药明康德有机合成讲义资料 有机氟化合物的合成共54页文档
- 格式:ppt
- 大小:4.64 MB
- 文档页数:54



经典化学合成反应标准操作药明康德新药开发有限公司化学合成部编写前言有机合成研究人员在做化学反应经常碰到常规的反应手边没有现成的标准操作步骤而要去查文献,在试同一类反应时,为了寻找各种反应条件方法也得去查资料。
为了提高大家的工作效率,因此化学合成部需要一份《经典合成反应标准操作》。
在这份材料中,我们精选药物化学中各类经典的合成反应,每类反应有什么方法,并通过实际经验对每类反应的各种条件进行点评,供大家在摸索合成条件时进行比较。
同时每种反应的标准操作,均可作为模板套用于书写客户的final report,这样可以大大节省研究人员书写final report的时间,也相应减少在报告中的文法错误。
另外本版是初版,在今后的工作中我们将根据需要修订这份材料。
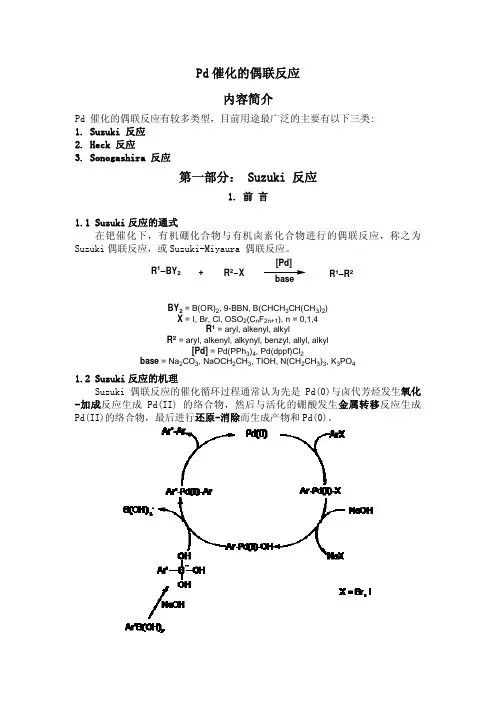
药明康德新药开发有限公司化学合成部2005-6-28目录1.胺的合成a)还原胺化b)直接烷基化c)腈的还原d)酰胺的还原e)硝基的还原f)叠氮的还原g)Hoffman降解h)羧酸通过Cris 重排2.羧酸衍生物的合成a)酰胺化的反应b)酯化反应c)腈转化为酯和酰胺d)钯催化的插羰反应e)酯交换为酰氨3.羧酸的合成a)醇氧化b)酯水解c)酰胺的水解d)腈的水解e)有机金属试剂的羰基化反应f)芳香甲基的氧化4.醛酮的合成a)Weinreb 酰胺合成醛酮b)醇氧化c)酯的直接还原d)有机金属试剂对腈加成合成酮5.脂肪卤代物的合成a)醇转化为脂肪溴代物通过PBr3 转化通过PPh3 与CBr4 转化HBr直接交换通过相应的氯代物或磺酸酯与LiBr交换、b)醇转化为脂肪氯代物通过SOCl2转化通过PPh3 与CCl4 转化HCl直接交换c)醇转化为脂肪碘代物通过PPh3 与I2 转化通过相应的氯代物或磺酸酯与NaI交换6.芳香卤代物的合成a)Sandermyyer 重氮化卤代b)直接卤代c)杂环的酚羟基或醚的卤代7.醇的合成a)羧酸或酯的还原b)醛酮的还原c)卤代烃的水解d)吡啶的氧化转位8.酚的合成a)Sandermayer 重氮化反应b)醚的水解c)Bayer-vigerlar 氧化d)硼酸的氧化9.腈的合成a)磺酸酯或卤代烃的取代b)酰胺的脱水c)芳卤代烃的氰基取代10.硝化反应11.醚的合成a)芳香醚的合成酚与烷基卤代烃的直接烷基化Mitsunobu 芳香醚化Buckwald芳香醚化b)脂肪醚的合成醇的醚化12.脲的合成a)胺与异腈酸酯的反应b)用三光气合成脲c)羰基二咪唑(CDI)合成脲d)对硝基苯酚碳酰胺合成脲13.烯烃的合成a)Wittig 反应b)羟基的消除c)Wittig-Horner 反应合成α,β-不饱和酯14.磺酸及磺酰氯的合成a)氯磺化反应合成磺酰氯b)从硫醇合成磺酰氯c)磺化反应15.氨基酸的合成a)Streck 反应合成b)手性氨基酸的合成16.偶联反应a)Suzuki Couplingb)Buckwald 芳胺化,芳酰胺化、c)Heck 反应17.Mitsunobu 反应a)醇的反转b)胺的取代18.脱羟基反应19.酮还原为亚甲基20.氨的保护及脱保护策略a)用碳酰胺作保护基b)苄基保护21.醇的保护及脱保护策略a)用硅醚进行保护b)其他醚类保护22.羧基的保护格氏反应---------------------------------------------------------------------------------------------------------1还原胺化---------------------------------------------------------------------------------------------------------2卤化反应---------------------------------------------------------------------------------------------------------2 Suzuki coupling-------------------------------------------------------------------------------------------------2磺化反应---------------------------------------------------------------------------------------------------------3酯化反应---------------------------------------------------------------------------------------------------------3水解反应---------------------------------------------------------------------------------------------------------3硝化反应---------------------------------------------------------------------------------------------------------4 n-BuLi------------------------------------------------------------------------------------------------------------4 LiAlH4还原-----------------------------------------------------------------------------------------------------4 POCl3的杂环氯代----------------------------------------------------------------------------------------------5 NaH---------------------------------------------------------------------------------------------------------------5 NBS---------------------------------------------------------------------------------------------------------------5m-CPBA ----------------------------------------------------------------------------------------------------------6EDC ---------------------------------------------------------------------------------------------------------------6用三光气成脲---------------------------------------------------------------------------------------------------7芳卤用n-BuLi 处理后与Weinreb 酰胺成酮-----------------------------------------------------------------7Boc 上保护OHH 2NHO OOOOOO OHN HO OHO O ABTo a solution of A (2.72 g, 13.9 mmol) and tetramethylammonium hydroxide pentahydrate (5.62 g, 31.0 mmol) in acetonitrile (270 mL) was added di-tert-butyldicarbonate (3.79 g; 17.4 mmol) and the resulting solution was allowed to stir 18 h at rt and concentrated. The residue was partitioned between Et2O/H2O; the phases were separated and the aqueous phase extracted twice more with Et2O. The aqueous phase was brought to pH 4 with solid citric acid and extracted with CHCl3 (3.x.100 mL). The organic extracts were combined, dried (Na2SO4) and concentrated to afford 2.58 g (63 percent) B as a white foam.ReturnBoc 脱保护OON HOOOOH 2NTert-Butyl 2-(2-methoxyphenoxy)ethylcarbamate (23.8 g, 89 mmol) in dichloromethane (10 ml) was cooled to 0 deg C and stirred as a mixture of trifluoroacetic acid: dichloromethane (1:1, 40 ml) was added dropwise. The mixture was allowed to warm to rt, stirred for 2 hours and concentrated in vacuo. The residue was taken back up in dichloromethane (100 ml) and the solution was washed with saturated aqueous sodium hydrogen carbonate (3*20 ml) and aqueous sodium hydroxide (10percent, 3*20 ml), dried (Na2SO4), filtered and concentrated in vacuo to provide 2-(2-methoxyphenoxy)ethylamine (13 g, 88percent yield) as a light yellow solid.Return格氏反应NCNNOA stirred mixture of magnesium turnings (23.6 g, 0.98 mol) and Et2O (200 mL) under nitrogen is treated with a crystal of iodine and about 5percent of a solution of bromoethane (56.3 ml, 0.75 mol) in Et2O (375 mL). When the reaction starts, the remainder of the bromoethane solution is added, dropwise at a rate sufficient to maintain a gentle reflux. After the addition, stirring is continued for 1 hour. To this solution of ethylmagnesium bromide was slowly added a solution of 4-cyanopyridine (39 g, 0.375 mol) in Et2O (750 ml). The reaction mixture was warmed at reflux for 12 hours, treated with concentrated H2SO4 (125 ml)/H2O (125 ml), and then washed three times with Et2O (250 ml). The aqueous portion was made basic (PH 9) with 15percent NaOH solution and extracted five times with 250 ml portions of Et2O. The combined Et2O extracts were dried (MgSO4), and the solvent was removed under reduced pressure to afford a brown oil (48.4 g, 95percent).Return还原胺化OHO H 2N+HON HA solution of 2-amino-4-ethylphenol (1.00 g. 7.28 mmol), 2-naphthaldehyde (1.13 g, 7.28 mmol), andp-toluenesulfonic acid (0.05 g) in methanol (50 ML) was stirred at room temp for 24 h. To the resultant solution, sodium borohydride (0.82 g, 22 mmol) was added in small portions. After addition was completed, the mixture was stirred at room temperature for 30 min and concentrated under vacuum. The residue was then subjected to column chromatography on silica gel eluted with 10percent ethyl acetate in hexane and followed by recrystallization (aqueous methanol) yielded 450 mg (22percent) of analytically pure product.Return卤化反应O2N O2NBrTo a stirred solution of 8-methyl-1-nitro-naphthalene (10.6g, 56.32 mmol) and iron (III) chloride (0.45 g, 2.77 mmo) in CCl4 (150 ml) heated to 60°C was added dropwise (3.0 ml, 58.23 mmol) of bromine. After one hour, the reaction mixture was poured into saturated NaHCO3 solution, and the layers were separated. The aqueous layer was re-extracted with CH2Cl2. The combined organic layers were dried (MgSO4) and the solvent was removed under reduced pressure. The crude residue was recrystallized from ethanol and the mother liquors were concentrated and then flash chromatographed on silica, eluding hexanes:ethyl acetate (12: 1).ReturnSuzuki couplingBrBOO NH+NH To a mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-1H-indole (2 g, 8.2 mnmol) and3-bromobenzene (0.87 ml, 8.3 mmol) in THF (28 ml) were added palladium catalyst Pd(PPh3)4 (284 mg, 0.25 mmol) and the freshly prepared sodium hydroxide solution (984 mg in 9 ml of water).The system was degassed and then charged with nitrogen for three times. The mixture was stirred under nitrogen at 70 °Coil bath for 6 hours. The reaction solution was cooled to room temperature, diluted with ethyl acetate and separated from water layer. The ethyl acetate solution was washed by brine, dried over Na2SO4 and concentrated. The residue was purified on a silica gel column eluding with hexanes: EtOAc 9:1 to give 1.38 g (78%yield) of 4-phenyl-1H-indole as a colorless liquid.Return磺化反应NOFFFNOFFFSOClOChlorosulfonic acid (4.66g, 40 mmol) is added dropwise to a cold (0°C) solution of2,3-dihydro-2-trifluoroacetyl-1H-Benz[de]isoquinoline (2.9g, 8 mmol) in chloroform (800 ml). The resulting solution is stirred at 0°C for 30 minutes. The cold bath is then removed and the solution is stirred at room temperature for 1 hour then cautiously poured into ice water. The organic layer is separated, dried over magnesium sulfate and concentrated to afford the title compound. The crude product is purified by column chromatography eluted with 10% acetic ether in petroleum ether (2.36 g, 81% yield).Return酯化反应HOHO O HOO OA mixture of 4-hydroxymethylnaphthoic acid (10 g, 50 mmol), methanol (300 ml), and concentrate H2SO4(2 ml) was refluxed overnight. The insolubles were filtered off and the filtrate was concentrated. The residue was taken up in ethyl acetate and washed with aqueous NaHCO3 (2*), brine, dried over MgSO4, and concentrated to give a yellow oil. Silica gel column chromatography using ethyl acetate/hexane (1/3) gave the desired product as a yellow oil (3.3 g, 35%yield).Return水解反应OO OHOA solution of 1-Methyl-naphthalene-2-carboxylic acid methyl ester (7.20g, 35mmol) and 2N sodium hydroxide (35ml) in tetrahydrofuran (130ml) was stirred under reflux for 18 hours. The mixture was neutralised using 2N hydrochloric acid, and extracted with dichloromethane (3x). The combined organic solutions were dried (MgSO4), and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gelusing an elution gradient of dichloromethane: methanol (100:0 to 97:3) to afford the title compound as a solid (3.11g, 47.8%yield).Return硝化反应NO 2To a cold (0°C) suspension of 1-methylnaphthalene (5 g, 35.2 mmol) in HNO3 was added H2SO4 (5 ml) dropwise. After stirring the reaction for one hour, the solution was diluted with ethyl acetate and washed with water (3*), aqueous saturated NaHCO3 (2*) and brine, dried over MgSO4, and concentrated. The product was purified by silica gel column chromatography using ethyl acetate: hexane (5: 95) and recrystallized from methanol to give yellow needles (0.22g, 33% yield).Returnn-BuLiEtOCF 3O CF 3O NCTo a dry three-necked round-bottomed flask with an addition funnel and at -78°C under inert atmosphere was charged with anhydrous THF (500 ml). A solution of n-butyllithium (2.5 M in hexane, 88ml, 220 mmol) was added dropwise followed by addition of a solution of acetonitrile (10.43 ml, 200 mmol) in anhydrous THF (100 ml). The internal temperature was maintained below -70°C during the entire addition process. After 2 hr at -78°C a solution of Trifluoro-acetic acid ethyl ester (14.2 g, 100 mmol) in anhydrous THF (30 ml) was added dropwise and the mixture was stirred for 1.5 hr. To the mixture was added acetic anhydride to quench the reaction. The reaction mixture was allowed to warm up to rt. A precipitate was filtered and the filtrate was concentrated to give a brown oil, which was used in the next step without purification.ReturnLiAlH4还原HOHO O OHOHOA solution of 2,3-naphthalenedicarboxylic acid (4.6 g, 0.023 mole) in dry THF (135 ml, warmed to 50° to maintain solution) is added dropwise over 15 minutes to a 1.15 M lithium aluminum hydride solution in THF (45 ml, 0.052 mole). The solution is stirred 3 hours after which TLC indicated consumption of diacid and formation of a new major product. The reaction is quenched carefully with THF-water, then 2N hydrochloric acid (40 ml) is added, and the resulting mixture is extracted 3 times with ether. The combined ether extracts are washed with water (2 times), with saturated sodium bicarbonate solution (1 time), with water, and are dried (sodium sulfate), filtered, and concentrated to give a tan solid (3.67 g). The solid is recrystallized from ethyl acetate giving the title compound (2.91 g, 67.3%yield) as a light tan crystalline material.ReturnPOCl3的杂环氯代NN HOOHN NClClTo a suspension of 2,4-dihydroxy-5,6-dimethylpyrimidine (6.2 g, 0.044 mol) in POCl3 (25 ml) was slowly added N,N-dimethylaniline (6.18 ml, 0.049 mol). The mixture was then refluxed at 125 °C for 3 hours. After this time, the starting material completely dissolved indicating that the reaction was completed.The reaction mixture was cooled and then poured slowly onto ice to quench the POCl3(caution[exothermic]). A precipitate formed, which was filtered and washed with ice-cold water. The precipitate was dried under high vacuum overnight to yield 2,4-dichloro-5,6-dimethyl-pyrimidine (7.2 g, 0.041 mol, 92%yield) as a yellow solid.ReturnNaHHSH 2N Cl +SNH 2Sodium hydride (50% in mineral oil, 5.5 g, 0.11 mol) was added portionwise at 0 °C under a nitrogen atmosphere to a solution of 2-aminobenzenethiol (12 ml, 0.1 mol) in DMF (120 ml). After 0.5 h, benzyl chloride (11.5 ml, 0.1 mol) in DMF (80 ml) was added in 0.5 h. The solution was stirred for 3 h while the temperature was allowed to rise to rt, then it was poured into ice/water (1000 g). The precipitate was filtered, dissolved in ethyl acetate and washed with brine. The organic layer was dried over Na2SO4 and evaporated. The solid obtained was ground in pentane (19.3 g, 90% yield).ReturnNBSNN FCl ClNBSN N FCl ClBrA mixture of 2,4-Dichloro-6-ethyl-5-fluoro-pyrimidine (27.46 g , 0.14mol), AIBN (1.32 g) and n-bromosuccinimide (27.02 g , 0.152mol) in CH2Cl2 (170 ml) was refluxed under a nitrogen atmosphere for 36 h. Then washed by water, the aqueous was extracted by CH2Cl2. The combined organic layer was washed by saturated Na2S2O3 and brine, dried over Na2SO4, and evaporated to give a white solid which was purified by column chromatography eluted with 50% acetic ether in petroleum ether (34 g, 88.6% yield).Return氢化反应O ONH OONH2Cl ClA mixture of ethyl 3-(N-benzylamino)-3-methylbutyrate hydrochloride (25g, 0.1 mol) and 10percent Pd-C (2g) in 250 ml of dried alcohol was hydrogenated under 55 psi H2 for four days. The reaction medium was then filtered and evaporated under reduced pressure to provide an amber oil which gradually crystallized upon standing (18 g, 100% yield).Returnm-CPBAS NH2SNH2OA solution of 85% m-chloroperoxybenzoic acid (19 g, 94 mmol) in CH2Cl2 (350 ml)was added at –5 –0 °C to a solution of 2-Benzylsulfanyl-phenylamine (19 g, 88 mmol) in CH2Cl2 (400 ml). The mixture was allowed to warm to rt in 3 h, then it was washed with a 5% Na2S2O3 solution, 10% NaHCO3 solution and brine. The organic layer was dried over Na2SO4, and evaporated. The solid was ground in pentane (19 g, 95% yield).ReturnEDCNH2OHNOO+HOHOHNOOTo a 0°C mixture of Boc-L-tyrosine (2.04 g, 7.26 mmol) and amylamine (0.63 gl, 7.26 mmol) in methylene chloride (30 ml) is added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) (1.53 g, 9.9 mmol). Thewhite mixture is stirred at 0°C for 5 min and at room temp for 23 hrs. The resulting solution is diluted with methylene chloride (30 ml) and washed successively with 0.5 M HCl (40 ml), water (20 ml) and sat aq sodium bicarbonate (25 ml). The organic phase is dried over magnesium sulfate and concentrated to a foam (1.84 g, 72.4%yield), sufficiently pure to carry into the next step. An analytical sample is obtained by HPLC.Return三光气成脲NH 2ONO 2Si O Cl Cl ClO O Cl Cl ClO 2NHN H NO OHOHNO 2+To a solution of 2-(tert-butyldimethylsilyloxy)-4-nitroaniline (200 mg, 0.75 mmol) in toluene (10 ml) triethylamine (0.13 ml, 1.64 mmol) and triphosgene (88.4 mg, 0.3 mmol) were added. The reaction mixture was stirred at 70 °C for 2 hours, then cooled to room temperature. Then more 2-(tert-butyldimethylsilyloxy)-4-nitroaniline (200 mg, 0.75 mmol) was added. The resulting mixture was allowed to stir at 70 °C for 48 hours then cooled to room temperature. The reaction mixture was partitioned between water and ethyl acetate. The combined organic phase was washed with brine, dried over MgSO4 and filtered. Removal of solvent at reduced pressure and chromatography of the resulting oil on silica gel (hexane: ethyl acetate, 10:1) gave 1,3-Bis-(2-hydroxy-4-nitro-phenyl)-urea (130 mg, 31%yield).Return芳卤用n-BuLi处理后与Weinreb酰胺成酮N FFFFNOO+FFFO NFTo a solution of diisopropylamine (17.69 ml, 0.135 mole) in THF (200 ml) at –78°C under argon was added n-butyllithium (54.0 ml, 2.5M in hexane, 0.135 mole), followed after 5 min by dropwise a solution of 2-fluoro-4-methylpyridine (10 g, 0.090 mole) in THF (20 ml). After stirring for 15 min at –78°C, a solution of N-methoxy-N-methyl-3-trifluoromethylbenzamide (23.08 g, 0.099 mole) in THF (10 ml) was added dropwise. After stirring for more 5 min, the reaction was allowed to warm to 0°C and quenched by pouring into water (400 ml) and ethyl acetate (400 ml). The layers were separated, and the aqueous layer washed with ethyl acetate (200 ml). The ethyl acetate extracts were combined, dried over anhydrous sodium sulfate, filtered, and concentrated to an oil which was chromatographed on silica gel with 20percent ethyl acetate in hexane to give 21.6 g of 2-(2-Fluoro-pyridin-4-yl)-1-(3-trifluoromethyl-phenyl)-ethanone (84.8%yield).Return。


经典化学合成反应标准操作有机氟化合物的合成目录1.前言 (2)2.通过不饱和C-C键的加成合成氟化合物 (4)3.通过重氮盐合成氟化合物 (7)3.1 Balz-Schiemann 反应 (7)3.2 从α-氨基酸合成α-氟代羧酸 (10)4.亲核氟代 (12)4.1.环氧开环合成氟化合物 (12)4.2.氧被氟取代合成氟化合物 (13)4.3. 硫被氟取代合成氟化合物 (20)4.4.磺酸酯被氟取代合成氟化合物 (22)5.亲电氟代 (24)5.1 芳环的亲电氟代 (25)5.2 通过烯醇,烯醇醚,烯醇酯及烯胺合成α-氟代羰基化合物 (27)5.3 有机金属化合物的氟代 (29)5.4 不对称亲电氟代 (31)6.三氟甲基的引入(Trifluoromethylation) (34)6.1 自由基三氟甲基化 (34)6.2 亲电三氟甲基化 (35)6.3 亲核三氟甲基化 (36)参考文献 (41)1.前言1771年Scheele 第一次报导了氟化氢,1836年Dumas 和Peligot 报导了第一个有机氟化合物:一氟甲烷的合成,而元素氟的制备则在50年后,1886年Henri Moissan 分离到了氟气。
Table 1 H,F,Cl的比较H F Cl电子排布1s1 2s22p5 3s23p53d0电负性 2.1 4.0 3.0电离能(kcal/mol) 315 403 300键能C-X (kcal/mol) 99 111 78键长C-X (Å) 1.09 1.32 1.77氟原子半径小,是电负性最强的元素,这种极强烈的电负性增加了氟与碳的亲和力。
因此它们所形成的C-F键要比C-H键能大得多,明显地增强了含氟有机化合物的稳定性。
如下面三个聚合物,聚乙烯和聚四氟乙烯都很稳定,而聚四氯乙烯则不稳定。
H2CH2nF2CCF2nCl2CCCl2nstable stable unstable氟原子的引入导致有机及无机化合物具有独特的物理、化学性能及生理活性。




培训目的: 理解立体化学的基本原理. 培训后有能力区分辨别镜像立体异构体(enantiomers)和非镜像立体异构体(diastereomers), 镜像立体异构体(enantiomers)和非镜像立体异构体(diastereomers)的物理化学性质, 镜像立体选择反应(enantioselective)和非镜像立体选择(diastereoselctive)反应等培训时间: 1.5 –2小时培训内容:1. 碳原子有机化合物的异构体种类(Isomerism in Carbon Compounds)2. 镜像立体异构体(enantiomers) 和非镜像立体异构体(diastereomers)的物理化学性质3. 立体选择反应(Stereochemistry in Chemical Reactions)参考文献:1. /nbauld/teach/stereo.html#stereo2. Organic Chemistry, Solomans&Fryhle, 4th editionProperties of Enantiomers and Diastereomers 手性分子使偏振光偏转对应的镜像异构体会向相反方向偏转相同的角度!!手性色谱(HPLC, GC, etc)Properties of Enantiomers and Diastereomers1.不是所有的外消旋化合物都是可以分开的2.外消旋化合物需要在手性柱上先被分开来测量反应ee%3.绝对构型无法通过手性色谱确立,除非同已知文献报道的结果相比NNH NNBocO NNH NNBocO 外消旋化合物(普通HPLC)NNH NNBoc O=+外消旋化合物(手性HPLC)OMeOMe: 你能将上图中的四组峰和下面四个化合物一一对应吗OH OMeOH OMeOH OMe OH OMe:51问题?OH。