药物构效关系
- 格式:pdf
- 大小:1.81 MB
- 文档页数:64
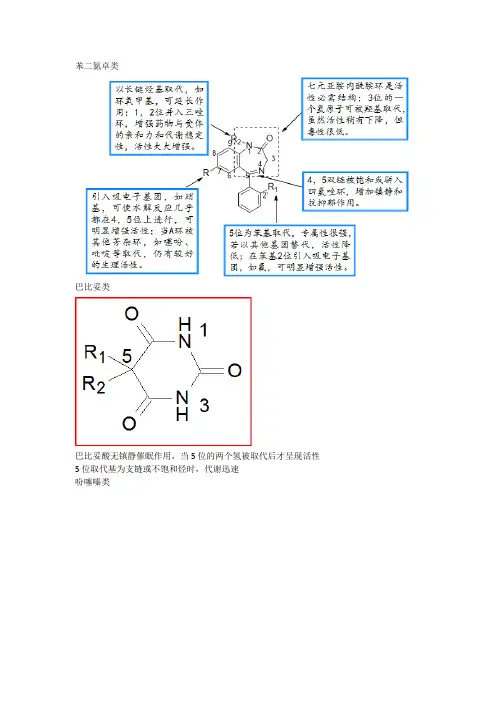
苯二氮卓类巴比妥类巴比妥酸无镇静催眠作用,当5位的两个氢被取代后才呈现活性5位取代基为支链或不饱和烃时,代谢迅速吩噻嗪类吗啡胆碱酯类M受体合成M受体拮抗剂R1和R2部分为较大基团,通过疏水性力或范德华力与M受体结合,阻碍乙酰胆碱与受体的接近和结合。
R3可以是H,OH,CH2OH或CONH2。
由于R3为OH或CH2OH时,可通过形成氢键使与受体结合增强,比R3为H时抗胆碱活性强,所以大多数M受体强效拮抗剂的R3为OH。
X一般是酯键-COO-,非活性必需。
氨基部分通常为季铵盐或叔胺结构。
R4、R5通常以甲基、乙基或异丙基等较小的烷基为好。
N上取代基也可形成杂环。
环取代基到氨基氮原子之间的距离,以n=2为最好,碳链长度一般在2~4个碳原子之间,再延长碳链则活性降低或消失。
局部麻醉药亲脂性部分可为芳烃、芳杂环,以苯环作用较强。
苯环上邻对位给电子取代基如氨基、烷氧基有利于增加活性;而吸电基会使活性下降。
中间部分-决定药物稳定性作用时间:-CH2CO->-CONH->-COS->-COO-作用强度:-COS->-COO-> -CH2CO-> -CONH-通常以n = 2-3碳原子为最好在苯环和羰基之间插入-CH2-,-O-,破坏了共轭体系,活性下降;插入-CH=CH-,则保持活性。
亲水性部分可为仲胺和叔胺,或脂环胺如吡咯烷、哌啶、吗啉等,以叔胺最为常见。
不可以是伯胺,不稳定而且毒性大。
青霉素喹诺酮吡啶酮酸的A 环是抗菌作用必需的基本药效基团,变化较小。
其中3位COOH和4位C=O 与DAN螺旋酶和拓扑异构酶Ⅳ结合,为抗菌活性不可缺少的部分。
B环可作较大改变,可以是并合的苯环(X=CH,Y=CH)、吡啶环(X=N,Y=CH)、嘧环(X=N,Y=N)等。
1位N上若为脂肪烃基取代时,以乙基或与乙基体积相似的乙烯基、氟乙基抗菌活性最好;若为脂环烃取代时其抗菌作用最好的取代基为环丙基、而且其抗菌活性大于乙基衍生物。

药物化学构效关系1.局部麻醉药的构效关系:①亲脂性部分:可变范围较大,可为芳环或芳杂环,但以苯环的作用较强,是局麻药物的必需部位。
当酯类药物苯环的邻位或对位引入给电子集团,如氨基、烷氧基时,局麻作用均较未取代得苯甲酸衍生物强;对氨基苯甲酸酯类苯环的邻位上若再有其他取代基如氯、氨基、烷氧基时,由于位阻作用而延长了酯的水解,因此活性增强,作用时间延长。
②中间连接部分:由羰基部分和烷基部分共同组成。
羰基部分与麻醉药持效时间及作用强度有关,作用持续时间为:酮﹥酰胺﹥硫代酯﹥酯;麻醉作用强度:硫代酯﹥酯﹥酮﹥酰胺。
烷基部分碳原子数以2~3个为好,当烷基部分为—CH2CH2CH2—时,麻醉作用最强。
③亲水性部分:大多数为叔胺,易形成可溶性的盐类。
氮原子上取代基的碳原子总和以3~5时作用最强,也可为酯环胺,其中以哌啶的作用最强。
2. 苯二氮卓类药物的构效关系:① 1、2位拼入三氮唑环,使代谢稳定性增加,提高与受体的亲和力,活性显著增加;② 3位引入手性碳,分子构想更稳定,对受体亲和力增强;③ 4、5位引入恶唑环,增强稳定性;④7位有吸电子取代基时,药物活性明显增强,且吸电子性越强,活性增加越明显,NO2>Br>CF3>Cl;⑤ 5位苯环的2’位引入体积较小的吸电子基团如F、Cl,可使活性增强。
①镇静作用的强度和起效快慢,与药物的理化性质有关。
【酸性解离常数pKa】巴比妥酸和5位取代的巴比妥类有较强的酸性,在生理pH=7.4几乎全都电离成离子状态,不易透过血脑屏障,无镇静催眠作用;5,5-二取代的巴比妥类,酸性减弱,生理pH条件下不易电离,易进入脑中发挥作用,显效快,作用强。
【脂水分配系数】5位无取代基时,分子有一定极性,亲脂性强,不易透过血脑屏障,无镇静催眠作用;5位取代基碳原子总数在7~8之间作用最强,若亲脂性过强,作用下降甚至出现惊厥。
药物有最适当的的脂溶性,有利于药物透过细胞膜和血脑屏障,起效快,作用强。

一、喹诺酮类构效关系:1、A环是必须的药效团,3羧和4酮为抗菌活性不可少的部分;2、B环可以是苯、吡啶、嘧啶;3、1位乙基及环丙基活性强,环丙基最佳(环丙沙星);4、2位取代活性低;5、5位氨基可增强活性.(司帕沙星)6、6位F改善细胞的通透性;7、7位引入杂环,增强抗菌活性,哌嗪最好;8、8位F、甲氧基或与1位成环,增强活性(左氧氟沙星),甲基、甲氧基光毒性减少二、苯二氮卓构效关系要点.1、3位引入羟基(奥沙西泮)降低毒性,并产生手性碳,右旋体作用强。
2、7位有吸电子基可增加活性,吸电子越强,作用越强,其次序为NO 2>Br>CF3>Cl3、5位苯是产生药效的重要基团,5位苯环的2’位引入体积小的吸电子基团.(如F、Cl )可使活性增强。
4、1,2位拼入三氮唑可提高稳定性,并提高与受体的亲和力,活性显著增加。
5、苯环用生物电子等排体噻吩杂环置换,保留活性。
6、1位取代基在体内代谢去烃基,仍有活性。
三、吩噻嗪类药物的构效关系:以氯丙嗪为先导化合物,对吩噻嗪类进行结构改造。
三方面:1、吩噻嗪环上的取代基:吩噻嗪环只有2位引入吸电子基团时可增强活性。
作用强度与吸电子性能成正比,CF3>Cl>COCH3>H>OH。
2位乙酰基可降低药物的毒性和副作用。
2、10位N上的取代基:母核上的10位N原子与侧链碱性氨基之间相隔3个直链碳原子时作用最强,是吩噻嗪类抗精神病药的基本结构。
侧链末端的碱性基团常为叔胺,也可为氮杂环,以哌嗪取代作用最强。
3、三环的生物电子等排体。
四、μ受体选择性激动剂构效关系1、芳环和碱性叔胺氮原子是μ受体激动剂的必要结构部分,二者通过2个或3个碳原子的碳链相连接。
2、芳环3位酚羟基的存在使活性显著增强。
氮原子上以甲基取代活性好,当N-取代基增大到3~5个碳原子时,如烯丙基(纳洛酮)、环丁基甲基时,由激动剂转变为拮抗剂。
3、μ受体选择性激动剂的药效构象相同,其芳环以直立键与哌啶环相连。

局部麻醉药构sheng效关系1.分类芳酸酯类、酰胺类、氨基醚类、氨基酮类、其他类2.构效关系亲酯部分中间链亲水部分⑴亲脂部分:芳烃或芳杂环,这一部分修饰对理化性质变化大,但苯环作用较强。
苯环上引入给电子取代基,麻醉作用增强,而吸电子取代基则作用减弱。
⑵中间部分:此部分决定药物稳定性,和局麻作用持续时间有关⑶亲水部分:常为仲胺和叔胺,仲胺刺激性较大;烃基链3~4个碳原子作用最强,杂环以哌啶环作用最强巴比妥类药构效关系(1)、分子中5位上应有两个取代基。
(2)、5位上的两个取代基的总碳数以4—8为最好(3)、5位上的两个取代基的总碳数以4—8为最好. (4)、在酰亚胺氮原于上引入甲基,可降低酸性和增加脂溶性。
(5)、将C2上的氧原子以硫原子代替,则脂溶性增加,起效快,作用时间短。
苯二氮卓类药物的构效关系(1)1,3-二氢-5-苯基-2H-1,4-苯二氮卓-2-酮是此类药物基本结构;(2)环A7位引入吸电子取代基活性增加(3)环B为七元亚胺-内酰胺结构是产生药理作用的必要结构(4)5位苯环上的取代基时产生药效的重要结构之一,(5)1,2位的酰胺键和4,5位的亚胺键在酸性条件下易水解开环.吩噻嗪类药构效关系R1 部分必须由三个成直链的碳原子组成,若为支链,与多巴胺受体B 部分立体上不匹配,抗精神病活性明显下降,抗组胺作用增强。
顺式吩噻嗪类药物与多巴胺的优势构象能部分重叠,活性高(当侧链与氯取代的苯环同侧时,成为顺式构象)。
丁酰苯类药物的构效关系(1)丁酰苯基为必需的基本骨架(2)侧链末端连一碱性叔胺(3)苯环的对位一般具有氟取代(4)侧链湠基于碱基之间以三个碳原子最好镇痛药的一般特征(1)分子中具有一个平坦的芳香结构(2)有一个碱性中心能在生理PH条件下大部分电离为阳离子(3)含有哌啶或类似于哌啶的空间结构吗啡的构效关系(半合成类镇痛药)叔胺是镇痛活性的关键基团,氮原子引入不同的取代基可使μ 受体激动剂转变为拮抗剂。

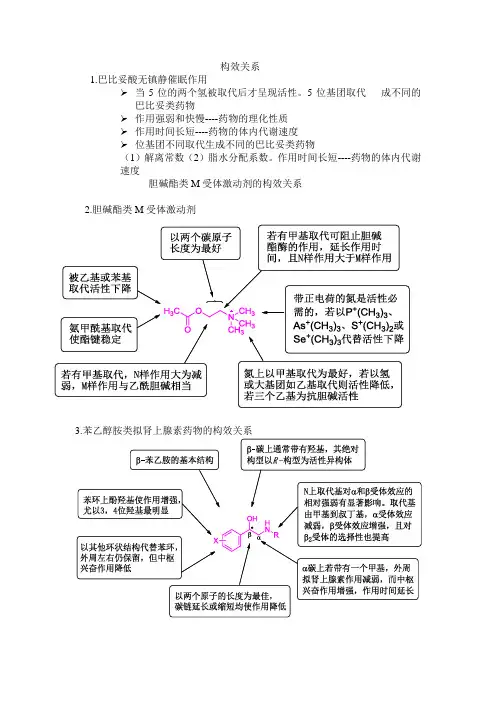
构效关系1.巴比妥酸无镇静催眠作用➢当5位的两个氢被取代后才呈现活性。
5位基团取代成不同的巴比妥类药物➢作用强弱和快慢----药物的理化性质➢作用时间长短----药物的体内代谢速度➢位基团不同取代生成不同的巴比妥类药物(1)解离常数(2)脂水分配系数。
作用时间长短----药物的体内代谢速度胆碱酯类M受体激动剂的构效关系2.胆碱酯类M受体激动剂3.苯乙醇胺类拟肾上腺素药物的构效关系4.局部麻醉药的构效关系亲脂性部分•可为芳烃、芳杂环,以苯环作用较强。
•苯环上邻对位给电子取代基如氨基、烷氧基有利于增加活性;而吸电基会使活性下降。
中间部分-决定药物稳定性•作用时间:-CH2CO->-CONH->-COS->-COO-•作用强度:-COS->-COO-> -CH2CO-> -CONH-•通常以n = 2-3碳原子为最好•在苯环和羰基之间插入-CH2-,-O-,破坏了共轭体系,活性下降;插入-CH=CH-,则保持活性。
亲水性部分•可为仲胺和叔胺,或脂环胺如吡咯烷、哌啶、吗啉等,以叔胺最为常见。
•不可以是伯胺,不稳定而且毒性大。
5.b受体阻滞剂的构效关系1,4-二氢吡啶环是必需结构,吡啶或六氢吡啶环则无活性,1位N不被取代为佳。
2,6-位取代基应为低级烷烃。
若C4有手性,立体结构有选择作用。
4位取代苯基上邻、间位有吸电子基团时活性较佳。
3,5-位取代基酯基是必要结构,-COCH3,-CN活性降低,硝基则激活钙通道。
7.组胺H2受体拮抗剂的构效关系(SAR)9.喹诺酮类抗菌药物的构效关系(1)吡啶酮酸的A 环是抗菌作用必需的基本药效基团,变化较小。
其中3位COOH和4位C=O与DAN螺旋酶和拓扑异构酶Ⅳ结合,为抗菌活性不可缺少的部分。
3位的羧基被磺酸基、乙酸基、磷酸基、磺酰氨基等酸性替团替代以及4位酮羰基被硫酮基、亚氨基等取代均使抗菌活性减弱。
(2)B环可作较大改变,可以是并合的苯环(X=CH,Y=CH)、吡啶环(X=N,Y=CH)、嘧环(X=N,Y=N)等。


巴比妥类药物巴比妥类药物属于结构非特异性药物.结构非特异性药物:药物的生物活性与药物的化学结构关系不大,与理化性质有关.结构特异性药:药物的作用依赖于药物分子的特异化学结构及空间相互排列.巴比妥类药物的作用强弱和起效时间的快慢与药物的解离常数,PKa,脂水分配系数有密切关系.解离常数:药物以分子的形式透过生物膜,以离子的形式产生作用.油水分配系数:药物既可以在体液中转运,又可以透过血脑屏障到达作用部位.该类药物5位上有两个取代基才有活性,当两个取代基的碳原子总数在4到8之间时,分配系数适中,活性最好.当碳原子总数超过8时,产生作用过强,易产生惊厥作用.结构中酰亚胺上的N原子上有甲基取代时可降低酸性和增加脂溶性,起效快.将C-2位的O用S替代时.脂溶性增加,易透过血脑屏障,起效快.巴比妥类药物的体内代谢过程与药物的代谢时间有关.二.苯二氮卓类药物(地西泮)A环为活性所必须.B环可以被其他芳杂环取代,仍保留其活性.1位一般为N-CH3.-CH3可在代谢中脱掉,但仍保留其活性1.2位可骈入杂环(三唑仑:稳定性增加活性增加)3位一个H原子可被-OH取代活性稍微降低,但毒性很低.4,5为双键被饱和.活性降低,并入恶唑环增加镇静和抗抑郁作用.5位-苯基的2位引入吸电子基(F,Cl,Br.....)活性增强.7位引入吸电子基,活性明显增强NO2>CF3>Br>Cl三.芳基丙酸类药物(布洛芬)苯环与羧基之间间距一个或一个以上的碳原子.羧基的a位又一个-CH3,限制了羧基的自由旋转,使其保持在适合与受体或酶结合的构像,以增加抗炎镇痛作用.由于羧基a位的-CH3的引入,使其产生了不对称中心,通常是S-构型的活性高于R-构型,在体内手性异构体之间可以相互转换,通常是无活性的R-构型转换为有活性的S-构型.芳环上可以引入另一个疏水基如,环己基,烯丙氧基等.在苯环羧基的间位引入一个吸电子基如F.Cl等.抗炎活性好.四.胆碱受体激动剂(氯贝胆碱)的构效关系1,季氨基.(1)带正电荷的氮原子是活性所必须.若以As,Se取代活性降低.(2)氮原子上以甲基取代为好,若以较大基团取代如乙基则活性降低,若为三个乙基则变为抗胆碱活性.2,乙酰氧基.(1)当乙酰或丁酰基等取代时活性降低,(2)乙酰基上的氢被芳环或较大基团取代时变为抗胆碱活性.(3)酯基的快速水解是乙酰胆碱作用时间短暂和不稳定的因素,因此用不易水解的基团取代乙酰基可以增加稳定性和作用时间.如用氨甲酰基取代乙酰基,由于氮上孤对电子的参与,羰基碳的亲电性较乙酰基为低,不易水解.3,亚乙基桥.(1)亚乙基桥的长度对活性有关键影响,两个碳为最好.随着碳链的延长,活性逐渐降低.(2)季氨氮原子的a位有甲基取代,整体活性降低,但N样作用大于M样作用.(3季氨氮原子的B位有甲基取代,可阻止胆碱酯酶的作用,延长作用时间,M 样作用于乙酰胆碱相当,N样作用大大减弱,成为选择性的M受体激动剂.肾上腺受体激动剂的构效关系苯乙胺的基本结构是活性所必须,碳链的延长或缩短均使作用减弱.苯环上的酚羟基可显著增强拟肾上腺素作用,尤其以3,4位最为明显,但作用时间短暂.以其他环状结构取代苯环,外周作用仍被保留,中枢兴奋作用降低.N上的取代基对a和B受体效应的相对强弱有显著影响.取代基从甲基到叔丁基,a受体效应减弱.B受体效应增强,且对B2受体的选择性提高.B-碳上通常连有羟基.其绝对构型以R-构型为活性体.局麻药的构效关系.(图自己想)邻对位给电子基取代,有利于两性离子的形成,活性增强.若有吸电子存在则活性下降.可以为芳环,芳杂环,此部分的修饰对活性的影响较大,活性顺序为苯环>吡咯>噻吩>呋喃通常以2-3个碳原子为最好有仲胺,叔胺或吡咯烷,哌啶.吗啉等,以叔胺最为常见.在苯环和羧基之间插入-CH2,-O-等基团,破坏了两性离子的形成.活性降低.若连入可以形成共轭的基团,如-CH2=CH2-等.活性可保持不变.酰胺也可形成两性离子.此部分决定药物的稳定性,按作用时间顺序羰基+-O->....+-S->....+-NH->....+-CH2-;按作用强度顺序:-S->-O->-CH2->-NH-B受体阻滞剂的构效关系(图自己想).苯乙醇胺类和芳氧丙醇胺类可以是苯,萘,杂环,稠环和脂肪性的不饱和杂环.可以有甲基,氯,硝基,甲氧基等取代基,在2,4和2,3,6位取代时活性最佳.用S,-CH2,-NCH3取代时,活性降低.S-构型异构体活性增加,R-构型异构体活性降低或消失.R-构型异构体活性增加,S-构型异构体活性降低或消失以叔丁基和异丙基取代时最好,甲基上的氢原子数小于3或N-N双取代时,活性降低.由于B 受体阻滞剂的结构组成自由度很大,所以其溶解度也有较大差异,这与其副作用和体位消除的位置有关.亲脂性-肝代谢-速率较快.亲水性-肾消除二氢吡啶类药物的构效关系(硝苯地平)二氢吡啶环为活性所必须.若变为吡啶环或六氢吡啶环则活性消失,环上的NH不被取代时,活性保持最佳.2,6位取代基为低级烷烃3,5位的羧酸脂为活性所必须,若变为乙酰基或氰基则活性降低,若变为硝基则激活钙通道. 3,5位羧酸脂不同,C4为手性中心,酯基的大小对活性影响不大,但不对称脂则影响作用部位. C4若为手性碳,具有立体选择性4位取代基与活性的关系:苯基或取代苯基>环烷基>甲基>H苯环上邻间位上有吸电子基取代活性较佳,对位取代时活性降低H2受体拮抗剂的构效关系(雷尼替丁).H2受体拮抗剂的结构有三部分组成:碱性的芳杂环结构,易曲饶的四原子链和平面极性基团.碱性的芳杂环和碱性的基团取代的芳杂环是活性所必须,咪唑环作为质子转移的机制,被异噻唑,恶唑置换后碱性下降,活性也随之降低.被亲脂性的芳杂环(苯环)取代时活性降低.被碱性基团取代,呋喃,噻唑置换后,是良好的H2受体拮抗剂.平面极性基团.是具有胍,脒基样的结构.在生理PH条件下,电离程度低的极性基团作为脒脲基团与受体形成一个以上的氢键保持活性.易曲饶的链或芳杂环:链长为4个原子.链长与其拮抗性有关.自由旋转受限使其活性下降,中间连接的链可被钢性环所取代.青霉素的构效关系(1)6位侧链的酰胺基团主要决定其抗菌谱,改变其极性,使其易于透过细胞膜,可扩大其抗菌谱.例如,在芳环乙酰胺基的a位引入极性-NH2,-COOH,-SO2等亲水性基团,扩大抗菌谱.增加亲水性.有利于对格兰阴性菌的抑菌作用,并能增强对青霉素结合蛋白的亲和力.在分子中的适当位置引入立体位阻基团.如在侧链引入立体位阻较大的基团和在6位引入甲氧基和甲酰胺基,因其立体位阻的效应降低了钝化酶的结构适应性,保护B-内酰胺环不被B-内酰胺酶进攻.因而得到耐酶的抗生素.青霉素的噻唑环上的羧基是基本活性基团,虽可被硫代酸和酰胺取代但活性降低.若羧基被还原为醇则失去抗菌活性.对于羧基可利用前药原理制成脂,改进口服吸收和药物代谢动力学性质.青霉烷酸分子中三个手性碳的构型对其活性至关重要.但青霉素的噻唑环上的两个甲基不是活性的必要因素.半合成头孢菌素的构效关系在7位侧链引入亲脂性的基团,如苯基,环稀基,噻吩和含氮的杂环.可增强抗菌活性,扩大其抗菌谱.同时改变3位取代基,引入杂环,可改进口服吸收分布也可扩大其抗菌谱.在7位酰胺的a位引入亲水性的-SO3H,-NH2,-COOH,等极性基团.可扩大抗菌谱同时改变3位取代基,引入-Cl,CH3,和含氮的杂环,可增强口服吸收扩大抗菌谱.带有7B为顺势”氨噻肟”的侧链可提高对B-内酰胺酶的稳定性,扩大抗菌谱.这主要是由于引入肟后,甲氧基占据了靠近B-内酰胺环的位置.阻止了酶分子对B-内酰胺环的靠近,因而使药物有耐酶,广谱的性质.5位的S用生物电子等排体O,CH2等取代,分别称为氧头孢菌素和碳头孢菌素,活性不降低.3位取代基的改造,如乙酰氧基可被甲基,氯等取代可扩大抗菌谱并且改变药物在体内的吸收分布和药物的渗透性的药物代谢动力学性质.2,3位的双键移位失活.2位-COOH可制成前药增加口服吸收.喹诺酮类药物的构效关系.A环(吡啶酮酸部分)是抗菌作用所必须的基本药效基团.其中3位的羧基和4位的酮基是与靶酶的结合位点,是抗菌活性必不可少的部分.B环部分可做较大改变,可并入苯环.吡啶环和嘧啶环等.N1位置取代基对抗菌活性的贡献较大,若由烃基,环烃基取代活性增加,尤以乙基,氟乙基和环丙基取代活性最佳.2位引入取代基活性减弱或消失,可能是由于空间位阻阻止了与受体的结合.3位的羧基和4位的酮基是抗菌活性中不可缺少的部分.被其他基团取代活性消失,与铁铝钙络合产生副作用.5位氨基取代活性增加.其他基团取代活性均降低.6位取代基对活性的影响很重要.活性取代顺序位:F>Cl>CN>=CH2>=H.引入F可比H的抗菌活性增强30倍.因为F代物与DNA螺旋酶亲和力增强2-17倍.对细菌细胞壁的穿透力增强1-70倍.7位引入取代基增强活性的顺序为:哌嗪基>二甲氨基>甲基>卤素>氢.其中以哌嗪基取代活性最好.8位以F,Cl,-OCH3,等取代活性增强,但以F取代光毒性也增强.若1,8位间成环,产生的光化学异构体之间也有较大差异.。


药物化学构效关系(第二版尤启冬主编)主要药物的构效关系应用抗肿瘤作用机理:1、药物在体内能形成缺电子活泼中间体(碳正离子)或其他具有活泼的亲电性基团的化合物,进而与肿瘤细胞的生物大分子(DNA,RNA,酶)中富电子基团(氨基,巯基,羟基等)发生共价结合,使其丧失活性,致肿瘤细胞死亡。
2、属细胞毒类药物,在抑制和毒害增生活跃的肿瘤细胞的同时,对其它增生较快的细胞产生抑制。
如骨髓细胞、肠上皮细胞、毛发细胞和生殖细胞等。
副作用大:影响造血功能和机体免疫功能,恶心、呕吐、骨髓抑制、脱发等。
氮芥类药物脂肪氮芥:氮原子的碱性比较强,在游离状态和生理PH(7.4)时,易和β位的氯原子作用生成高度活泼的亚乙基亚胺离子,为亲电性的强烷化剂,极易与细胞成分的,亲核中心发生烷基化反应。
脂肪族氮芥:烷化历程是双分子亲核取代反应(SN2),反应速率取决于烷化剂和亲核中心的浓度。
脂肪氮芥属强烷化剂,对肿瘤细胞的杀伤能力也较大,抗肿瘤谱较广;但选择性比较差,毒性也较大。
芳香族氮芥:氮原子与苯环共轭,减弱了碱性,碳正离子中间体,单分子的亲核取代反应。
氮芥类药物及大多数烷化剂主要是通过和,DNA上鸟嘌呤或胞嘧啶碱基发生烷基化,产生DNA链内、链间交联或DNA蛋白质交联而抑制,DNA的合成,阻止细胞分裂。
β-内酰胺类抗生素的化学结构特点:1分子内有一个四元的β-内酰胺环,除了单环β-内酰胺外,该四元环通过N原子和邻近的第三碳原子与另一个五元环或六元环相稠合。
2除单环β-内酰胺外,与β-内酰胺环稠合的环上都有一个羧基。
3所有β-内酰胺类抗生素的β-内酰胺环羰基α-碳都有一个酰胺基侧链。
4β-内酰胺环为一个平面结构,但两稠环不共平面β-内酰胺类药物可抑制粘肽转肽酶的活性和青霉素结合蛋白青霉素构效关系(1)6位的侧链酰胺基团决定其抗菌谱。
改变其极性,使之易于透过细胞膜可以扩大抗菌谱。
例如,在芳环乙酰氨基的α位上引入-NH2、-COOH、和-SO3H等亲水性基团,可以扩大抗菌谱,增强亲水性有利于对革兰阴性菌的抑制作用并能增强对青霉素结合蛋白的亲和力。

药物的构效关系药物的构效关系(Structure-activity relationship, SAR)是指药物的结构与其生物活性之间的关系。
通过研究不同化合物的结构特征和生物活性数据,可以揭示药物分子的作用机制,指导药物设计和优化,提高研发效率和成功率。
药物的构效关系研究对于药物化学、药理学和药代动力学等领域都有重要的意义。
以下是一些常见的构效关系的参考内容:1. 功能团对药效的影响:研究表明,药物分子中的特定功能团如羟基、酰胺、酯等,可以影响药物的生物活性。
例如,对于抗菌药物,羟基和酰胺基团通常与细菌靶标结合,从而发挥药效。
2. 结构类似性对药效的影响:药物分子的结构类似性对于药效也有重要的影响。
通常来说,结构相似的化合物可能具有相似的生物活性。
因此,通过对已知药物结构进行改良和优化,可以获得具有更高活性和选择性的新化合物。
3. 空间构型对药效的影响:药物分子的空间结构对于其与靶标的相互作用和选择性也起着重要作用。
例如,药物分子的立体异构体可能具有不同的生物活性。
研究不同空间构型的药效差异,有助于设计和优化具有更好活性和选择性的药物。
4. 电子结构对药效的影响:电子结构指的是药物分子中原子和键的电荷分布和云密度。
电子结构的差异可以影响药物分子与靶标的相互作用和药效。
例如,芳香环的电子密度与药物的溶解度、生物利用度和靶标的亲和性有关。
5. 氢键和离子键对药效的影响:氢键和离子键是药物分子与靶标相互作用的常见方式。
氢键的强度和方向性可以影响分子的亲和性和选择性。
离子键的形成可以改变药物分子的溶解度和稳定性。
6. 毒性与构效关系:药物的构效关系研究中还要考虑药物的毒性和副作用。
通过研究药物结构与毒性之间的关系,可以优化药物的安全性和耐受性,减少不良反应。
总的来说,药物的构效关系研究可以从多个角度考察药物分子的结构与生物活性之间的关系。
通过深入理解药物分子的作用机制,可以为药物设计和优化提供重要的理论指导。
一、喹诺酮类构效关系:1、A环是必须的药效团,3羧和4酮为抗菌活性不可少的部分;2、B环可以是苯、吡啶、嘧啶;3、1位乙基及环丙基活性强,环丙基最佳(环丙沙星);4、2位取代活性低;5、5位氨基可增强活性.(司帕沙星)6、6位F改善细胞的通透性;7、7位引入杂环,增强抗菌活性,哌嗪最好;8、8位F、甲氧基或与1位成环,增强活性(左氧氟沙星),甲基、甲氧基光毒性减少二、苯二氮卓构效关系要点.1、3位引入羟基(奥沙西泮)降低毒性,并产生手性碳,右旋体作用强。
2、7位有吸电子基可增加活性,吸电子越强,作用越强,其次序为NO 2>Br>CF3>Cl3、5位苯是产生药效的重要基团,5位苯环的2’位引入体积小的吸电子基团.(如F、Cl )可使活性增强。
4、1,2位拼入三氮唑可提高稳定性,并提高与受体的亲和力,活性显著增加。
5、苯环用生物电子等排体噻吩杂环置换,保留活性。
6、1位取代基在体内代谢去烃基,仍有活性。
三、吩噻嗪类药物的构效关系:以氯丙嗪为先导化合物,对吩噻嗪类进行结构改造。
三方面:1、吩噻嗪环上的取代基:吩噻嗪环只有2位引入吸电子基团时可增强活性。
作用强度与吸电子性能成正比,CF3>Cl>COCH3>H>OH。
2位乙酰基可降低药物的毒性和副作用。
2、10位N上的取代基:母核上的10位N原子与侧链碱性氨基之间相隔3个直链碳原子时作用最强,是吩噻嗪类抗精神病药的基本结构。
侧链末端的碱性基团常为叔胺,也可为氮杂环,以哌嗪取代作用最强。
3、三环的生物电子等排体。
四、μ受体选择性激动剂构效关系1、芳环和碱性叔胺氮原子是μ受体激动剂的必要结构部分,二者通过2个或3个碳原子的碳链相连接。
2、芳环3位酚羟基的存在使活性显著增强。
氮原子上以甲基取代活性好,当N-取代基增大到3~5个碳原子时,如烯丙基(纳洛酮)、环丁基甲基时,由激动剂转变为拮抗剂。
3、μ受体选择性激动剂的药效构象相同,其芳环以直立键与哌啶环相连。
构效关系的名词解释
构效关系的名词解释是:药物或其他生理活性物质的化学结构与其生理活性之间的关系,是药物化学的主要研究内容之一。
狭义的构效关系研究的对象是药物,广义的构效关系研究的对象则是一切具有生理活性的化学物质,包括药物、农药、化学毒剂等。
最早期的构效关系研究以直观的方式定性推测生理活性物质结构与活性的关系,进而推测靶酶活性位点的结构和设计新的活性物质结构,随着信息技术的发展,以计算机为辅助工具的定量构效关系成为构效关系研究的主要方向,定量构效关系也成为合理药物设计的重要方法之一。
药物的构效关系:
例如拟胆碱药的化学结构与乙酰胆碱相似,都有季胺或叔胺基团,都能与胆碱受体结合,形成具有活性的复合物,因而表现出相似的作用。
又如,磺胺药与对氨基苯甲酸化学结构相似,因而能与对氨基苯甲酸竞争二氢叶酸合成酶而影响细菌叶酸的代谢。
在具有基本结构的任何一类药物中,药理作用的类型是由它们的基本结构决定的,而它们的药理作用的相对强度,是由基本结构上各个取代基团的性质决定的,如磺胺类药物。
但也有化学结构相似而作用相拮抗的情况,如磺胺与对氨基苯甲酸、氨丙啉与硫胺等。
同时,也有化学结构不同而药理作用相似的情况,如麻黄碱与茶碱。
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表本人。
本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。
如发现本站有涉嫌抄袭侵权/违法违规的内容请联系客服!。
药物分子的合成和构效关系研究药物是现代医学的重要组成部分,药物分子的合成和构效关系研究则是药物开发研究领域的重要方向。
药物分子是指由一个或多个元素组成的化学物质,它们与生物体内的分子相互作用,改变生物体的代谢或功能,从而达到治疗疾病的目的。
药物分子的合成和构效关系研究是药物研发的关键环节,下面就该主题进行分析和探讨。
一、药物分子的合成方法药物分子的合成方法通常包括几个步骤:分子设计、反应物选择、化学反应、物质分离和纯化、小分子合成和工艺优化。
其中分子设计是整个药物开发研究的重要一环,它涉及到分子的选择、功能结构设计、化合物的活性和副作用测试等。
反应物选择则是药物分子合成的关键,需要针对分子设计的需求选择合适的反应物,不能过度耗费,一定要可行且经济有效。
化学反应则是药物分子合成的核心部分,必须进行简便有效的反应,同时需要考虑反应速度和立体化学效应等因素。
物质分离和纯化是药物合成过程中不可缺少的环节,需要采用多种分离技术,如结晶、萃取、色谱、凝胶电泳等。
小分子合成则是将药物分子按照工艺流程进行逐步合成,从而得到最终的合成产物。
工艺优化则是合成过程的最后一步,需要对合成过程进行改进,以提高合成产品的纯度和产量。
二、药物分子的构效关系研究药物分子的构效关系研究是药物研发的重要一环,它可以从物理化学角度揭示药物分子与生物体的相互作用机理,为药物设计提供重要的理论支持。
商品药品大多数都是有机分子化合物,其分子结构和药效作用之间存在着密切的联系,因此需要通过构效关系研究来推断分子结构与药效之间的联系,从而提高药物的疗效和降低毒性。
药物分子的构效关系研究主要涉及药效、代谢、毒性、光学活性等方面,需要遵循以下原则:一、基于分子结构的比较分析;二、考虑分子结构与生物活性之间的密切联系;三、基于现代计算机科学的技术手段;四、通过分子修饰和合成新药分子进行实验验证。
三、药物分子的未来发展趋势近年来,随着科学技术的进步和人们对健康意识的不断提高,药物分子的研究也将越来越重要。
药物构效关系数字化教材院〔部〕食品药品学院教研室药品技术教师潍坊职业学院药物构效关系受体是一种细胞膜上或细胞内能特异识别生物活性分子并与之结合,进而引起生物学效应的特殊蛋白质〔少数也可是糖脂〕。
影响药物产生作用的因素主要包括药物与受体的作用及药物到达作用部位的浓度大小,显然这两者均与药物的化学结构和理化性质密切相关。
本节讨论药物根本结构与药效关系、药物立体结构与药效关系、药物理化性质与药效关系。
药物根本结构与药效关系根据药物在体内的作用方式,可将药物分为结构非特异性药物和结构特异性药物。
结构非特异性药物的生物活性主要受药物的理化性质的影响,而对化学结构无特异要求,如全身麻醉药中的吸入麻醉药,其药效主要受药物的脂水分配系数的影响。
临床上使用的绝大对数药物属于结构特异性药物,药物生物活性除与理化性质相关外,主要受药物的化学结构影响,其与特异性受体相互作用形成复合物产生生物活性。
化学结构的微小变化,都可能导致药物活性大为降低甚至丧失。
被受体识别的药物可以与受体结合,生成药物和受体复合物,激活受体兴奋或抑制,产生一系列特定的生理生化反响,到达治疗疾病的目的。
受体对药物的识别主要表现在结构互补和立体化学的选择性方面,因此凡与受体结合的药物均为结构特异性药物。
在构效关系研究中,把具有相同药理作用的药物中相同局部的化学结构称为药物的根本结构。
具有根本结构的药物都是结构特异性药物,都能被受体识别并与之结合。
许多药物都具有根本结构,如含有对氨基苯甲酸酯根本结构的局部麻醉药、含有对氨基苯磺酰胺根本结构的磺胺类药物、含有苯乙胺根本结构的拟肾上腺素药物及含有吩噻嗪根本结构的抗精神病药等。
药物根本结构确实定有助于该类药物的结构优化,从而得到疗效好、毒性低的药物。
如在确定对氨基苯磺酰胺为磺胺类药物的根本结构后,通过嘧啶环或噻唑环等含氮杂环取代磺酰胺基氮原子上的氢,结果发现可使药物活性大为增强,进而得到N-单取代且以杂环取代疗效最正确的磺胺类药物;假设在苯环上再引入取代基后,抑菌作用那么降低或丧失,从而提示在苯环上其余四个位置上无需引入任何取代基。