原料药分析方法开发流程
- 格式:docx
- 大小:11.30 KB
- 文档页数:4


原料药药物分析流程English Answer:Drug Substance Analytical Workflow.1. Incoming Material Inspection.Verify Certificate of Analysis (CoA) and documentation.Inspect packaging and labeling for integrity and compliance.Perform visual inspection for any apparent defects or contamination.2. Raw Material Testing.Identification: Confirm the identity of the rawmaterial using spectroscopic techniques (e.g., IR, UV, MS)。
Assay: Determine the potency and purity of the raw material using quantitative analytical methods (e.g., HPLC, GC)。
Water Content: Measure the amount of water present in the raw material using Karl Fischer titration or other techniques.Residual Solvents: Analyze for volatile organic impurities that may be present as residual solvents using headspace GC.Elemental Impurities: Determine the presence of heavy metals and other elemental impurities using atomic absorption spectroscopy or inductively coupled plasma mass spectrometry.Microbiological Testing: Perform microbial limit tests to ensure the absence of unacceptable levels of bacteria, fungi, and yeast.3. In-Process Testing.Identity: Verify the identity of intermediates and final product at critical stages of the manufacturing process.Assay: Monitor the potency of the drug substance throughout the synthesis.Impurity Profiling: Identify and quantify impurities that may form during the manufacturing process.Residual Solvents: Monitor for the presence of residual solvents at various stages of the process.4. Final Product Testing.Identity: Confirm the identity of the final drug substance using a combination of analytical techniques.Assay: Determine the potency and purity of the final product to ensure it meets specifications.Impurity Profiling: Conduct comprehensive impurity analysis to identify and quantify all potential impurities.Residual Solvents: Ensure compliance with regulatory limits for residual solvents.Water Content: Measure the amount of water present in the final product.Elemental Impurities: Determine the presence of heavy metals and other elemental impurities.Microbiological Testing: Perform microbial limit tests to meet pharmacopoeial and customer requirements.5. Stability Testing.Conduct long-term and accelerated stability studies to monitor the stability of the drug substance under various storage conditions.Evaluate the effects of temperature, humidity, light,and other factors on the potency, purity, and impurity profile of the drug substance.6. Release Testing.Review all analytical data and ensure it meets regulatory and customer requirements.Approve or reject the release of the drug substance for further processing or distribution.Chinese Answer:原料药药物分析流程。

化学原料药制备的生产工艺分析摘要:化学原料的制备对化学生产及正常生活影响重大,且对后续药品生产有诸多影响。
原料药的生产是医药产业的根源,在医药产业中地位重要。
对原料药的生产采取审批制度支持,原料药物须有批文才可生产。
虽然我国化学原料药制备工艺较为成熟,但是和国际发达国家还不能相提并论。
故以下对化学原料药制备生产工艺分析,以此为推动国家化学原料生产行业进一步发展提供一定支持。
关键词:化学;原料;制备;生产;工艺化学原料药是医学药品发挥药理作用的主要活性成分,其在辅料配合下,用于临床治疗[1]。
在国家原料药注册审批制度支持下,原料药需和制剂经重返的临床前研究后才可投入使用。
国家高度重视化学原料药制备及生产,构建《化学用药物原料药制备和结构确证研究的技术指导原则》,积极推动国家原料药生产向国际化方向发展[2]。
但在原料药物的研究及评价中仍然需要了解生产工艺的细节,便于后续对化学原料药制备进行创新分析。
1.研究背景伴随国家经济水平不断提高,相关医学也得到新的发展[3]。
与此同时,引进西药是医疗系统的重要基础,国家积极研制各项新药品、试剂。
新药研发中需要用到各式各样的化学原料药,但随着制药企业增多,相关制药硬件及软件技术也需要不断完善。
制药工艺技术要求高、操作复杂,若生产工艺稍有不慎,就会威胁工作人员生命健康、安全[4]。
国家出台《化学用药物原料药制备和结构确证研究的技术指导原则》,为化学原料制备工艺提供技术指导规范支持,可供研发人员参考、借鉴,提高药品研发效率,促进制药企业长久发展。
2.化学原料药制备生产过程化学原料药制备的生产过程如表1所示。
上述几阶段可代表化学原料药制备一般过程,但由于化学原料不同、生产药物不同等,其对应的生产阶段也有所差异,实际制备生产中,需结合实际情况反复调整,甚至需要推倒全过程实现目的[5]。
故在设计初期,应及时评价后续各阶段的工作难度,分析各阶段的可行性,避免生产进行到一半无法继续向前[6]。

原料药中试生产方案原料药中试生产方案的目的是进行中试生产,验证和优化药物的合成路线和工艺参数,确定最适合的工艺条件,并获取足够的中试生产样品,用于药物的进一步研发和评价。
下面是一个基本的原料药中试生产方案的步骤:1. 前期准备:确认药物合成路线和工艺参数,收集和准备所需的原料和试剂,准备所需的设备和工艺辅助材料。
2. 反应条件确定:根据药物的合成路线和工艺参数,确定反应的温度、时间、pH值等条件。
通过小规模试验和实验室仪器进行初步验证。
3. 冶炼剂的制备:根据反应条件确定的温度和时间,制备所需的冶炼剂。
通过小规模试验和实验室仪器进行验证。
4. 中试反应器操作:将制备好的冶炼剂和原料按照一定的比例加入中试反应器中。
通过控制反应温度、时间和搅拌速度等参数,进行中试反应。
5. 反应结束和产物分离:根据反应的终点和产物的性质,确定反应结束的时间。
通过过滤、结晶、萃取、蒸馏等方法,将反应产物与副产品分离。
6. 产物纯化和结晶:将分离得到的产物进行进一步纯化。
通过溶剂、蒸馏、结晶等方法,提高产物的纯度和产率。
7. 产品检测和分析:对纯化后的产物进行产品检测和分析。
通过质量分析和性能测试等方法,确定产物的质量和合格性。
8. 中试样品制备:将纯化和分析合格的产物,按照一定的规格和数量,制备成中试样品。
用于进一步研发和评价,包括药理学、毒理学和临床试验等环节。
9. 数据分析和总结:对中试生产过程的数据进行分析和总结。
评估生产过程的可行性和有效性,为进一步工业化生产提供数据支持。
10. 环境和安全控制:在中试生产过程中,要严格遵守环境和安全规定。
保护环境,确保工作人员的安全和健康。
以上是一个基本的原料药中试生产方案的步骤。
具体方案的细节和操作流程,应根据不同的药物和工艺条件进行调整和优化。

药品及生物制品的分析方法和方法验证指导原则目录1.介绍...................... (1)2.背景..................... .. (2)3.分析方法开发. ..................... . (3)4.分析程序内容.............................................. ......... ..................................... .. 3A.原则/范围 (4)B.仪器/设备............................................. . (4)C.操作参数.............................................. .. (4)D.试剂/标准............................................. . (4)E.样品制备.............................................. .. (4)F.标准对照品溶液的制备............................................ .. (5)G.步骤......... ....................................... (5)H.系统适应性..... (5)I.计算 (5)J.数据报告 (5)5.参考标准和教材............................................ (6)6分析方法验证用于新药,仿制药,生物制品和DMF (6)A.非药典分析方法............................................. (6)B.验证特征 (7)C.药典分析方法............................................. .. (8)7.统计分析和模型 (8)A.统计 (8)B.模型 (8)8.生命周期管理分析程序 (9)A.重新验证 (9)B.分析方法的可比性研究............................................ . (10)1.另一种分析方法............................................... .. (10)2.分析方法转移的研究 (11)C.报告上市后变更已批准的新药,仿制药,或生物制品 (11)9.美国FDA方法验证............................................... . (12)10.参考文献前言本指导原则草案,定稿后,将代表美国食品和药物管理局(FDA)目前关于这个话题目前的想法。

原料药分析方法开发流程精品范本开发一种原料药分析方法需要经历以下步骤:1.收集文献资料:首先需要收集与该原料药相关的文献资料,了解已有的分析方法以及已知的方法的优缺点。
这些信息可以帮助我们制定开发方向和策略。
2.确定目标:接下来,需要明确分析方法的目标。
这包括确定分析参数,例如精确度、准确度、线性范围、灵敏度、选择性、重复性等。
同时需要考虑实际生产中可能遇到的问题,例如药物纯度、杂质水平等。
3.选择适当方法:根据收集到的文献资料和目标,选择适当的方法进行开发。
这可能涉及到物理化学性质测试、色谱法、质谱法、荧光法等不同的技术方法。
4.优化方法参数:在选择方法后,需要对方法进行优化。
这通常涉及到尝试不同的实验条件,例如流速、洗脱剂浓度、柱温等。
通过不断调整这些参数,找到最佳方法。
5.验证方法:一旦找到最佳方法,需要对其进行验证。
这包括验证方法的准确度、精确度、线性范围、选择性等。
一般使用已知含量样品和不同浓度的样品进行验证。
6.稳定性研究:分析方法的稳定性也是一个关键因素。
在稳定性研究中,可以评估方法在不同时间点、不同储存条件下的性能。
这有助于确定方法的可靠性和稳定性。
7.编写方法标准操作程序(SOP):完成验证后,需要编写方法的标准操作程序。
SOP应包括详细的实验步骤、操作条件、仪器使用指南等。
这将作为使用该方法的指导手册。
8.培训和推广:最后,需要对方法进行培训和推广。
在制药工业中,开发的方法通常需要被多个实验室广泛应用。
因此,培训其他实验室的人员使用该方法是非常重要的。
以上是一份关于原料药分析方法开发流程的精品范本。
这个流程可以帮助研究人员系统地进行原料药分析方法的开发,保证方法的准确性和可靠性。
当然,实际开发中可能还需要根据具体的情况进行调整和修改。

介绍原料药研发全文共四篇示例,供读者参考第一篇示例:原料药是指用作制备制剂和制品或进行药物疗法的药物成分。
原料药的研发是药物研发的第一步,是现代医药工业中最重要的环节之一。
在原料药研发过程中,研究人员需要对原料药的化学结构、药理学特性、制备方法等方面进行深入研究,以确保药物的疗效和安全性。
原料药研发的过程通常包括以下几个关键步骤:1. 发现新药物:这是原料药研发的第一步,研究人员通过高通量筛选、计算机辅助设计等方法发现具有潜在药理活性的化合物。
2. 药物设计:在发现新药物后,研究人员需要对化合物进行结构优化,以提高药物的活性、选择性和药代动力学性质。
3. 合成制备:在确定了最终的化合物结构后,研究人员需要设计出合成路线,合成原料药的中间体和最终产物。
4. 药物评价:研究人员需要对合成的原料药进行生物药学、药效学和毒理学评价,以确保药物的有效性和安全性。
5. 临床研究:在通过前期的药物评价后,研究人员需要进行临床试验,评估药物在人体内的疗效和安全性。
原料药研发是一项复杂而精细的工作,需要团队间的密切合作和高度的技术水平。
研究人员通常需要具备化学、药理学、药学和生物学等多学科知识,以应对原料药研发中的各种挑战。
原料药研发还需要大量的资金投入和长期的时间支持,因此很多公司会选择与其他企业或研究机构进行合作,共同推动原料药的研发进程。
在原料药研发中,国际上有很多先进的技术和方法可供选择。
计算机辅助药物设计可以加速新药物的发现和设计过程;高通量筛选技术可以快速筛选出具有潜在活性的化合物;凝胶滴定法和液相色谱-质谱联用技术可以提高原料药的纯度和稳定性。
原料药研发是一项具有挑战性和前景广阔的工作。
随着科技的不断发展和创新,我们相信在未来会有更多更好的原料药问世,为人类的健康和福祉作出更大的贡献。
第二篇示例:原料药研发是制药行业中非常重要的一环,它直接影响到药品的质量和效果。
原料药是药品中最主要的成分,其质量和纯度直接关系到药品的疗效和安全性。

原料药分析方法开发流程原料药的分析方法开发是一个以实验为基础的过程,旨在确定原料药中特定成分的含量、结构和纯度等信息。
下面是一般的原料药分析方法开发流程:1.确定分析目标:首先需要明确分析的目的,包括确定主要成分的含量、确定杂质的种类和含量、验证方法等。
根据目标的不同,选择不同的分析方法,如色谱法、光谱法、质谱法等。
3.初步试验和参数优化:通过初步试验,确定一些基本参数,如试剂的选择和浓度、仪器的选择和条件等。
然后进行参数优化,通过修改和调整参数,以获得更准确和稳定的结果。
4.方法开发和验证:根据前期试验的结果,在此阶段进行方法的设计和开发。
包括决定试剂的用量和浓度、建立适当的标准曲线、选择合适的色谱柱和检测器等。
5.方法验证和准确性评估:通过验证试验,对开发的方法进行准确性、精确性、线性范围、检测限、可重复性和选择性等方面的评估。
可以进行重复性试验和对标准物质的回收率试验。
6.方法优化和修正:在实际应用中,可能会遇到一些问题或优化的空间,例如峰的分离不清晰、分析时间太长等。
根据实际需求,进行方法的修正和优化,以改进方法的性能。
7.编写方法文件和报告:在方法开发完成后,需要编写相应的方法文件和开发报告。
方法文件应包括方法的名称、原理、应用范围、试剂和仪器的规格等内容。
报告则应详细描述方法的开发过程、参数优化、验证结果和方法的应用范围等。
8.方法转移和培训:根据具体情况,将开发好的方法转移到实际应用环境中,并向相关人员进行培训,使其能够熟练掌握和运用该方法。
以上是一般的原料药分析方法开发流程。
实际操作时,还需要根据具体的需求和实验条件进行一些个性化的调整和改进。
分析方法的开发是一个不断优化和改进的过程,需要通过实验和实践不断探索,以获得更准确和可靠的结果。
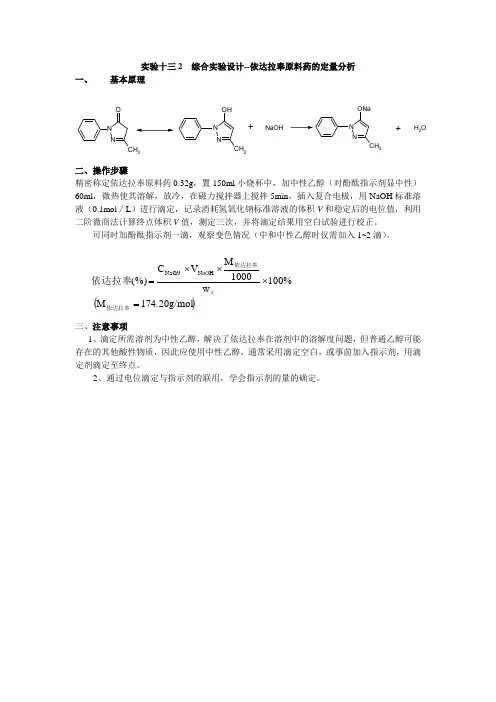
实验十三2 综合实验设计--依达拉奉原料药的定量分析一、 基本原理二、操作步骤精密称定依达拉奉原料药0.32g ,置150ml 小烧杯中,加中性乙醇(对酚酞指示剂显中性)60ml ,微热使其溶解,放冷,在磁力搅拌器上搅拌5min ,插入复合电极,用NaOH 标准溶液(0.1mol ∕L )进行滴定,记录消耗氢氧化钠标准溶液的体积V 和稳定后的电位值,利用二阶微商法计算终点体积V 值,测定三次,并将滴定结果用空白试验进行校正。
可同时加酚酞指示剂一滴,观察变色情况(中和中性乙醇时仅需加入1~2滴)。
三、注意事项1、滴定所需溶剂为中性乙醇,解决了依达拉奉在溶剂中的溶解度问题,但普通乙醇可能存在的其他酸性物质,因此应使用中性乙醇,通常采用滴定空白,或事前加入指示剂,用滴定剂滴定至终点。
2、通过电位滴定与指示剂的联用,学会指示剂的量的确定。
NN 3O N N 3OH +NaOH N N CH 3ONa +H 2O ()4.20g/mol 71M 100%w 1000M V C (%)H N NaO =⨯⨯⨯=依达拉奉依达拉奉依达拉奉s aO H样品-01--滴定及其导数曲线E(mV)V(mL)10.6-311.815.0016.930dE/dV168.3终点1 体积:16.465mL 电位:-256.6mV浓度:5.392e+3mg/L, 102.64254%样品-02--滴定及其导数曲线E(mV)V(mL)4.7-314.615.0016.09dE/dV184.7终点1 体积:15.610mL 电位:-256.5mV浓度:5.112e+3mg/L, 102.69095%四、数据及结果五、讨论对于在50~100ml的溶液中加入0.1%酚酞指示剂2~3滴,pH=9时出现红色,又因为指示剂本身为弱酸或弱碱,故控制指示剂的用量。
在本实验中加了1滴酚酞中和中性乙醇,同时通过电位滴定与指示剂的联用,学会判断指示剂的用量,所以不宜加过多酚酞。

化学原料药的分析方法验证、转移及确认化学原料药的分析方法是指通过制药的相关步骤,对药品的生产流程进行全方位的分析,它包括样品对比参照物以及药品的相应试剂。
研究者需要通过一定的仪器和计算公式,对药品的成分和药效进行科学合理的分析。
通常情况下,分析方法有不同的种类,可以根据化学原料药的成分进行含量测试,也可以对化学原料药的杂质进行分析,其中包括定量测试和限度测试。
另外还可以测试化学原料药的化学性能,例如,原料药的溶出度测试、效价测试、含量均匀度测试,还可以进行化学原料药的鉴别测试。
二、分析方法验证(一)分析方法验证的相关概念根据《美国药典》中的相关内容,我们可以得知按照已经成立的实验室研究证明方法,可以对化学原料药的生产流程和技术要求进行验证。
ICHQ2中提到了分析方法验证的目标是测验化学原料药的作用是否能够达到预期的目的,从而保证药品能够满足实验需求。
在《工业指南》中,根据定义分析法可以得知,化学原料药的阐述分析适用于化学原料药的使用目的及过程。
这种分析方法需要实验者明确药品的使用目的,从而进行深入分析和验证。
CFDA中提到的分析方法验证的定义是证明化学原料药的操作过程和使用规范,是否能够达到相应的预期结果,从而保证实验者在实验过程中能够规范使用药品,保证实验效果。
通常情况下,分析方法验证是根据实际的测验需要,设定一定的测验内容,实验者根据合理的测验方案来验证分析方法是否能够满足药品检测的需求。
在一些情况下,如果化学原料药的合成工艺发生了改变,配料及配方有所调整,需要将分析方法也相应的进行修改,保证方法的可行性,适用于调整后的药品检测。
实验者一定要根据药品的配方情况,选择相应的分析方法验证。
保证验证结果的准确性和真实性。
分析方法验证的目的是实验者通过一定的实验操作证明该分析方法能够起到化学原料药检测的作用。
(二)分析方法验证的项目实验者在应用要点分析方法对实验药品进行分析验证的过程中,应当查询该药品或原料药的相关资料,明确相应的验证项目,从而有针对性的利用实验进行药品性质的测验。

原料药小试阶段流程
原料药的小试阶段流程通常包括以下几个步骤:
1. 实验设计,在小试阶段,首先需要设计实验方案,确定实验的具体目的、方法和步骤。
这包括确定需要使用的原料和试剂,实验条件和参数等。
2. 原料准备,在进行小试阶段实验之前,需要准备好所需的原料和试剂。
这可能涉及到原料的采购、检验和储存,确保原料的质量和纯度符合实验要求。
3. 反应试验,根据实验设计,进行原料药的小试反应实验。
这可能涉及到不同的合成方法、反应条件的优化和控制,以及对反应过程中产物的分析和检测。
4. 产物分离与纯化,在反应完成后,需要对产物进行分离和纯化。
这可能涉及到溶剂萃取、结晶、色谱等技术,以获得目标产物并提高其纯度。
5. 产物分析,对分离纯化后的产物进行分析,确定其结构和纯
度。
这可能包括质谱分析、核磁共振等技术,以确保产物符合预期
的质量标准。
6. 实验数据处理与报告撰写,对实验数据进行处理和分析,撰
写实验报告,总结实验结果和结论,为进一步的工艺开发和放大生
产提供参考。
总的来说,原料药的小试阶段流程涉及到实验设计、原料准备、反应试验、产物分离与纯化、产物分析以及实验数据处理与报告撰
写等多个环节,需要严谨的操作和全面的数据记录,以确保实验结
果的可靠性和可重复性。
原料药分析方法开发流程分析方法在药物的研发过程中起到的是“灯塔"的作用,是原料药及制剂开发、质量控制的标尺及眼睛,因此分析方法在药物开发过程中起到了领航员的作用。
下面简单的介绍一下原料药分析方法的开发流程.原料药的分析方法开发一般分为两大部分:1、起始物料的分析方法开发;2、中间体及API 的分析方法开发.按照正常的逻辑顺序,应该是起始物料的分析方法开发先行,但是一般在实际操作过程中,往往是中间体及API的分析方法先行开发。
主要是因为,在打通工艺路线时期或者是文献调研的阶段,主要是针对中间体及API的分析方法的工作.只有在工艺优化的中期或者中后期,对起始物料厂家基本选定时才会有针对性的启动起始物料的分析方法开发工作。
虽然如此,考虑到逻辑顺序,还是按照起始物料、中间体、API这样的顺序进行逐一介绍。
3、分析方法的开发二、中间体中间体分为过程控制及质量控制,过程控制主要监控反应进行的程度,质量控制是制定中间体的中控标准。
1、过程控制方法1)过程控制方法的开发根究反应液的具体情况以及涉及物料自身的性质,中间体的过程控制方法可以选择TLC 或者HPLC的手段进行控制.一般在反应过程中主要关心的是原料的剩余及产物的生成情况,所以确保在原料及产物峰周围没有干扰。
如果有需要特殊关注的杂质,也需确保杂质的分离度、峰纯度等。
2)过程控制方法的验证如果过程控制的方法只是定性的检测,在方法验证时一般只需进行:专属性、检测限、耐用性等方面的验证工作。
如果涉及定量检测的方法,则需要进行全面的分析方法验证工作.2、质量控制方法1)质量控制方法的开发对中间体的质量控制,一方面根据工艺优化的结果制定质量控制的限度,另一方面需要根据API质量的要求对中间体所涉及杂质的限度进行定量的控制.当然,质量控制方面不只是杂质限度的控制,如果接下来的反应是无水反应,上一步的中间体依然需要对水分的限度进行严格的控制。
中间体的制备过程中,也会涉及GTI、手性杂质及重金属等杂质的研究。
化学药cmc流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!化学药 CMC 流程一、项目启动阶段。
在开展化学药的 CMC(化学、生产和控制)工作之前,首先要明确项目目标和需求。
一种测定原料药中哌啶残留的检测方法与流程
测定原料药中哌啶残留的常用方法是高效液相色谱法(HPLC)。
以下是一种测定原料药中哌啶残留的检测方法与流程:
1. 样品制备:将原料药样品加入适量的溶剂,如甲醇或乙醇,使其完全溶解,并经过适当的稀释。
2. 色谱柱选择:选择适合哌啶的固定相柱,如C18柱。
3. 流动相配制:根据实验要求,根据估计的保留时间和峰分离度,选择一种或多种有机溶剂与缓冲溶液的混合物作为流动相。
典型的流动相组成包括甲酸/乙腈、磷酸盐缓冲液/乙腈等。
4. 仪器设置:将待测样品注入HPLC仪器,并设置适当的流速、柱温和检测波长。
根据哌啶的紫外吸收特性,一般选择波长为200-250nm。
5. 校准曲线绘制:准备一系列已知浓度的哌啶标准溶液,并进行HPLC分析,得到哌啶峰的峰面积与浓度之间的线性关系。
根据线性关系绘制校准曲线。
6. 样品检测:将待测样品进行HPLC分析,测量哌啶峰的峰
面积。
7. 计算结果:利用校准曲线计算样品中的哌啶浓度。
根据待测
样品的净峰面积比例与标准曲线的回归方程得出。
8. 结果分析和报告生成:根据检测结果的准确性和可靠性进行结果分析,撰写实验报告。
需要注意的是,以上流程仅为一种常用的方法,实际操作中可能会根据实验要求和样品的特性进行相应的调整。
因此,在进行实验之前,需要根据具体情况设计和优化方法和流程。
同时,为了确保结果的准确性和可靠性,实验中需要进行合适的质量控制措施,如空白试验、精密度和准确度控制等。
原料药工艺开发1:路线设计和选择A)经济(原料价格,商业化资源多少;收率高低;路线是否是线形)B)安全(物料的使用,单元反应操作)C)注册起始原料的初步估计(如果路线起始原料就是注册起始原料,此项归为A因素关于注册起始原料的选择请参考ICH Q11并关注Org Process Res Dev 2014 18 (5) pp 587-600)。
D)基因毒(原料、中间体或者必须试剂是否涉及基因毒,如果涉及能否避开,基因毒不可怕,可怕的是分析方法的开发)在QBD(quality by design)理念指导下,通过对产品杂质概况的初步了解,综合上面因素确定工艺路线。
二:工艺优化QBD是一种风险降低工具,目的保证每批API产品质量都是一样的合格的。
他的运用主要是三个阶段,第一阶段是目标API的明确(QTPP),第二阶段是设计和理解(产品的设计和理解;工艺的设计和理解),第三阶段是实现阶段(控制策略和持续改进)。
在工艺研发中到底什么是QBD,如果你能通过理解和控制工艺的关键变量(CPP和CMA),进而控制产品的质量(CQA),这就是QBD。
(Org. Process Res. Dev. 2015,19(11), 1634–1654)一个完整的工艺优化应该包括六部分:第一部分:优化试验得到正常操作范围(NOR:normal operation range),此时CPP和CMA应该包含在NOR中。
只要工艺参数不超出范围,工艺可以产出一致的合格的产品。
如果公司有条件可以运用DOE设计实验。
第二部分:宽度试验得到证明可以接受的范围(PAR:proven acceptable range),该工作可以帮助我们确认CPP和CMA,我把这部分工作理解为文献经常提到的FMEA(failure mode and effect analysis),有时候因为时间的关系我们很难把所有的工艺参数都进行宽度实验。
第三部分:延长工艺时间试验得到副反应或者降解等关乎产品质量的信息;A)延长反应时间(考察副反应和降解杂质,尤其高温反应,充分考虑到生产时反应釜降温时间以及IPC监控时间,特指HPLC监控时运行时间较长)B)延长后处理浓缩反应液或者有机层溶液的时间(考察浓缩稳定性)C)延长后处理酸化或者碱化后反应体系的搅拌时间(考察产品在此条件下的稳定性)D)延长干燥时间(考察产品干燥条件下的稳定性)这部分工作同样可以帮助我们确认CPP和CMA。
原料药分析方法开发流程
分析方法在药物的研发过程中起到的是灯塔”的作用,是原料药及制剂开发、质量控制
的标尺及眼睛,因此分析方法在药物开发过程中起到了领航员的作用。
下面简单的介绍一下
原料药分析方法的开发流程。
原料药的分析方法开发一般分为两大部分:1、起始物料的分析方法开发;2、中间体及API
的分析方法开发。
按照正常的逻辑顺序,应该是起始物料的分析方法开发先行,但是一般在
实际操作过程中,往往是中间体及API的分析方法先行开发。
主要是因为,在打通工艺路
线时期或者是文献调研的阶段,主要是针对中间体及API的分析方法的工作。
只有在工艺
优化的中期或者中后期,对起始物料厂家基本选定时才会有针对性的启动起始物料的分析方法开发工作。
虽然如此,考虑到逻辑顺序,还是按照起始物料、中间体、API这样的顺序进
行逐一介绍。
一、起始物料
1、合成路线的获取
在启动分析方法开发工作之前,一定要获得起始物料厂家提供的合成路线,需要包括以
下几点:起始物料、中间体、反应溶剂、后处理溶剂及关键催化剂等。
如果厂家提供反应步骤过长,一般选择3-5步即可。
2、杂质分析
结合起始物料厂家提供的工艺路线,对可能存在或者产生的各种杂质进行详细的分析,
为分析方法开发的方向奠定一个基础,也好对所需要的仪器耗材有提前的准备。
一般涉及杂
质如下:
3、分析方法的开发
起始物料路线中所涉及的杂质种类,进行对应分析方法的开发。
以厂家COA及提供的
分析方法为基础,结合自身工艺对起始物料质量的要求,建立适合自己的起始物料内控方法。
4、分析方法的验证
在API工艺进行逐级放大前,取得起始物料供应商提供的不小于3批的中试生产批量
样品,使用一批次进行分析方法验证工作,其余批次进行分析方法重现工作,同时也是为了验证厂家提供起始物料质量的稳定和可控。
5、杂质限度制定的依据
根据多批次起始物料的检测结果,以及API杂质限度的要求,结合工艺路线对杂质的
二、中间体
中间体分为过程控制及质量控制,过程控制主要监控反应进行的程度,质量控制是制定
中间体的中控标准。
1、过程控制方法
1)过程控制方法的开发
根究反应液的具体情况以及涉及物料自身的性质,中间体的过程控制方法可以选择TLC
或者HPLC的手段进行控制。
一般在反应过程中主要关心的是原料的剩余及产物的生成情
况,所以确保在原料及产物峰周围没有干扰。
如果有需要特殊关注的杂质,也需确保杂质的
分离度、峰纯度等。
2)过程控制方法的验证
如果过程控制的方法只是定性的检测,在方法验证时一般只需进行:专属性、检测限、
耐用性等方面的验证工作。
如果涉及定量检测的方法,则需要进行全面的分析方法验证工作。
2、质量控制方法
1)质量控制方法的开发
对中间体的质量控制,一方面根据工艺优化的结果制定质量控制的限度,另一方面需要
根据API质量的要求对中间体所涉及杂质的限度进行定量的控制。
当然,质量控制方面不只是杂质限度的控制,如果接下来的反应是无水反应,上一步的中间体依然需要对水分的限
度进行严格的控制。
中间体的制备过程中,也会涉及GTI、手性杂质及重金属等杂质的研究。
一般情况,此类杂质最好放在中间体的质量标准中进行,如果控制有难度或者在之后的反应过程中仍然会又引入及生成,放在之后的步骤控制或者API控制均可。
2)质量控制方法的验证
一般情况下,中间体的质量控制均会涉及杂质的定量检测,因此中间体质控方法的验证
均需要按照定量检测的方法进行全面的分析方法验证工作。
三、API
同起始物料分析方法开发基本一致,在API分析方法的开发前期,首先需要进行有关
化合物或者相似化合物分析方法的调研工作,其次进行杂质的分析工作。
针对不同类型的杂
质选择有针对性的文献报道的方法为方法开发的基础,结合工艺本身,对分析方法进行逐步
的优化。
有关物质的制定,可以根据靠近终反应步骤的杂质谱以及最后一步反应的杂质体系进行制定。
溶剂残留的制定需要结合起始物料所涉及的溶剂,如果有与起始物料有差异的溶剂,那
么差异溶剂需要在起始物料中进行控制;若能包含起始物料所涉及溶剂,可以将所用溶剂在
终产品中进行控制,起始物料中只需进行干燥失重的检测。
同时,还需要关注一些潜在的溶
剂,例如:反应中涉及苯的衍生物,也需要关注苯的残留控制等。
3、分析方法的开发
根据不同的杂质种类,API的分析方法涉及到普通杂质、手性杂质、残留溶剂、基因毒性杂质、重金属等。
普通杂质:一般采用HPLC法,在进行分析方法开发之前,首先要进行充分的文献调研工作,查询各国药典标准、文献、专利中是否收载了相同或同类化合物,以此为基础进行
分析方法的开发,结合API的理化性质以及对各步反应机理的分析,进行分析方法的逐步优化。
在分析方法优
化过程中,应同时对API进行强制降解实验,评估分析方法对潜在降
解产物的分离能力。
手性杂质:采用HPLC法,选用手性色谱柱或手性流动相。
对有多个手性中心的,对映异构体需采用手性色谱进行分析,非对映异构体,一般可与普通杂质采用同一方法进行控
制。
残留溶剂:残留溶剂采用GC法,根据溶剂的种类,选择不同极性的色谱柱进行分离。
除要关注合成工艺中使用的溶剂外,还需要关注潜在的残留溶剂,如使用了甲苯,需要对苯
进行检测与控制,使用了甲基叔丁基醚,需要关注叔丁醇的残留,酯键断裂过程中产生的醇
类等。
基因毒性杂质:由于基因毒性杂质的限度较低,对方法的灵敏度要求较高,一般要用到
HPLC-MS或GC-MS,在分析方法开发中,需重点关注检测限是否能满足质量控制的要求。
重金属:对于一般的重金属,采用药典通则的检测方法即可,对于pd等重金属需要特
殊研究。
4、分析方法的验证
在进行分析方法验证前,需要对质量标准中所涉及的分析方法进行一个汇总,合理安排
验证工作,以免进行重复的工作,例如:含量和有关物质方法一致,有些重复的验证项目就
只进行一次验证即可。
一般API需要进行验证的方法有:有关物质(包括异构体、分别控制的有关物质)方法、含量方法、溶剂残留方法等。