MDCK细胞培养基本技术方法 -2011本
- 格式:docx
- 大小:20.40 KB
- 文档页数:2
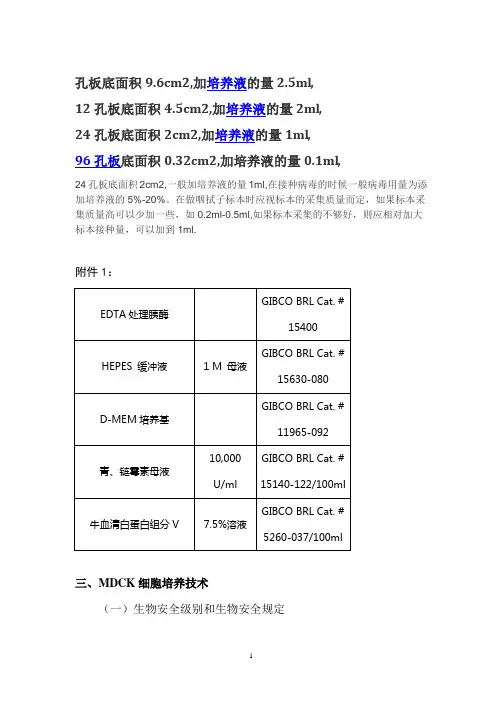
孔板底面积9.6cm2,加培养液的量2.5ml,12孔板底面积4.5cm2,加培养液的量2ml,24孔板底面积2cm2,加培养液的量1ml,96孔板底面积0.32cm2,加培养液的量0.1ml,24孔板底面积2cm2,一般加培养液的量1ml,在接种病毒的时候一般病毒用量为添加培养液的5%-20%。
在做咽拭子标本时应视标本的采集质量而定,如果标本采集质量高可以少加一些,如0.2ml-0.5ml,如果标本采集的不够好,则应相对加大标本接种量,可以加到1ml.附件1:三、MDCK细胞培养技术(一)生物安全级别和生物安全规定1. MDCK细胞培养实验室生物安全级别:正常MDCK细胞培养在生物安全二级实验室进行。
2. MDCK细胞培养实验室生物安全规定:遵守生物安全实验室相应的生物安全规定。
(二)MDCK细胞的传代培养步骤1. 细胞培养所需的材料(部分试剂在此列出生产厂家货号仅供参考,可自行选择):(1)生长成片的MDCK细胞;(2)T-75细胞培养瓶,颈口有倾角;(3)D-MEM培养液;(4)青、链霉素母液(10,000U/mL青霉素G;10,000µg/mL硫酸链霉素),分装后保存于-20℃;(5)HEPES缓冲液,1M母液;(6)胎牛血清;(7)EDTA-胰酶(0.05%胰酶;0.53mMEDTA·4Na)(Gibco CatNo. 25300-062),分装后保存于-20℃;(8)牛血清白蛋白组分V,7.5%溶液(GIBCO CatNo. 15260);(9)1mL和10mL无菌移液管等;(10)70%~75%的酒精。
建议:经常检查试剂使用的有效期和MDCK细胞的代数。
2. 培养基和试剂的准备:(1)500mL的D-MEM液中加入a) 青、链霉素母液5mL(终浓度达:100U/mL青霉素和100µg/mL 链霉素);b) HEPES缓冲液12.5mL(终浓度:25mM);c) 7.5%牛血清白蛋白组分V12.5mL。

实习六幼狗肾(MDCK)传代细胞的换液与继代
目的:认识MDCK传代细胞的形态并掌握其换液与继代操作技术。
材料:MDCK传代细胞、0.02% EDTA、MEM营养液、小牛血清、注射器(2ml、10ml)、吸管、细胞瓶等。
内容与方法:
1.普通光学显微镜下观察MDCK传代细胞的显微形态。
2.每90ml MEM水解乳蛋白液加10ml小牛血清。
3.换液。
将长有细胞的瓶壁转向上方,用注射器从瓶底吸去旧营养液,再加入新营养液,盖紧胶塞,然后将培养瓶反转复原位放置37℃继续培养。
4.继代。
吸(倒)去旧营养液,加入2ml EDTA液漂洗,吸去,再加入5滴EDTA,使其与细胞接触,放置37℃作用30秒--2分钟不等,在显微镜下观察,待细胞变园后吸去EDTA,然后加入20ml新营养液,用注射器反复冲散细胞,最后分为3瓶(6~7ml/瓶),在瓶上方做好标记,置37℃CO2培养箱内培养。

MDCK细胞培养MEM细胞生长液配制500ml MEM液+5ml双抗(青霉素+链霉素)+5ml L-谷氨酰胺+7.5ml碳酸氢钠+50ml小牛血清90%MEM液1%双抗1% L-谷氨酰胺 1.5%碳酸氢钠10%小牛血清0.25%EDTA胰酶(从冰箱取出后放入培养箱中待用)(1)细胞传代将细胞瓶中的生长液倒去,加入MEM液(大概6ml左右)清洗,倒去;加入EDTA-胰酶(根据细胞消化情况而加量),倒去;再加入EDTA-胰酶,放置培养箱,进行消化(大概10分钟左右),待其细胞开始脱落后(在细胞瓶中呈毛玻璃);吸干EDTA-胰酶倒去,放置培养箱一段时间(细胞呈逐个完整),取出;【细胞生长好,多,可1传3或者1传4】将配好的细胞生长液吸入,吹打混匀,待完全脱落至过长液中;将其分装入细胞瓶和细胞板中。
6ml/瓶37℃培养1ml/孔CO2培养箱(用于标本接种)(2)标本处理复融,从-70℃冰箱取出,常温下溶解;充分震荡,后取出试导,至4℃冰箱中,静置20分钟左右;取螺旋管,在其加入0.2ml的双抗(20%),后加入1.8ml的病毒液,放置4℃,过夜上毒。
(3)细胞上毒(需培养一周)洗细胞,将细胞板(前一天)上的液体(生长液)吸干,加入1ml的MEM,清洗,重复一次,然后吸干;加入处理后的病毒液180ul;放置CO2培养箱中1.5h。
将细胞板取;1ml/孔加入病毒过长液后,放置CO2培养箱,培养一周;并将上周上毒的细胞液取出,放置-70℃冰箱,隔天鉴定。
配制病毒过长液:100mlMEM中加入1ml双抗,1ml L-谷氨酰胺1ml TPCK-胰酶 1.5ml碳酸氢钠血凝试验:病毒经系列稀释,与一定浓度的红细胞混合,混合液放置在专为血凝试验设计的板的圆底中,未出现凝集的红细胞可在孔底自由滚动,形成浓密的,极易识别的细胞团,而凝集红细胞不能在池底自由滚动,均匀地覆盖池底。
(4)1%人O型红细胞洗涤首先应选择无乙肝病毒携带者的O型血,待其自然沉降后;吸取红细胞层的红细胞置入离心管中;加入无菌生理盐水至离心管2/3处,两管平衡后,1800r/10min;用无菌吸管吸去生理盐水,再加入生理盐水、混匀;重新离心,重复3次,离心取出后;取细胞瓶,吸取9ml生理盐水;加入1ml的离心好的红细胞,混匀即可。
![[教学]MDCK细胞培养技术](https://uimg.taocdn.com/538dfcd583d049649b6658e9.webp)
[教学]MDCK细胞培养技术孔板底面积9.6cm2,加培养液的量2.5ml,培养液的量2ml, 12孔板底面积4.5cm2,加24孔板底面积2cm2,加培养液的量1ml,96孔板底面积0.32cm2,加培养液的量0.1ml,24孔板底面积2cm2,一般加培养液的量1ml,在接种病毒的时候一般病毒用量为添加培养液的5%-20%。
在做咽拭子标本时应视标本的采集质量而定,如果标本采集质量高可以少加一些,如0.2ml-0.5ml,如果标本采集的不够好,则应相对加大标本接种量,可以加到1ml.附件1:GIBCO BRL Cat. # EDTA处理胰酶 15400GIBCO BRL Cat. # HEPES 缓冲液 1 M 母液 15630-080GIBCO BRL Cat. # D-MEM培养基 11965-092GIBCO BRL Cat. # 青、链霉素母液 10,000 U/ml 15140-122/100mlGIBCO BRL Cat. # 牛血清白蛋白组分V 7.5%溶液 5260-037/100ml三、MDCK细胞培养技术(一)生物安全级别和生物安全规定1. MDCK细胞培养实验室生物安全级别:正常MDCK细胞培养在生物安全二级实验室进行。
2. MDCK细胞培养实验室生物安全规定:遵守生物安全实验室相应的生物安全规定。
(二)MDCK细胞的传代培养步骤1. 细胞培养所需的材料(部分试剂在此列出生产厂家货号仅供参考,可自行选择):(1) 生长成片的MDCK细胞;(2) T-75细胞培养瓶,颈口有倾角;(3) D-MEM培养液;(4) 青、链霉素母液(10,000U/mL青霉素G;10,000µg/mL硫酸链霉素),分装后保存于-20?;(5) HEPES缓冲液,1M母液;(6) 胎牛血清;(7) EDTA-胰酶(0.05,胰酶;0.53mMEDTA?4Na)(Gibco CatNo.25300-062),分装后保存于-20?;(8) 牛血清白蛋白组分V,7.5,溶液(GIBCO CatNo. 15260);(9) 1mL和10mL无菌移液管等;(10) 70,,75,的酒精。
MDCK细胞培养MEM细胞生长液配制500ml MEM液+5ml双抗(青霉素+链霉素)+5ml L-谷氨酰胺+7.5ml碳酸氢钠+50ml小牛血清90%MEM液1%双抗1% L-谷氨酰胺 1.5%碳酸氢钠10%小牛血清0.25%EDTA胰酶(从冰箱取出后放入培养箱中待用)(1)细胞传代将细胞瓶中的生长液倒去,加入MEM液(大概6ml左右)清洗,倒去;加入EDTA-胰酶(根据细胞消化情况而加量),倒去;再加入EDTA-胰酶,放置培养箱,进行消化(大概10分钟左右),待其细胞开始脱落后(在细胞瓶中呈毛玻璃);吸干EDTA-胰酶倒去,放置培养箱一段时间(细胞呈逐个完整),取出;【细胞生长好,多,可1传3或者1传4】将配好的细胞生长液吸入,吹打混匀,待完全脱落至过长液中;将其分装入细胞瓶和细胞板中。
6ml/瓶37℃培养1ml/孔CO2培养箱(用于标本接种)(2)标本处理复融,从-70℃冰箱取出,常温下溶解;充分震荡,后取出试导,至4℃冰箱中,静置20分钟左右;取螺旋管,在其加入0.2ml的双抗(20%),后加入1.8ml的病毒液,放置4℃,过夜上毒。
(3)细胞上毒(需培养一周)洗细胞,将细胞板(前一天)上的液体(生长液)吸干,加入1ml的MEM,清洗,重复一次,然后吸干;加入处理后的病毒液180ul;放置CO2培养箱中1.5h。
将细胞板取;1ml/孔加入病毒过长液后,放置CO2培养箱,培养一周;并将上周上毒的细胞液取出,放置-70℃冰箱,隔天鉴定。
配制病毒过长液:100mlMEM中加入1ml双抗,1ml L-谷氨酰胺1ml TPCK-胰酶 1.5ml碳酸氢钠血凝试验:病毒经系列稀释,与一定浓度的红细胞混合,混合液放置在专为血凝试验设计的板的圆底中,未出现凝集的红细胞可在孔底自由滚动,形成浓密的,极易识别的细胞团,而凝集红细胞不能在池底自由滚动,均匀地覆盖池底。
(4)1%人O型红细胞洗涤首先应选择无乙肝病毒携带者的O型血,待其自然沉降后;吸取红细胞层的红细胞置入离心管中;加入无菌生理盐水至离心管2/3处,两管平衡后,1800r/10min;用无菌吸管吸去生理盐水,再加入生理盐水、混匀;重新离心,重复3次,离心取出后;取细胞瓶,吸取9ml生理盐水;加入1ml的离心好的红细胞,混匀即可。

附件7甲型H1N1流感病毒的MDCK细胞分离方法一、目的流感病毒的MDCK细胞分离方法是流感实验的关键技术,用于流感病毒的分离、培养。
本SOP是为确保MDCK细胞分离甲型H1N1病毒的操作准确、规范和可靠而制定。
二、程序(一)生物安全要求甲型H1N1病毒的MDCK细胞分离培养实验室生物安全级别:BSL-3。
实验操作人员需进行BSL-3防护。
甲型H1N1病毒的MDCK细胞分离操作必须在BSL-3级实验室的生物安全柜中进行。
(二)材料1、75%~90%成片的MDCK细胞,选用合适大小的细胞培养瓶和细胞培养板来用做病毒分离2、TPCK处理胰酶(牛胰腺来源Ⅷ型)3、HEPES缓冲液,1M母液(GIBCO Cat No 15630)4、D-MEM培养基(高糖含有L-谷氨酰胺 GIBCO Cat No 11965),Hank's液(病毒所配液室配制)5、青、链霉素母液(10000 U/mL 青霉素G;10000 µg/mL硫酸链霉素(GIBCO Cat No 15140)6、牛血清白蛋白组分Ⅴ,7.5%溶液(GIBCO Cat No 15620)7、临床样品0.5mL8、1mL无菌移液管9、10mL无菌移液管10、15mL无菌离心管(三)实验步骤1、准备病毒生长液1)细胞维持液准备500mL D-MEM液中加入下列试剂:(1)青、链霉素母液5mL(终浓度达:100U/mL 青霉素;100µg/mL链霉素)(2)牛血清白蛋白组分Ⅴ 12.5mL(终浓度:0.2%)(3)HEPES缓冲液12.5mL(终浓度:25mM)2)病毒生长液每500mL细胞维持液中加入0.5mL的TPCK-胰酶(母液浓度为2mg/mL)使TPCK-胰酶的终浓度为2µg/mL。
2、甲型H1N1病毒MDCK细胞分离步骤1)75%~90%成片细胞的准备,以选取T25细胞瓶为例。
①用40×物镜观察细胞生长状态。

MDCK细胞培养一、目的MDCK细胞培养是分离流感病毒及相关研究实验的基本技术。
疾控中心的所有技术人员,必须按照本文件相关的操作规程进行操作。
二、适用范围适用于疾控中心所有技术人员。
三、程序(一)生物安全要求实验室生物安全级别:BSL-1所有操作必须在BSL-1实验室的生物安全柜里进行。
(二)材料1.生长成片的MDCK细胞2.无菌的T25细胞培养瓶3.D-MEM培养液(含有L-谷氨酰胺)4.青、链霉素母液(10000U/mL青霉素G;10000µg/mL硫酸链霉素),分装后保存于-20℃5.HEPES缓冲液,1M母液6.胎牛血清7.EDTA-胰酶(0.05%胰酶,0.53mMEDTA-4Na),分装后保存于-20℃8.7.5%牛血清白蛋白组分V9.1mL、10mL无菌移液管10.70%~75%的酒精注意事项:经常检查试剂使用的有效期。
(三)实验步骤这里以T75细胞瓶的单层细胞培养为例,叙述MDCK细胞的培养程序。
如果细胞瓶的规格有变,MDCK细胞悬液的量必须做相应的调整。
1.D-MEM培养液的准备500mLD-MEM液中加入:青、链霉素母液5mL(终浓度达:100U/mL青霉素;100µg/mL链霉素),HEPES缓冲液12.5mL(终浓度:25mM)。
7.5%牛血清白蛋白组分Ⅴ12.5mL2.细胞生长液的准备胎牛血清10mL加到90mL的上述(1)的液体中,使胎牛血清的终浓度为10%。
3.首先将细胞培养瓶中的培养液弃去,加入5mL在37℃水浴中预热的EDTA-胰酶。
4.温和地摇动细胞瓶1min,使EDTA-胰酶均匀分布在整个细胞薄层。
然后用移液管吸去EDTA胰酶。
5.重新加入5mL在37℃水浴中预热的EDTA-胰酶重复上述步骤。
6.加入1mLEDTA-胰酶使其均匀分布在整个细胞薄层,37℃孵育细胞瓶直至细胞从塑料细胞瓶的表面分离(约5~10min)。
必要时可以摇动或吹打来分离细胞。

MDCK细胞介绍(MDCK Cells introduction)MDCK细胞系(MDCK Cell Lines)由Madin和Darby于1958年从美国Cocker Spaniel母曲架犬的肾脏组织分离培育建立,通常是以贴壁方式生长的上皮样细胞。
犬肾上皮连续细胞系MDCK (Madin-Daby canine kidney cells) 细胞株购买信息:ATCC MDCK cell lines。
目前,MDCK细胞系(MDCK Cell Lines)广泛用于多种病毒的扩增和纯化,如:呼肠孤病毒)、腺病毒、犬细小病毒、猫粒细胞缺乏症病毒)及禽流感病毒等。
由于其病毒感染效率高、增殖快,且不易变异,MDCK细胞系(MDCK Cell Lines)被公认为最适于甲、乙型流感病毒疫苗生产的3种细胞系之一。
传统的MDCK细胞培养大多采用有血清贴壁培养方式。
血清是由血浆去除纤维蛋白而形成的一种复杂混合物,其含有细胞生长所需的生长因子、激素、载体蛋白、贴壁因子、微量元素以及其他营养物质,可以有效地促进细胞生长和产物表达。
然而,血清的应用也存在许多问题:易受病毒、支原体或其他病原体的污染;批间差异造成产品批次间的质量难以严格控制;大量血清蛋白的存在增加了下游分离纯化的难度,部分蛋白难以通过分离纯化手段彻底去除,影响了产品的最终质量;此外,血清来源困难、价格昂贵,大规模动物细胞培养过程中使用血清将会大大增加生产成本。
犬肾细胞MDCK无血清培养基 UltraMDCK Serum-free Medium 产品介绍MDCK无血清培养基是一种在经过优化的基础培养基中只添加了重组人胰岛素蛋白和牛转铁蛋白的低蛋白含量的成分配制而成的SFM培养基。
与在含有血清生长环境中相比,在MDCK 无血清培养基中生长的MDCK细胞较小并紧密成团。
在不更换培养基的条件下,MDCK细胞可以持续旺盛生长至少2周时间。
经过MDCK细胞的持续生长,从单层细胞形成球形结构,通常我们称这种球形结构为“漂浮物”。

激流式生物反应器培养MDCK细胞条件优化余彬辉;王斌卿;胡志航;朱炳林;李肖梁;方维焕【摘要】应用激流式生物反应器培养MDCK细胞,从培养基种类、接种密度、pH 和溶氧量等关键参数进行优化.结果表明,激流式生物反应器培养MDCK细胞,细胞接种密度为1.5×106个/mL、pH7.2、溶氧量50%~60%、循环流量400mL/min和振荡频率60 r/min条件下培养5d可达到最大细胞密度(1.02×10 7个/mL)及最高糖耗量(3.1 g/L),为激流生物反应器大规模培养细胞及病毒疫苗生产奠定基础.【期刊名称】《动物医学进展》【年(卷),期】2012(033)010【总页数】5页(P16-20)【关键词】激流式生物反应器;MDCK细胞;培养条件【作者】余彬辉;王斌卿;胡志航;朱炳林;李肖梁;方维焕【作者单位】浙江大学动物预防医学研究所,浙江省动物预防医学重点实验室,浙江杭州310058;杭州荐量兽用生物制品有限公司,浙江杭州310018;杭州荐量兽用生物制品有限公司,浙江杭州310018;杭州荐量兽用生物制品有限公司,浙江杭州310018;浙江大学动物预防医学研究所,浙江省动物预防医学重点实验室,浙江杭州310058;浙江大学动物预防医学研究所,浙江省动物预防医学重点实验室,浙江杭州310058;浙江大学动物预防医学研究所,浙江省动物预防医学重点实验室,浙江杭州310058【正文语种】中文【中图分类】Q813.1当今病毒性疫苗多利用转瓶培养,生产工艺不仅费时费力,而且占用空间大,更重要的是病毒滴度不稳定,批次之间病毒效价差异大。
随着疫苗市场的迅速发展,其生产技术将经历一场深刻的技术革新,从转瓶培养向生物反应器培养转变。
这种转变符合世界生物制药发展趋势,也表明我国疫苗生产技术将逐步与国际疫苗生产技术接轨。
动物细胞大规模培养在疫苗生产领域发挥着越来越重要的作用[1]。
动物细胞大规模培养和表达对生产疫苗有明显的优势,一方面活性高、稳定性好,是最重要的表达系统,但动物细胞大规模培养生产疫苗还存在一定问题[2-3],培养密度和产物浓度较低,在技术和设备上远没有原核细胞培养成熟。

MDBK(牛肾细胞)细胞培养方法MDBK(牛肾细胞)细胞培养方法传代步骤:弃去旧的生长液-->用无钙镁水(CMF)冲洗贴壁细胞数次(视细胞瓶的大小,3--5次)-->剩少许CMF,滴入 ATV 2--3滴-->摇匀细胞瓶中的液体,使ATV均匀的分布到瓶里的没个地方-->放入37度二氧化碳温箱中3--5min消化-->弃去ATV,加入生长液,拍打吸吹下细胞-->镜下观察-->分瓶-->作好记号,放入37度二氧化碳温箱生长细胞特点:该细胞生长力强,而且快速一般24h就可以张到80%,48h不传就会变的很老,所以该细胞传时要勤点;我感觉MDBK还是很好传的,比以前我传的ST、Vero细胞等要好传一些,且不那么容易死亡,所以是我的一个比较好的选择!该细胞适合接种疱疹病毒(如:伪狂犬病毒)!BHK-21:弃除旧的培养液后---用缓冲液冲洗一遍----用0.22%的胰酶消化液消化约两分钟左右可以看到细胞像沙子一样脱落-------赶紧倒掉胰酶加入含10%的牛血清培养液终止消化。
但目前国内能找到的BHK-21细胞大部分都有支原体感染,如果是好的BHK-21细胞能做到1比20的分种率。
补充一点关于BHK-21的培养,先用少量胰酶冲洗一下,弃去,再用胰酶消化1min左右,效果会好一些皮肤成纤维细胞和THP-1细胞:皮肤成纤维细胞是贴壁生长的。
THP-1细胞是悬浮的。
由于我们实验室养细胞的时间也不是很长,所以也只能说说自己平时的操作了。
先说皮肤成纤维细胞。
当细胞铺满瓶底,也就是细胞与细胞之间没有间隙时,就可以传代了。
一般十天左右传一代。
先吸弃旧培养液,用D-Hanks液洗两遍,再加入0.25%的胰酶,加入的量以能覆盖瓶底为准。
加入后轻轻摇晃培养瓶使液体覆盖瓶底,然后静置两分钟左右,最好是能在显微镜下观察,当细胞之间出现间隙且有一部分细胞呈现圆形后即可。
吸去胰酶,加入含血清培养液,摇晃瓶子使培养液覆盖瓶底就行了。
MDCK细胞培养一、目的MDCK细胞培养是分离流感病毒及相关研究实验的基本技术。
疾控中心的所有技术人员,必须按照本文件相关的操作规程进行操作。
二、适用范围适用于疾控中心所有技术人员。
三、程序(一)生物安全要求实验室生物安全级别:BSL-1所有操作必须在BSL-1实验室的生物安全柜里进行。
(二)材料1.生长成片的MDCK细胞2.无菌的T25细胞培养瓶3.D-MEM培养液(含有L-谷氨酰胺)4.青、链霉素母液(10000U/mL青霉素G;10000µg/mL硫酸链霉素),分装后保存于-20℃5.HEPES缓冲液,1M母液6.胎牛血清7.EDTA-胰酶(0.05%胰酶,0.53mMEDTA-4Na),分装后保存于-20℃8.7.5%牛血清白蛋白组分V9.1mL、10mL无菌移液管10.70%~75%的酒精注意事项:经常检查试剂使用的有效期。
(三)实验步骤这里以T75细胞瓶的单层细胞培养为例,叙述MDCK细胞的培养程序。
如果细胞瓶的规格有变,MDCK细胞悬液的量必须做相应的调整。
1.D-MEM培养液的准备500mLD-MEM液中加入:青、链霉素母液5mL(终浓度达:100U/mL青霉素;100µg/mL链霉素),HEPES缓冲液12.5mL(终浓度:25mM)。
7.5%牛血清白蛋白组分Ⅴ12.5mL2.细胞生长液的准备胎牛血清10mL加到90mL的上述(1)的液体中,使胎牛血清的终浓度为10%。
3.首先将细胞培养瓶中的培养液弃去,加入5mL在37℃水浴中预热的EDTA-胰酶。
4.温和地摇动细胞瓶1min,使EDTA-胰酶均匀分布在整个细胞薄层。
然后用移液管吸去EDTA胰酶。
5.重新加入5mL在37℃水浴中预热的EDTA-胰酶重复上述步骤。
6.加入1mLEDTA-胰酶使其均匀分布在整个细胞薄层,37℃孵育细胞瓶直至细胞从塑料细胞瓶的表面分离(约5~10min)。
必要时可以摇动或吹打来分离细胞。
MDCK细胞培养
一、目的MDCK细胞培养是分离流感病毒及相关研究实验的基本技术。
二、适用范围适用于疾控中心所有技术人员。
三、程序
(一)生物安全要求实验室生物安全级别:BSL-1所有操作必须在BSL-1实验室的生物安全柜里进行。
(二)材料
1.生长成片的MDCK细胞
2.无菌的T25细胞培养瓶
3.D-MEM培养液(含有L-谷氨酰胺)
4.青、链霉素母液(10000U/mL青霉素G;10000µg/mL硫酸链霉素),分装后保存于-20℃
5.HEPES缓冲液,1M母液
6.胎牛血清
7.EDTA-胰酶(0.05%胰酶,0.53mMEDTA-4Na),分装后保存于-20℃8.7.5%牛血清白蛋白组分V9.1mL、10mL无菌移液管10.70%~75%的酒精注意事项:经常检查试剂使用的有效期。
(三)实验步骤这里以T75细胞瓶的单层细胞培养为例,叙述MDCK细胞的培养程序。
如果细胞瓶的规格有变,MDCK细胞悬液的量必须做相应的调整。
1.D-MEM培养液的准备
500mLD-MEM液中加入:青、链霉素母液5mL(终浓度达:100U/mL青霉素;100µg/mL链霉素),HEPES缓冲液12.5mL(终浓度:25mM)。
7.5%牛血清白蛋白组分Ⅴ12.5mL
2.细胞生长液的准备
胎牛血清10mL加到90mL的上述(1)的液体中,使胎牛血清的终浓度为10%。
3.首先将细胞培养瓶中的培养液弃去,加入5mL在37℃水浴中预热的EDTA-胰酶。
4.温和地摇动细胞瓶1min,使EDTA-胰酶均匀分布在整个细胞薄层。
然后用移液管吸去EDTA胰酶。
5.重新加入5mL在37℃水浴中预热的EDTA-胰酶重复上述步骤。
6.加入1mLEDTA-胰酶使其均匀分布在整个细胞薄层,37℃孵育细胞瓶直至细胞从塑料细胞瓶的表面分离(约5~10min)。
必要时可以摇动或吹打来分离细胞。
然后加入1mL胎牛血清灭活残余的胰酶。
7.加9mL已经配置好的含有L-谷氨酸的D-MEM培养液,轻轻用移动移液管来吹散细胞团。
8.取10mL混合物加到90mL细胞生长液(细胞悬液的浓度大约为每毫升含105细胞)
9.每个T25细胞培养瓶加入6mL(6×105/mL)细胞悬液,剩余的细胞悬液可以加到T75细胞瓶用于细胞传代。
通常6mL细胞悬液2~3日可生长成片(80%~90%)的单层细胞。
10.于37℃,5%CO2培养箱里培养细胞,每天观察细胞状态,以供进一步实验用。