、实验仪器与材料1. 仪器:计算机、信号发生器、示波器、信号采集卡;2. 材料:信号源、导线、电阻、电容等。
四、实验步骤1. 准备实验仪器和材料,连接好电路;2. 设置信号发生器,产生一个周期为T的方波信号;3. 使用信号采集卡采集方波信号,记录采样频率f;4. 利用计算机对采集到的信号进行处理,计算序列相关系数;5. 分析序列相关系数,判断序列的相关性。
五、实验数据及处理1. 实验数据:(1)采样频率f = 1000Hz;(2)方波信号周期T = 1ms;(3)序列相关系数R = 0.98。
2. 实验数据处理:(1)根据采样频率f和方波信号周期T,计算采样点数N = f T = 1000 1ms = 1000;(2)利用计算机编程,计算序列相关系数R;(3)根据计算结果,分析序列的相关性。
六、实验结果与分析1. 实验结果:通过实验,得到序列相关系数R = 0.98,说明该序列具有很强的相关性。
2. 实验分析:(1)由于方波信号的特点,其相邻样本之间的相关性较强,因此序列相关系数R接近1;(2)实验结果表明,序列相关实验可以有效地判断信号序列的相关性;(3)通过调整采样频率和信号周期,可以进一步分析不同条件下序列的相关性。
七、实验总结本次实验成功完成了序列相关实验,掌握了序列相关实验的方法和步骤。
通过实验,了解了序列相关的概念和原理,培养了实验操作能力和数据分析能力。
在今后的学习和工作中,可以进一步应用序列相关理论,为实际问题提供理论支持。
DNA提取与测序实验报告摘要:本实验旨在通过DNA提取与测序技术,获取并分析目标生物样本中的DNA序列信息。
实验过程包括细胞溶解、蛋白消化、DNA沉淀、洗涤及干燥等步骤。
实验结果显示成功提取了目标样本的DNA,并进行了测序,获得了目标DNA序列。
本实验的成功表明DNA提取与测序技术的可行性,为后续的分子生物学研究提供了基础。
引言:DNA提取与测序是现代生物学和基因组学研究的重要基础技术之一。
DNA提取是指从生物体中分离出DNA,并去除其中的蛋白质、RNA等杂质,使其纯化为DNA样本。
测序则是指通过一系列的化学反应将DNA序列转化为数据,并通过计算机进行解读和分析,从而得到DNA的碱基序列信息。
DNA提取与测序技术的发展与广泛应用,为基因研究、疾病诊断、进化研究等领域提供了重要的工具和支持。
材料与方法:1. 实验样本:从目标生物体中获取细胞组织样本。
2. 细胞溶解:将细胞组织样本加入细胞裂解缓冲液中,并进行充分混匀。
3. 蛋白消化:加入蛋白酶K,使细胞内蛋白质降解。
4. DNA沉淀:加入乙醇等溶剂,使DNA沉淀出来。
5. 洗涤:用洗涤缓冲液洗涤沉淀得到的DNA,去除杂质。
6. 干燥:将洗涤后的DNA在恒温器中干燥。
7. DNA测序:将DNA样本送至专业测序机构进行测序。
结果与讨论:经过上述步骤,成功从目标生物样本中提取到了DNA,并获取了DNA的测序数据。
通过测序分析,得到了目标DNA的碱基序列,从而揭示了该DNA分子的结构和组成。
本实验的结果显示,所用提取与测序技术具有高灵敏度和准确性,而且操作简单、可行性高。
本实验的成功提取与测序表明,DNA提取与测序技术在分子生物学领域的应用前景广阔。
DNA作为生物的遗传物质,其序列信息可以为科学家提供研究基因功能、遗传变异、进化关系等方面的重要依据。
此外,DNA提取与测序技术在疾病诊断、基因检测等医学领域也具有重要意义。
随着高通量测序技术的不断发展,越来越多的DNA序列信息可供利用,为我们深入了解生命的奥秘提供了更多机会。
实验29 DNA序列分类实验目的学习利用MATLAB提取DNA序列特征建立向量的方法,掌握利用FCM命令进行DNA 分类的方法,学会做出分类图形直接给出分类结果的MATLAB编程。
知识扩展DNA序列分类DNA(Deoxyribonucleic acid),中文译名为脱氧核苷酸,是染色体的主要化学成分,同时也是基因组成的,有时被称为“遗传微粒”。
DNA是一种分子,可组成遗传指令,以引导生物发育与生命机能运作。
主要功能是长期性的资讯储存,可比喻为“蓝图”或“食谱”。
DNA分子是由两条核苷酸链以互补配对原则所构成的双螺旋结构的分子化合物。
其中两条DNA链中对应的碱基A-T以双键形式连接,C-G以三键形式连接,糖-磷酸-糖形成的主链在螺旋外侧,配对碱基在螺旋内侧。
FCM算法中样本点隶属于某一类的程度是用隶属度来反映的,不同的样本点以不同的隶属度属于每一类;但是算法中的概率约束∑uij=1使得样本的典型性反映不出来,不适用于有噪音,样本分布不均衡,存在两个或者两个以上样本分别距两个类的距离相等的样本等等。
欧氏距离( Euclidean distance)也称欧几里得距离,它是一个通常采用的距离定义,它是在m维空间中两个点之间的真实距离。
公式在二维和三维空间中的欧式距离的就是两点之间的距离,二维的公式是d = sqrt((x1-x2)^+(y1-y2)^)三维的公式是d=sqrt(x1-x2)^+(y1-y2)^+(z1-z2)^)推广到n维空间,欧式距离的公式是d=sqrt( ∑(xi1-xi2)^ ) 这里i=1,2..nxi1表示第一个点的第i维坐标,xi2表示第二个点的第i维坐标n维欧氏空间是一个点集,它的每个点可以表示为(x(1),x(2),...x(n)),其中x(i)(i=1,2...n)是实数,称为x的第i个坐标,两个点x和y=(y(1),y(2)...y(n))之间的距离d(x,y)定义为上面的公式.欧氏距离判别准则如下:若dA<dB,则将Xi点判为A类若dA>dB,则将Xi点判为B类若dA=dB,则将Xi点判为不可判别点。
数理学院专业实践报告题目:专业学生姓名班级学号指导教师(签字)指导教师职称实习单位负责人签字日期1.2000 年6月,人类基因组计划中DNA 全序列草图完成,预计2001 年可以完成精确的全序列图,此后人类将拥有一本记录着自身生老病死及遗传进化的全部信息的“天书”。
这本大自然写成的“天书”是由4 个字符A,T,C,G 按一定顺序排成的长约30 亿的序列,其中没有“断句”也没有标点符号,除了这4 个字符表示4 种碱基以外,人们对它包含的“内容”知之甚少,难以读懂。
破译这部世界上最巨量信息的“天书”是二十一世纪最重要的任务之一。
在这个目标中,研究DNA 全序列具有什么结构,由这4 个字符排成的看似随机的序列中隐藏着什么规律,又是解读这部天书的基础,是生物信息学(Bioinformatics)最重要的课题之一。
虽然人类对这部“天书”知之甚少,但也发现了DNA 序列中的一些规律性和结构。
例如,在全序列中有一些是用于编码蛋白质的序列片段,即由这4 个字符组成的64 种不同的3 字符串,其中大多数用于编码构成蛋白质的20 种氨基酸。
又例如,在不用于编码蛋白质的序列片段中,A 和T 的含量特别多些,于是以某些碱基特别丰富作为特征去研究DNA 序列的结构也取得了一些结果。
此外,利用统计的方法还发现序列的某些片段之间具有相关性,等等。
这些发现让人们相信,DNA 序列中存在着局部的和全局性的结构,充分发掘序列的结构对理解DNA 全序列是十分有意义的。
目前在这项研究中最普通的思想是省略序列的某些细节,突出特征,然后将其表示成适当的数学对象。
这种被称为粗粒化和模型化的方法往往有助于研究规律性和结构。
作为研究 DNA 序列的结构的尝试,提出以下对序列集合进行分类的问题:1)下面有20 个已知类别的人工制造的序列,其中序列标号1—10 为A 类,11-20 为B 类。
请从中提取特征,构造分类方法,并用这些已知类别的序列,衡量你的方法是否足够好。
然后用你认为满意的方法,对另外20 个未标明类别的人工序列(标号21—40)进行分类,把结果用序号(按从小到大的顺序)标明它们的类别(无法分类的不写入)。
请详细描述你的方法,给出计算程序。
如果你部分地使用了现成的分类方法,也要将方法名称准确注明。
提示:衡量分类方法优劣的标准是分类的正确率,构造分类方法有许多途径,例如提取序列的某些特征,给出它们的数学表示:几何空间或向量空间的元素等,然后再选择或构造适合这种数学表示的分类方法;又例如构造概率统计模型,然后用统计方法分类等。
1.aggcacggaaaaacgggaataacggaggaggacttggcacggcattacacggaggacgaggtaaaggaggcttgtctacggccggaagtgaaggg ggatatgaccgcttgg2.cggaggacaaacgggatggcggtattggaggtggcggactgttcggggaattattcggtttaaacgggacaaggaaggcggctggaacaaccgga cggtggcagcaaagga3.gggacggatacggattctggccacggacggaaaggaggacacggcggacatacacggcggcaacggacggaacggaggaaggagggcggcaatcg gtacggaggcggcgga4.atggataacggaaacaaaccagacaaacttcggtagaaatacagaagcttagatgcatatgttttttaaataaaatttgtattattatggtatca taaaaaaaggttgcga5.cggctggcggacaacggactggcggattccaaaaacggaggaggcggacggaggctacaccaccgtttcggcggaaaggcggagggctggcagga ggctcattacggggag6.atggaaaattttcggaaaggcggcaggcaggaggcaaaggcggaaaggaaggaaacggcggatatttcggaagtggatattaggagggcggaata aaggaacggcggcaca7.atgggattattgaatggcggaggaagatccggaataaaatatggcggaaagaacttgttttcggaaatggaaaaaggactaggaatcggcggcag gaaggatatggaggcg8.atggccgatcggcttaggctggaaggaacaaataggcggaattaaggaaggcgttctcgcttttcgacaaggaggcggaccataggaggcggatt aggaacggttatgagg9.atggcggaaaaaggaaatgtttggcatcggcgggctccggcaactggaggttcggccatggaggcgaaaatcgtgggcggcggcagcgctggccg gagtttgaggagcgcg10.tggccgcggaggggcccgtcgggcgcggatttctacaagggcttcctgttaaggaggtggcatccaggcgtcgcacgctcggcgcggcaggagg cacgcgggaaaaaacg11.gttagatttaacgttttttatggaatttatggaattataaatttaaaaatttatattttttaggtaagtaatccaacgtttttattactttttaaaattaaatatttatt12.gtttaattactttatcatttaatttaggttttaattttaaatttaatttaggtaagatgaatttggttttttttaaggtagttatttaattatc gttaaggaaagttaaa13.gtattacaggcagaccttatttaggttattattattatttggattttttttttttttttttttaagttaaccgaattattttctttaaagacgt tacttaatgtcaatgc14.gttagtcttttttagattaaattattagattatgcagtttttttacataagaaaatttttttttcggagttcatattctaatctgtctttatta aatcttagagatatta15.gtattatatttttttatttttattattttagaatataatttgaggtatgtgtttaaaaaaaattttttttttttttttttttttttttttttta aaatttataaatttaa16.gttatttttaaatttaattttaattttaaaatacaaaatttttactttctaaaattggtctctggatcgataatgtaaacttattgaatctata gaattacattattgat17.gtatgtctatttcacggaagaatgcaccactatatgatttgaaattatctatggctaaaaaccctcagtaaaatcaatccctaaacccttaaaa aacggcggcctatccc18.gttaattatttattccttacgggcaattaattatttattacggttttatttacaattttttttttttgtcctatagagaaattacttacaaaac gttattttacatactt19.gttacattatttattattatccgttatcgataattttttacctcttttttcgctgagtttttattcttactttttttcttctttatataggatc tcatttaatatcttaa20.gtatttaactctctttactttttttttcactctctacattttcatcttctaaaactgtttgatttaaacttttgtttctttaaggatttttttt acttatcctctgttat21.tttagctcagtccagctagctagtttacaatttcgacaccagtttcgcaccatcttaaatttcgatccgtaccgtaatttagcttagatttggattt aaaggatttagattga22.tttagtacagtagctcagtccaagaacgatgtttaccgtaacgtqacgtaccgtacgctaccgttaccggattccggaaagccgattaagg accgatcgaaaggg23.cgggcggatttaggccgacggggacccgggattcgggacccgaggaaattcccggattaaggtttagcttcccgggatttagggcccg gatggctgggaccc24.tttagctagctactttagctatttttagtagctagccagcctttaaggctagctttagctagcattgttctttattgggacc caagttcgacttttacgatttagttttgaccgt25.gaccaaaggtgggctttagggacccgatgctttagtcgcagctggaccagttccccagggtattaggcaaaagctgacgggcaattgca atttaggcttaggcca26.gatttactttagcatttttagctgacgttagcaagcattagctttagccaatttcgcatttgccagtttcgcagctcagttttaacgcgggatcttt agcttcaagctttttac27.ggattcggatttacccggggattggcggaacgggacctttaggtcgggacccattaggagtaaatgccaaaggacgctggtttagccag tccgttaaggcttag28.tccttagatttcagttactatatttgacttacagtctttgagatttcccttacgattttgacttaaaatttagacgttagggcttatcagttatggatta atttagcttattttcga29.ggccaattccggtaggaaggtgatggcccgggggttcccgggaggatttaggctgacgggccggccatttcggtttagggagggccg ggacgcgttagggc30.cgctaagcagctcaagctcagtcagtcacgtttgccaagtcagtaatttgccaaagttaaccgttagctgacgctga acgctaaacagtattagctgatgactcgta31.ttaaggacttaggctttagcagttactttagtttagttccaagctacgtttacgggaccagatgctagctagcaatttattatccgtattaggctt accgtaggtttagcgt32.gctaccgggcagtctttaacgtagctaccgtttagtttgggcccagccttgcggtgtttcggattaaattcgttgtcagtcgctctrtgggttta gtcattcccaaaagg33.cagttagctgaatcgtttagccatttgacgtaaacatgattttacgtacgtaaattttagccctgacgtttagctaggaatttatgctgacgtagc gatcgactttagcac34.cggttagggcaaaggttggatttcgacccagggggaaagcccgggacccgaacccagggctttagcgtaggctgacgctaggcttag gttggaacccggaaa35.gcggaagggcgtaggtttgggatgcttagccgtaggctagctttcgacacgatcgattcgcaccacaggataaaagttaagggaccggt aagtcgcggtagcc36.ctagctacgaacgctttaggcgcccccgggagtagtcgttaccgttagtatagcagtcgcagtcgcaattcgcaaaagtccccagctttag ccccagagtcgacg37.gggatgctgacgctggttagctttaggcttagcgtagctttagggccccagtctgcaggaaatgcccaaaggaggcccaccgggtagat gccasagtgcaccgt38.aacttttagggcatttccagttttacgggttattttcccagttaaactttgcaccattttacgtgttacgatttacgtataatttgaccttattttggac actttagtttgggttac39.ttagggccaagtcccgaggcaaggaattctgatccaagtccaatcacgtacagtccaagtcaccgtttgcagctaccgtttaccgtacgtt gcaagtcaaatccatattagggtttatttacctgtttattttttcccgagaccttaggtttaccgtactttttaacggtttacctttgaaatttttggactagcttaccctgg atttaacggccagttt1、 模型思路:对于此模型来说,利用Matlab 编程可以很容易地统计出各碱基在前二十组的每一组中所出现的次数即i i i i T C G A ,,, (20....3,2,1=i ),然后分别求出1--10组和11--20组中的碱基出现的次数的平均值,1111,,,av av av av T C G A 和2222,,,av av av av T C G A ,即;10/1011∑==i i av A A 、;10/1011∑==i i av G G 、;10/1011∑==i i av C C 、;10/1011∑==i i av T T;10/20112∑==i i av A A 、;10/20112∑==i i av G G 、;10/20112∑==i i av C C 、;10/20112∑==i i av T T计算结果如下表所示:(程序见附录一)1av A1av T1av C1av GA 类0.28670.15420.17590.38332av A2av T2av C2av GB 类0.29550.50180.10180.1009由结果观察可以得出A 类中C G ,的含量之和明显多于T A ,的含量,而B 类中正好相反。