OOS实验室调查报告
- 格式:docx
- 大小:56.07 KB
- 文档页数:4

文件内容:1、目的—————————————————— 22、范围—————————————————— 23、职责—————————————————— 24、定义—————————————————— 25、原则—————————————————— 36、一般程序———————————————— 37、参考文献、文件————————————— 58、关键词————————————————— 69、附录—————————————————— 610、培训—————————————————— 611、文件历史变更—————————————— 6颁发部门:分发部门:执行日期:第1页共6页1、目的:作为判断产品是否放行于市场或从市场中可能的召回的依据,并指导实验室发现实验过程的缺陷,进行整改并采取相应的措施。
2、范围:本规程适用于本企业所有在质量控制实验室以及中间控制实验室发生的任何对物料、中间产品及成品的检验。
3、职责:3.1质量管理部负责本规程的起草、修订、审核与颁布,并监督本规程的实施情况;3.2一旦出现OOS/OOT/AD情况,QC实验室以及中控实验室应发起实验室结果调查;3.3如非实验室差错,质量部与生产部应依照偏差处理程序等方法调查,其他部门应全面配合结果调查工作;3.4质量部携相关部门对调查结果分析评估,并监督整改措施与预防措施的实施,如为上市批次,应上报权威机构,以采取相应措施。
4、定义:4.1超出标准(OOS—Out of Specification)的结果:包括所有超出标准或由法规、法定方法或制造商规定的可接受限度的所有可疑的结果。
4.2超出趋势(OOT—Out of trend)的结果:结果虽然在质量标准之内但仍然比较反常,与长期观察到的趋势或者预期结果不一致。
4.3异常数据(AD—abnormal data):指超出标准与超出趋势外的异常数据或来自异常测试过程中的数据或事件。
4.4实验室差错(Laboratory Error):因仪器故障和实验室操作相关的差错(如标识错误、计算、称量、稀释等)而产生的差错。



质量管理体系那回事之三——OOS调查有人说了,OOS明明是发生在实验室里的东西,为什么不写在“检验那回事”系列中,而放在“QMS那回事”中呢。
这主要是由于,虽然问题发现在实验室,但问题的根本原因可能在实验室、生产车间、物料仓储、采购、公用工程等各个方面,所以放在这里提可能更合适些,而且这本身也是偏差的一种特例,放在质量管理体系下也更合适。
当然我并不是说大家公司OOS调查文件必须要放在QA文件目录下,放在QC文件目录下在国内企业可能更多,这也很大程度上说明国内QA对于QC了解的程度还不太够。
但无论放在哪里,尤其是放在QC体系下,OOS调查文件必须完全覆盖整个调查的始末。
比如举个例子,前几天去一个企业看了他们的体系,发现他们OOS只是调查了实验室的部分,而几乎没有QA参与,更没有生产调查这部分内容。
l 问——你没有生产调查的内容,那你实验室调查完没有调查出原因怎么办?l 答——那是生产的事,生产来调查。
l 问——那生产调查的指令在哪里,他怎么调查?l 答——那按偏差调查呗。
l 问——现在没有偏差发现呢,他怎么按偏差调查?问题不一定出现在他们那里。
l 答……所以OOS调查必须是一个完整的体系,当然其中可能会涉及到偏差或CAPA,但一方面你可以转到相应的表格,也可以继续使用OOS调查表格来说明,就像我在第二部分偏差调查里说的一样,即使是偏差,也并不一定非得使用“偏差调查表格”来完成偏差调查,如果可能,没有必要表格转来转去。
看过我写的东西人可能都知道,我一般写东西基本上是不看什么指南的,可能有关物质那篇有点例外,那是由于当时的内容主要是用于当年的职称答辩,必须要有文献,所以可能引用的药典和指南最多;在一篇文章我也有必要引入一篇指南,因为这是一个我所接触过的我认为最接地气的指南之一,当然这也可能与我看过的指南不是太多有关,尤其是国内的,这篇指南就是FDA在2006年发布的Investigating Out-of-Specification (OOS) Test Resultsfor Pharmaceutical Production,它基本上涵盖了OOS调查的所有要素,这在里,我大多数的概念均出自于此,但我觉得有一些程序上的顺序我会有一些调整,这是因为我们所发生的OOS 的原因的最大比例是由于实验室造成的,调整也是为了尽可能地确保生产的顺利进行,文章中我还会举一些案例和分析来帮助大家理解。


文件内容:1、目的—————————————————— 22、范围—————————————————— 23、职责—————————————————— 24、定义—————————————————— 25、原则—————————————————— 36、一般程序———————————————— 37、参考文献、文件————————————— 58、关键词————————————————— 69、附录—————————————————— 610、培训—————————————————— 611、文件历史变更—————————————— 6颁发部门:分发部门:执行日期:第1页共6页1、目的:作为判断产品是否放行于市场或从市场中可能的召回的依据,并指导实验室发现实验过程的缺陷,进行整改并采取相应的措施。
2、范围:本规程适用于本企业所有在质量控制实验室以及中间控制实验室发生的任何对物料、中间产品及成品的检验。
3、职责:3.1质量管理部负责本规程的起草、修订、审核与颁布,并监督本规程的实施情况;3.2一旦出现OOS/OOT/AD情况,QC实验室以及中控实验室应发起实验室结果调查;3.3如非实验室差错,质量部与生产部应依照偏差处理程序等方法调查,其他部门应全面配合结果调查工作;3.4质量部携相关部门对调查结果分析评估,并监督整改措施与预防措施的实施,如为上市批次,应上报权威机构,以采取相应措施。
4、定义:4.1超出标准(OOS—Out of Specification)的结果:包括所有超出标准或由法规、法定方法或制造商规定的可接受限度的所有可疑的结果。
4.2超出趋势(OOT—Out of trend)的结果:结果虽然在质量标准之内但仍然比较反常,与长期观察到的趋势或者预期结果不一致。
4.3异常数据(AD—abnormal data):指超出标准与超出趋势外的异常数据或来自异常测试过程中的数据或事件。
4.4实验室差错(Laboratory Error):因仪器故障和实验室操作相关的差错(如标识错误、计算、称量、稀释等)而产生的差错。

检验结果超标情况的调查及处理操作规程内容:1、定义:检验结果超标:超出质量标准的检验结果(简称OOS结果)和超出趋势的检验结果(OOT结果)的统称。
超出质量标准的检验结果(简称OOS结果):不符合既定的质量标准或接受标准的检验结果。
任何有OOS结果的产品都不能被放行。
超出趋势的结果(简称OOT结果):此结果没有超出既定的质量标准或接受标准的限度,但已产生的适当数量的数据不符合正常的结果分布。
任何出现OOT结果的产品仍将要被放行。
OOT 只是用于内部管理控制的目的。
有效结果:在适当、科学并经批准的条件下所产生的检验结果,不管是否在质量标准或接受标准的限度之内。
无效结果:若依照科学的依据,如果某项检验结果在不正确或未获准的条件下产生,该结果即视为无效结果,需摒弃。
局外检验结果:由不明确原因导致的检验结果。
通过统计学分析,它往往超出其它数据的范围之外,可作为被抛弃的结果。
一般用于微生物检验而不是化学检验结果的分析。
实验室的调查:在实验室内展开的调查,(比如,调查是与试剂制备/仪器和分析方法等等有关的)以找出导致异常检验结果的原因。
再分析:对准备的同种样品进行的再次检验,例如可导致异常检验结果的部分样品或稀释液。
再取样:从物料原始的包装和批次中获取额外的物料。
这直接涉及到返回到物料的原包装/批次,并对物料进行额外取样。
再检验:同一样品的不同部分的重复分析。
对照样品:以前曾经检测合格的或具有良好特性的样品。
实验室错误:在实验室发生的,由检验员、仪器或设备的某部分、或物料所引起的错误。
包括使用错误的检验标准;不正确的样品或对照品/标准品的制备程序;使用校准有误的天平,有误差的设备或仪器;计算错误。
2.职责:QA经理:QA经理应确保本规程符合当地和政府的有关药品生产管理规范的要求;对由任何实验物料/产品产生的OOS结果做出质量决定;对调查进行监督,以确保进行了正确的调查及记录,且通知了相关部门。
QC经理:QC经理有责任就本规程的内容要求对检验员进行培训;确保检验员无论何时遇到检验结果超标情况,都要遵守本规程的要求;指导检验员按本规程的要求进行实验室内部调查;协助实施实验室以外的调查;确保进行了正确的调查及记录;确保报告所有的调查结果,且通知了相关部门。

International Journal of Generic DrugsG U I D A N C E F O R I N D U S T R YOUT-OF-SPECIFICATIONTEST RESULTS‘…A n O u t -o f -S p e c i f i c a t i o n T e s t R e s u l t i s n o t n e c e s s a r ya p r o d u c t f a i l u r e - a n d n e e d s t ob e q u a l i f i e d …FDA's NEW OOS GUIDANCEGUIDANCE FOR INDUSTRYInvestigating Out-of-Specification(OOS) Test Results for Pharmaceutical Production.I. INTRODUCTIONhis guidance has been prepared by the Office of Compliance /Division of Manufacturing and Product 1 Quality, Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration. This guidance document represents the Agency’s current thinking on evaluating OOS test results. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute,regulations, or both.T his guidance for industry provides the Agency’s current thinking on how to evaluate suspect, or out of specification (OOS), test results.F or purposes of this document, the term OOS results includes all suspect results that fall outside the specifications or acceptance criteria established in new drug applications,official compendia, or by the manufacturer.T his guidance applies to laboratorytesting during the manufacture of active pharmaceutical ingredients,excipients, and other components and the testing of finished products to the extent that current good manufacturing practices (CGMP)regulations apply (21 CFR parts 210and 211). S pecifically, the guidance discusses how to investigate suspect,or OOS test results, including the responsibilities of laboratory personnel, the laboratory phase of the investigation, additional testing that may be necessary, when to expand the investigation outside the laboratory, and the final evaluation of all test results.4TABLE OF CONTENTSI. INTRODUCTION II. BACKGROUNDIII. IDENTIFYING AND ASSESSING OOS TEST RESULTSA. Responsibility of the AnalystB. Responsibilities of the SupervisorIV. INVESTIGATING OOS TEST RESULTS A. General Investigational Principles B. Laboratory Phase of an Investigation V. CONCLUDING THE INVESTIGATION A. Interpretation of Investigation Results B. Reporting4TII. BACKGROUNDFDA considers the integrity of laboratory testing and documentation records to be important during drug manufacturing. Laboratory testing, which is required by the cGMP regulations (§ 211.165), is necessary to confirm that components, containers-closures, in-process materials; finished products, conform to specifications, including stability.T esting also supports analytical and process validation efforts. General CGMP regulations covering laboratory operations can be found in part 211, subparts I (Laboratory Controls) and J (Records and Reports).T hese regulations provide for the establishment of scientifically sound and appropriate specifications, standards, and test procedures that are designed to ensure that components and containers of drug products conform to the established standards. Section 211.165(f) of the CGMP regulations specifies that products that fail to meet established standards and other relevant quality control criteria will be rejected. 4 III. IDENTIFYING AND ASSESSING OOS TEST RESULTSFDA regulations require that an investigation be conducted whenever an OOS test result is obtained. The purpose of the investigation is to determine the cause of the OOS. Even if a batch is rejected based on an OOS result, the investigation is necessary to determine if the result is associated with other batches of the same drug product or other products.Every Failure (OOS)MUST be investigatedand its impacts on related batches evaluated B atch rejection does not negate the need to perform the investigation. The regulations require that a written record of the investigation be made including the conclusions of the investigation and follow-up (211.192). T o be meaningful, the investigation should be thorough, timely, unbiased, well-documented, and scientifically defensible.T he first phase of the investigation includes an initial assessment of the accuracy of the laboratory's data, before test solutions are discarded, whenever possible. Investigate, Concludeand Follow-up EverySpecification FailureT his way, hypotheses regarding laboratory error or instrument malfunctions may be tested using the same test solutions. If this initial assessment indicates that no errors were made in the analytical process used to arrive at the data, a complete failure investigation should follow.A. Responsibility of the AnalystT he first responsibility for achieving accurate laboratory testing results lies with the analyst who is performing the test. The analyst should be aware of potential problems that could occur during the testing process and should watch for problems that could create OOS results.I n accordance with the CGMP regulations1 the analyst should ensure that only those instruments meeting established specifications are used and that all instruments are properly calibrated. 1(§ 211.160 (b)(4)),C ertain analytical methods have system suitability requirements, and systems not meeting such requirements should not be used. For example, in chromatographic systems, reference standard solutions may be injected at intervals throughout chromatographic runs to measure drift, noise, and repeatability.I f reference standard responses indicate that the system is not functioning properly, all of the data collected during the suspect time period should be properly identified and should not be used.T he cause of the malfunction should be identified and corrected before a decision is made whether to use any data prior to the suspect period.Track AnalyticalFailures Back toTheir Origin PointB efore discarding test preparations or standard preparations, analysts should check the data for compliance with specifications. When unexpected results are obtained and no obvious explanation exists, test preparations should be retained and the analyst should inform the supervisor.An assessment of the accuracy of the results should be started immediately.I f errors are obvious, such as the spilling of a sample solution or the incomplete transfer of a sample composite, the analyst should immediately document what happened.Analysis Developinga Fault MUST beStopped ImmediatelyA nalysts should not knowingly continue an analysis they expect to invalidate at a later time for an assignable cause (i.e., analyses should not be completed for the sole purpose of seeing what results can be obtained when obvious errors are known). These same responsibilities extend to analysts at contract testing laboratories.B. Responsibilities of the Supervisor O nce an OOS result has been identified, the supervisor's assessment should be objective and timely. There should be no preconceived assumptions as to the cause of the OOS result.D ata should be assessed promptly to ascertain if the results may be attributed to laboratory error, or whether the results could indicate problems in the manufacturing process.Is the OOS a Laboratory or Production Error? An immediate assessment could include re-examination of the actual solutions, test units, and glassware used in the original measurements and preparations, which would allow more credibility to be given to laboratory error theories.S teps should be taken as part of the supervisor's assessment:Key:- [D E C I D E D][1]. D iscuss the test method with the analyst; confirm analyst knowledge of and performance of the correct procedure.[2]. E xamine the raw data obtained in the analysis, including chromatograms and spectra, and identify anomalous or suspect information.[3]. C onfirm the performance of the instruments.[4]. D etermine that the appropriate reference standards, solvents, reagents, and other solutions were used and that they meet quality control specifications. [5]. E valuate the performance of the testing method to ensure that it is performing according to the standard expected based on method validation data.[6]. D ocument and preserve evidence of this assessment.T he assignment of a cause for OOS results will be greatly facilitated if the retained sample preparations are examined promptly. Hypotheses regarding what might have happened (e.g. dilution error, instrument malfunction) can be tested. Examination of the retained solutions can be performed as part of the laboratory investigation. Examples:n Solutions can be re-injected as partof an investigation where a transient equipment malfunction is suspected. T his could occur, if bubbles were introduced during an injection on a chromatographic system, which other tests indicated was performing properly. Such theories are difficult to prove.H owever, a re-injection can provide strong evidence that the problem should be attributed to the instrument, rather than the sample or its preparation.n For release rate testing of certain specialized dosage forms, where possible, examination of the dosage unit tested might determine whether it was damaged in a way that affected its performance. Such damage would provide evidence to invalidate the OOS test result, and a retest would be indicated.n Further extraction of a dosage unit can be performed to determine whether it was fully extracted during the original analysis. Incomplete extraction could invalidate the test results and should lead to questions regarding validation of the test method (i.e. the extraction procedure).I t is important that each step in the investigation be fully documented. The supervisor should ascertain not only the reliability of the individual value obtained, but also the significance these OOS results represent in the overall quality assurance program. Supervisors should be especially alert to developing trends.L aboratory error should be relatively rare. Frequent errors suggest a problem that might be due to inadequate training of analysts, poorly maintained or improperly calibrated equipment, or careless work. Whenever laboratory error is identified, the firm should determine the source of that error and take corrective action to ensure that it does not occur again.T o ensure full compliance with the CGMP regulations, the manufacturer also should maintain adequate documentation of the corrective action.OOS Rules:Clear Error- Invalidate Unclear Failure - Investigate Do not assume anything!I n summary, when clear evidence of laboratory error exists, laboratory testing results should be invalidated. When evidence of laboratory error remains unclear, a failure investigation should be conducted to determine what caused the unexpected results.I t should not be assumed that failing test results are attributable to analytical error without performing and documenting an investigation. Both the initial laboratory assessment and the following failure investigation should be documented fully.4IV. INVESTIGATING OOS TEST RESULTS Full scale investigationsW hen the initial assessment does not determine that laboratory error caused the OOS result and testing results appear to be accurate, a full-scale failure investigation using a predefined procedure should be conducted.T he objective of such an investigation should be to identify the source of the OOS result. Varying test results could indicate problems in the manufacturing process, or result from sampling problems.Such investigations present a challenge both to employees and to management and should be given the highest priority. T he investigation should be conducted by the quality control unit and should involve all otherdepartments that could be implicated, including manufacturing, process development, maintenance, and engineering. Other potential problems should be identified and investigated.QC Unit Investigates: Production a nd Laboratory Systems & Documentation T he records and documentation of the manufacturing process should be fully investigated to determine the possible cause of the OOS results.[A]. General Investigational PrinciplesA failure investigation should consist of a timely, thorough, and well-documented review.The written record should reflect that the following general steps have been taken. [ I-T R A C][1]. The reason for the I nvestigation has been clearly identified.[2].T he overall manufacturing process sequences that may have caused the problem should be summarized.[3].R esults of the documentation review should be provided with the assignment of actual or probable cause.[4].A review should be made to determine if the problem has occurred previously.[5].C orrective actions taken should be described.T he general review should include a list of other batches and products possibly affected and any required corrective actions taken including any comments and signatures of appropriate production and quality control personnel regarding any material that may have been reprocessed after additional testing. [B]. Laboratory Phase of an InvestigationA number of practices are used during the laboratory phase of an investigation. These include:[1]. Retesting a portion of the original sample [2]. Testing a specimen from the collection of a new sample from the batch[3]. Re-sampling testing data[4]. Using outlier testing.1. RETESTINGP art of the investigation may involve retesting of a portion of the original sample. The sample used for the retesting should be taken from the same homogeneous material that was originally collected from the lot, tested, and yielded the OOS results.F or a liquid, it may be from the original unit liquid product or composite of the liquid product; for a solid it may be an additional weighing from the same sample composite that had been prepared by the analyst.S ituations where retesting is indicated include investigating testing instrument malfunctions or to identify a possible sample handling integrity problem, for example, a suspected dilution error. Generally, retesting is neither specified nor prohibited by approved applications or by the compendia.Investigation Retesting: First Performed onthe original sampleby a Second AnalystD ecisions to retest should be based on the objectives of the testing and sound scientific judgement. Retesting should be performed by an analyst other than the one who performed the original test.T he CGMP regulations require the establishment of specifications, standards, sampling plans, test procedures, and other laboratory control mechanisms (§ 211.160).T he establishment of such control mechanisms for examination of additional specimens for commercialor regulatory compliance testing must be in accordance with "predetermined guidelines or sampling strategies" (USP 23, General Notices and Requirements, p.9). S ome firms have used a strategy of repeated testing until a passing result is obtained (testing into compliance), then disregarding the OOS results without scientific justification. Testing into compliance is objectionable under the CGMPs.Multiple Retesting:Into ComplianceIs a GMP ViolationT he number of retests to be performed on a sample should be specified in advance by the firm in the SOP.The num ber may vary depending upon the variability of the particular test method employed, but should be based on scientifically sound, supportable principles. The number should not be adjusted depending on the results obtained.T he firm's predetermined testing procedures should contain a point at which the testing ends and the product is evaluated. If, at this point, the results are unsatisfactory, the batch is suspect and must be rejected or held pending further investigation (§211.165(f)).I n the case of a clearly identified laboratory error, the retest results would substitute for the original test results. The original results should be retained, however, and an explanation recorded.This record should be initialed and dated by the involved persons and include a discussion of the error and supervisory comments.If no laboratory or statistical errors are identified in the first test, there is no scientific basis for invalidating initial OOS results in favor of passing retest results. All test results, both passing and suspect, should be reported and considered in batch release decisions. Consider OOS ++ RetestResult - if theInvestigated OOScan not be Invalidated2. RE-SAMPLINGW hile retesting refers to analysis of the original sample, re-sampling involves analyzing a specimen from the collection of a new sample from the batch. The establishment of control mechanisms for examination of additional specimens for commercial or regulatory compliance testing should be in accordance with predetermined procedures and sampling strategies (§ 211.165(c)).I n some cases, when all data have been examined, it may be concluded that the original sample was prepared improperly and was therefore not representative of the batch (§211.160(b)(3)).Re-sample Only ifO riginal Sampleis Provedas UnrepresentativeA re-sampling of the batch should be conducted if the investigation shows that the original sample was not representative of the batch.This would be indicated, for example, by widely varied results obtained from several aliquots of the original composite (after determining there was no error in the performance of the analysis).R e-sampling should be performed by the same qualified, validated methods that were used for the initial sample.H owever, if the investigation determines that the initial sampling method was in error, a new accurate sampling method must be developed,qualified, and documented. (§§ 211.160 and 165(c)).3. AVERAGINGA veraging test data can be a valid approach, but its use depends upon the sample and its purpose. For example, in an optical rotation test, several discrete measurements are averaged to determine the optical rotation for a sample, and this average is reported as the test result. If the sample can be assumed to be homogeneous (i.e., an individual sample preparation designed to be homogeneous), using averages can provide a more accurate result.I n the case of microbiological assays, the USP prefers the use of averages because of the innate variability of the biological test system.Reliance on averages has the disadvantage of hiding variability among individual test results. For this reason, unless averaging is specified by the test method or adequate written investigation procedures, all individual test results should be reported.I n some cases, a statistical treatment of the variability of results should be reported. For example, in a test for dosage form content uniformity, the standard deviation (or relative standard deviation) is also reported. A veraging also can conceal variations in the different portions of the sample. For example, the use of averages is inappropriate when performing powder blend/mixture uniformity or dosage form content uniformity determinations.I n these cases, the testing is intended to measure variability within the product, and the individual results should be reported.I t should be noted that a test might consist of replicates to arrive at a result. For instance, an HPLC assay result may be determined by averaging the peak responses from a number of consecutive, replicate injections from the same preparation (usually 2 or 3).T he assay result would be calculated using the peak response average. This determination is considered one test and one result. This is a distinct difference from the analysis of different portions from a lot, intended to determine variability within the lot. T he use of replicates should be included in the written, approved, test methodology. Unexpected variation in replicate determinations should trigger investigation and documentation requirements (21 CFR 211.192).I n some cases, a series of assay results may be a part of the test procedure. If some of the results are OOS and some are within specification and all are within the documented variation of the method, the passing results should be given no more credence than the failing results, in the absence of documented evidence that analytical error had occurred.R elying on test data averaging in such a case can be particularly misleading. For example, in an assay with a given range of 90 to 110 percent, test results of 89 percent, 89 percent, and 92 percent would produce an average of 90 percent even though two of the assay values represent failing results. T o use averaged results for assay reporting, all test results should conform to specifications. Although the above average of 90 percent may be useful in terms of an overall assessment of process capabilities, the individual assay results indicate non-conformance because two of the three results are outside of the range.A low assay value should also trigger concerns that the batch was not formulated properly because the batchmust be formulated with the intent to provide not less than 100 percent of the labeled or established amount of active ingredient (21 CFR 211.101(a)). T he above example does not necessarily require the manufacturer to fail the batch, but indicates that an immediate investigation should be conducted for batch disposition decisions.4. OUTLIER TESTST he CGMP regulations require that statistically valid quality control criteria include appropriate acceptance and/or rejection levels (§ 211.165(d)). On rare occasions, a value may be obtained that is markedly different from the others in a series obtained using a validated method. Such a value may qualify as a statistical outlier. An outlier may result from a deviation from prescribed test methods, or it may be the result of variability in the sample.I t should never be assumed that the reason for an outlier is error in the testing procedure, rather than inherent variability in the sample being tested. O utlier testing is a statistical procedure for identifying from an array those data that are extreme. The possible use of outlier tests should be determined in advance. This should be written into SOPs for data interpretation and be well documented.T he SOPs should include the specific outlier test to be applied with relevant parameters specified in advance.The SOPs should specify the minimum number of results required to obtain a statistically significant assessment from the specified outlier test.F or biological assays having a high variability, an outlier test may be an appropriate statistical analysis to identify those results that are statistically extreme observations.T he USP describes outlier tests in the section on 'Design and Analysis of Biological Assays'(USP 23, p. 1705). In these cases, the outlier observation is omitted from calculations. The USP also states that "arbitrary rejection or retention of an apparently aberrant response can be a serious source of bias. . .the rejection of observations solely on the basis of their relative magnitudes is a procedure to be used sparingly" (USP 23, p. 1705).F or validated chemical tests with relatively small variance, and if the sample being tested can be considered homogeneous (for example, an assay of composited dosage form to determine strength), an outlier test is only a statistical analysis of the data obtained from testing and retesting.It will not identify the cause of an extreme observation and, thus should not be used to invalidate the data.A n outlier test may be useful as part of the evaluation of the significance of that result for batch evaluation, along with other data.Don't UseOutlier Testing inDissolutionContent Uniformity(i.e. where variability exists)O utlier tests have no applicability in cases where the variability in the product is what is being assessed, such as for content uniformity, dissolution, or release rate determinations.I n these applications, a value perceived to be an outlier may in fact be an accurate result of a non-uniform product.V. CONCLUDING THE INVESTIGATION T o conclude the investigation, the results should be evaluated, the batch quality should be determined, and a release decision should be made. The SOPs should be followed in arriving at this point. Once a batch has been rejected, there is no limit to further testing to determine the cause of the failure so that a corrective action can be taken. [A]. Interpretation of Investigation Results A n OOS result does not necessarily mean the subject batch fails and must be rejected.OOS ResultsDo Not AutomaticallyFail the Batch (investigate & interpret fully) T he OOS result should be investigated, and the findings of the investigation, including retest results, should be interpreted to evaluate the batch and reach a decision regarding release or rejection (§ 211.165).I n those instances where an investigation has revealed a cause, and the suspect result is invalidated, the result should not used to evaluate the quality of the batch or lot.A proven invalid result is always discarded I nvalidation of a discrete test result is done only upon the observation and documentation of a test event that can reasonably be determined to have caused the OOS result.I n those cases where the investigation indicates an OOS result is caused by a factor affecting the batch quality (i.e., an OOS result is confirmed), the result should be used in evaluating the quality of the batch or lot.A confirmed OOS result indicates that the batch does not meet established standards or specifications and should result in the batch's rejection, in accordance with § 211.165(f), and proper disposal.F or inconclusive investigationsi.e. in cases where an investigation:-[1] does not reveal a cause for the OOS test result and[2] does not confirm the OOS result —the OOS result should be retained in the record and given full consideration in the batch or lot disposition decision.A un proven OOS result is always includedS tatistical treatments of data should not be used to invalidate a discrete chemical test result.I n very rare occasions and only after a full investigation has failed to reveal the cause of the OOS result, a statistical analysis may be valuable as one assessment of the probability of the OOS result as discordant, and for providing perspective on the result in the overall evaluation of batch quality R ecords must be kept of complete data derived from all tests performed to ensure compliance with established specifications/standards (21 CFR 211.194).[B]. ReportingF or those products that are the subject of applications, regulations require submitting within three working days a Field Alert Report (FAR) of information concerning any failure of a distributed batch to meet any of the specifications established in an application (21 CFR 314.81(b)(1)(ii).OOS test results not invalidated on distributed batches/lots for this class of products are considered to be one kind of “information concerning any failure” described in this regulation.T his includes OOS results that are considered to be discordant and of low value in batch quality evaluation. In these cases, a FAR should be submitted.DECISIONTREE . (IJGD Vol. 02 #07 1998)。

⽆菌检查实验阳性结果的OOS调查报告实例15.9 案例分析以下是某公司发⽣⼀起⽆菌检查实验阳性结果的OOS调查报告实例。
⽬录[隐藏]1 1. 偏差概述2 2. 偏差调查o 2.1 2.1 实验室初步调查o 2.2 2.2 ⽣产过程调查o 2.3 2.3 实验室深⼊调查3 3. 污染菌来源分析o 3.1 3.1 ⽆菌⼯艺药品污染菌来源分析o 3.2 3.2 最终灭菌⼯艺药品污染菌来源分析4 4. 产品⽆菌风险评估o 4.1 4.1 ⽆菌⼯艺药品×××(批号:×××1)的⽆菌风险评估o 4.2 4.2 最终灭菌⼯艺药品××××(批号:××××2)的⽆菌风险评估5 5. 基于风险评估的产品微⽣物学质量评价1. 偏差概述××年××⽉××⽇,微⽣物实验室对3批⽆菌⼯艺药品和3批最终灭菌⼯艺药品进⾏⽆菌检查初试,其中有1批⽆菌⼯艺药品和1批最终灭菌⼯艺药品的检验结果呈阳性。
有关偏差情况见表15-11。
2. 偏差调查2.1 实验室初步调查2.1.1 检验过程调查××年××⽉××⽇进⾏⽆菌检查试验初检期间,检验⽤设施、设备运⾏正常,未发⽣故障;检验设施和样品的消毒⼯作按SOP进⾏;在样品培养过程中未发现明显的⼈为性差错。
2.1.2 培养结果调查在××年××⽉××⽇所进⾏的⽆菌检查实验中,共有2只培养瓶呈阳性结果。
当天各样品的检查结果见表15-12。
阴性对照试验结果全部呈阴性。
2.1.3 初检当⽇⽆菌检查试验环境监测数据调查在××年××⽉××⽇对⽆菌检查试验室进⾏了环境监测,其中包括空⽓悬浮粒⼦14个样品、空⽓浮游菌14个样品、空⽓沉降菌19个样品、设施设备表⾯微⽣物43个样品、瓶表⾯微⽣物10个样品、⼈员卫⽣状况监测50个样品,所有空⽓悬浮粒⼦监测结果合格,检出污染微⽣物的样品仅为地板表⾯(未超过警戒限度,葡萄球菌,见2.3.1.5.1)。

III.鉴定和评估OOS检验结果—阶段I:实验室调查FDA法规(§ 211.192)要求,当得到OOS检验结果时,应予调查。
调查的目的是确定引起OOS的原因。
应确定是检验过程的异常还是生产工艺异常导致的OOS结果。
即使因OOS结果判定了不合格批,仍必须进行调查以确定该结果是否影响到同种产品其它批号或其它产品,一批不合格也不能否定调查的必要。
法规(§ 211.192)要求要有调查的书面记录,包括结论和跟进措施。
为了使调查有意义,调查应是彻底的,及时的,没有偏见的,形成文件和经得起科学推敲的。
调查的第一阶段应包括对实验室数据的准确性的初步评价。
如果可能,这应在丢弃试验溶液(包括被测样品复合的或同质的来源)之前进行。
这样,假定是实验室错误或仪器故障,可以使用原溶液测定。
如果初步评估显示在得到该数据的分析过程中没有发生错误,应进行全面的OOS调查。
与实验室约定,实验室应把它的数据、缺陷、辅助文件移交给生产公司的QCU,QCU之后开始全面的OOS调查。
A.分析员的责任得到准确的实验数据,是进行检验的分析员的首要责任。
分析员应意识到在检验过程中可能发生的问题,并应注意可能产生不准确结果的问题。
按照CGMP § 211.160 (b)(4),分析员应确保只有符合已制定性能规范的仪器才能使用,并且所有的仪器应恰当的校准。
特定分析方法有系统适应性要求,不符合这些要求的系统不以能使用。
比如,在色谱系统中,在进行色谱检测期间内间隔一段时间进样对照品溶液去测定漂移、噪声和重复性.如果对照品响应值表明该系统功能不正常,在可疑的时间内收集的所有数据应该被适当标识并不能使用。
应鉴别故障的原因,如果可能,在决定是否使用可疑期之前的数据前,应予以纠正。
分析员在丢弃检验溶液或标准溶液之前,应核对数据是否符合检验规范。
当获得意想不到的结果且没有明显的理由时,应该保留样品制备液且分析员应该通知主管.应该立即开始评估检验结果的正确性如果错误是明显的,如:样品溶液有洒出或样品成分的未完全转移,分析员应该立即记录所发生的情况。
简述oos结果调查流程下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by theeditor. I hope that after you download them,they can help yousolve practical problems. The document can be customized andmodified after downloading,please adjust and use it according toactual needs, thank you!In addition, our shop provides you with various types ofpractical materials,such as educational essays, diaryappreciation,sentence excerpts,ancient poems,classic articles,topic composition,work summary,word parsing,copy excerpts,other materials and so on,want to know different data formats andwriting methods,please pay attention!OOS 结果调查流程是指对超出质量标准的结果进行调查和分析的过程。
OOS总结
《OOS与OOT》做为偏差部分内容的延续,主要发生在实验室,在质量体系中具有一定的独立性,调查方式上也与常规的偏差调查略有不同。
OOS/OOT调查分为两个阶段,
《OOS第一阶段调查》主要目的是确认OOS和OOT是否为明显的分析错误产生,计算、样品错误以及分析过程是产生明显分析错误的几个常见因素。
《OOS第二阶段调查》往往是由于原因不明显,从而需要重新对样品进行制备甚至是重新取样,第二阶段调查通常由更有经验的人员完成,以排除人员操作的问题,但建议在原产生OOS数据的设备上进行,需要预先制订重新测试方案,不允许测试直到通过(Test into Compliance)。
生产调查可参见之前的《Major/Critical偏差报告要求》,因为OOS/OOT的结果一旦确认,肯定会是一个高级别的偏差。
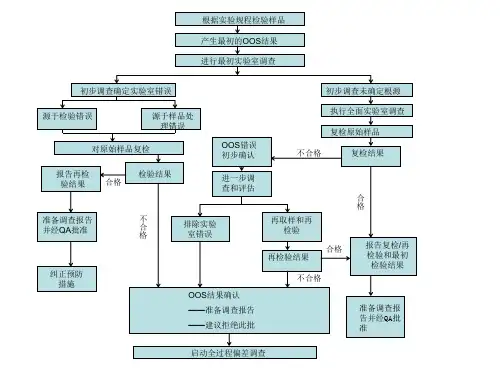
通过一个流程图---《OOS调查流程图》,也可以将整个OOS/OOT的流程串起来,不同的部门在OOS调查过程中的职责可以参见《OOS调查中的职责》。
OOS或者OOT产生的相关的行动项也需要进行有效性检查与确认,同时企业应该结合质量管理评审,对OOS和OOT进行《趋势追踪》,以不断完善和健全质量体系。
预告:
明天的推送文章已经写好,可是局限于微信公众平台每天只能发一篇的限制,需要先留个悬念,个人觉得值得小小期待一下,喜欢的话请明天多多转发。
小长假后两天可能没有图文推送,我也需要休息调整一下,祝大家端午小长假快乐!。
关于药品检验结果OOS的调查Guidance for Industry,Investigating Out of Specification (OOS) Test Results for PharmaceuticalProduction行业指南:药品检验结果OOS的调查DRAFT GUIDANCE指南草案U.S. Department of Health and Human ServicesFood and Drug AdministrationCenter for Drug Evaluation and Research (CDER)September 1998CP #TABLE OF CONTENTS名目I. INTRODUCTION 序言. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . .. . . . .. . . .. . . 1II. BACKGROUND . 背景. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ... . . . . . . 1 III. IDENTIFYING AND ASSESSING OOS TEST RESULTS OOS检验结果的判定和评估. . .. 2A.Responsibility of the Analyst 检验员的责任. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. 2B. Responsibilities of the Supervisor主管的责任. . . . . . . . . . . . . . . .. . . . . .. . . . . . . . . . . . . . . .. . . . .3 IV. INVESTIGATING OOS TEST RESULTS OOS 检验结果的调查. . . . . . . . . . . . . . . . . . . .. 5A. General Investigational Principles . . 一样调查原那么. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5B. Laboratory Phase of an Investigation . 实验室的调查时期. . . . . . . . . . . . . . . .. . . .. . . . . . . . . . . . 6 V. CONCLUDING THE INVESTIGATION . .调查结论. . . . . . . . . . . . . . . . . . . . . . . . .. . .. . . . . . . 10A. Interpretation of Investigation Results . 调查结果的说明. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10B. Reporting . . .报告. . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11This guidance has been prepared by the Office of Compliance/Division of Manufacturing and ProductQuality, Center for Drug Evaluation and Research (CDER) at the Food and Drug Administration. This guidance document represents the Agency’s current thinking on evaluating OOS test results. Itdoes not create or confer any rights for or on any person and does not operate to bind FDA or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute, regulations, or both.本指南由FDA的CDER的达标办公室/制造、产品、质量分部起草,本指南阐明了机构关于评估OOS 检验结果的现行的方法。