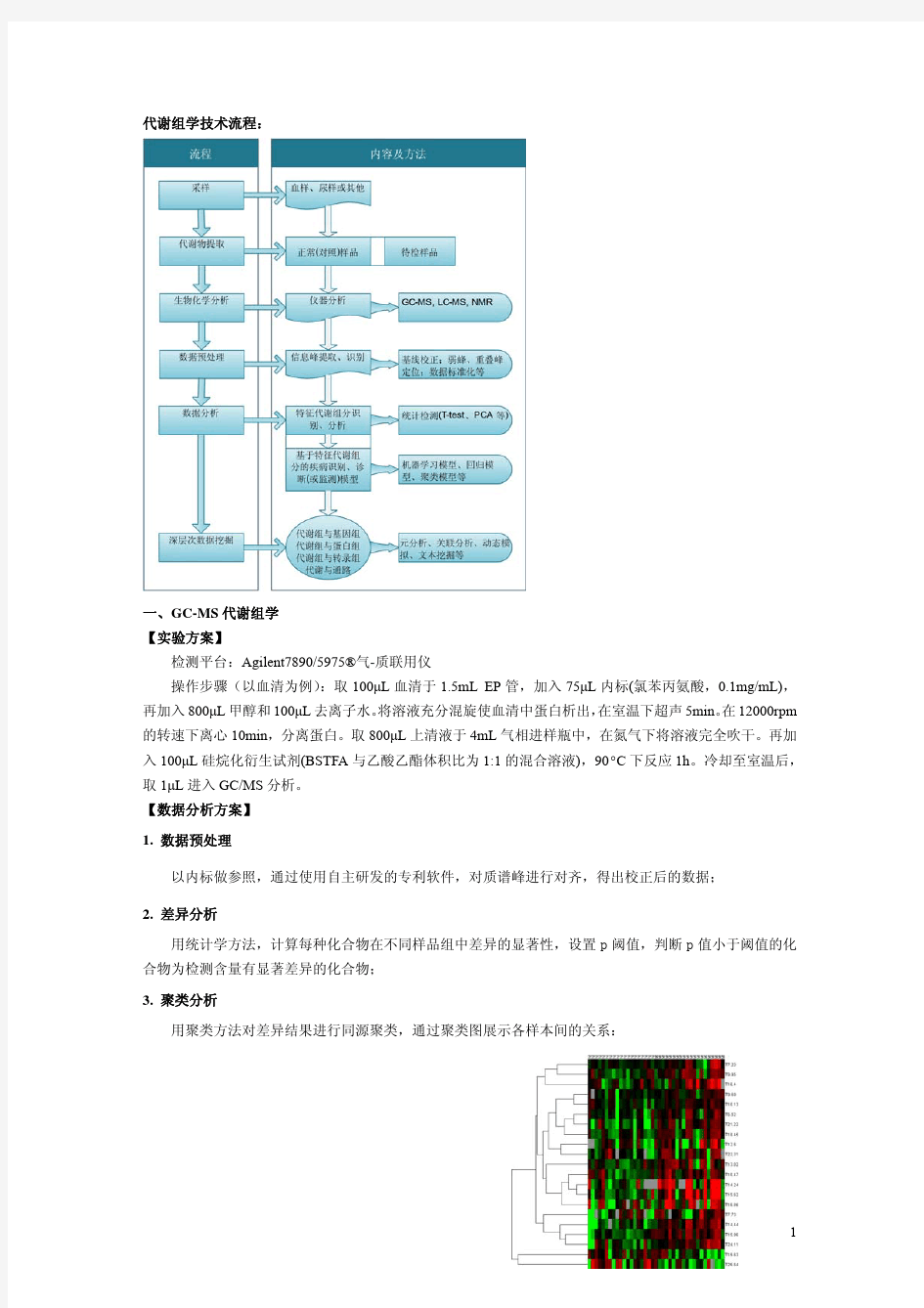

代谢组学技术流程:
一、GC-MS 代谢组学 【实验方案】 检测平台:Agilent7890/5975?气-质联用仪 操作步骤(以血清为例) :取 100μL 血清于 1.5mL EP 管,加入 75μL 内标(氯苯丙氨酸,0.1mg/mL), 再加入 800μL 甲醇和 100μL 去离子水。 将溶液充分混旋使血清中蛋白析出, 在室温下超声 5min。 12000rpm 在 的转速下离心 10min,分离蛋白。取 800μL 上清液于 4mL 气相进样瓶中,在氮气下将溶液完全吹干。再加 入 100μL 硅烷化衍生试剂(BSTFA 与乙酸乙酯体积比为 1:1 的混合溶液),90°C 下反应 1h。冷却至室温后, 取 1μL 进入 GC/MS 分析。 【数据分析方案】 1. 数据预处理 以内标做参照,通过使用自主研发的专利软件,对质谱峰进行对齐,得出校正后的数据; 2. 差异分析 用统计学方法,计算每种化合物在不同样品组中差异的显著性,设置 p 阈值,判断 p 值小于阈值的化 合物为检测含量有显著差异的化合物; 3. 聚类分析 用聚类方法对差异结果进行同源聚类,通过聚类图展示各样本间的关系:
1
4. PCA 分析(principal component analysis) 通过对有显著性差异的化合物数据做 PCA 分析,将样品的特征量压缩,在低维度空间反映样品之间的 关系。用 score plot 展示 PCA 分析结果。
5. ROC 诊断分析 ROC 曲线(receiver operating characteristic curve)又 称受试者工作特征曲线该方法广泛用于医学诊断性能的评价, 将灵敏度设为纵轴,特异性设在横轴,该曲线下的积分面积大 小与诊断试验的优劣密切相关:
ROC curve
1 0.9
T e p sitive ra (se sib ru o te n ility)
二、LC-MS 代谢组学 【实验方案】 检测平台:Waters ACQUITY? UPLC 系统 操作步骤 (以尿液为例) 在室温下对冻存的尿液样 : 本进行解冻,然后添加 2 倍 4℃去离子水稀释,蜗旋混 【数据分析方案】 1. 数据预处理 对原始数据进行提取, 对齐, 归一化分析, 通过原始基峰离子(BPI)色谱图, 初步观察仪器 的保留时间重现性,所测得的物质数量,以及 不同组之间的色谱图是否具有较明显的差异。
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0 0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
False positive rate (1-specificity)
合后过 0.22 微米滤膜,对所得滤液进行液质分析。进样量为 5 微升,样本运行按照随机顺序进行。
2. 聚类分析
用聚类方法对差异结果进行同源聚类,通过 聚类图展示各样本间的关系,并观察组间的分离 关系。 3. PCA 分析(principal component analysis) 通过对有显著性差异的化合物数据做 PCA 分析,将样品的特 征量压缩,在低维度空间反映样品之间的关系。在正式多维统计 前, 先对所有归一化的数据进行平均中心化和 Pareto-scaling 预处
2
理,用 score plot 展示 PCA 分析结果。
4. PLS-DA 分析 一种用偏最小二乘法回归作为核心算法的分类计算方法,其 最终结果返回的是回归因子,根据其返回结果进行类别判断。我 们能根据 PLS-DA 对所建模型的对外预测能力进行评价。
5. 差异化合物筛选 根据 PLS-DA 模型的 VIP 值大于 1,且的 p 值小于 0.05 的原则获得差异性物质,如下表所示: 均值对数比 保留时间(min) 0.67 4.24 7.51 0.71 ... 几个代表性物质的 Box plot 如下所示: 质荷比 203.0522 447.0923 373.2736 114.0655 … p‐value?(ttest) 3.69E‐10 0.002677 0.001979 0.00023 … (B/A) 1.616 1.345 ‐0.567 ‐0.582 … 中值对数比 (B/A) 1.751 1.934 ‐0.744 ‐0.648 …?
6. 差异化合物鉴定 我们搜索 Metlin 数据库(Mass tolerance < 0.1 Da) ,获得一些可能的物质信息。 目前对液质分析获得的化合物的定性方法还主要依靠与标准品的比对来判定,因此定性工作将面临巨 大的挑战。我公司可对化合物进行进一步定性,如有需要可联系公司相关区域销售代表。 三、NMR 代谢组学 【实验方案】 检测平台:NMR Varian Inova600MHz? (配置超低温探头)/ NMR Bruker AVANCE II 600MHz?。 操作步骤(以血清为例) :样品预处理所有血清解冻后在4℃、12000g离心10min。取0.4 mL上层血清加 至5mm NMR测试管中,再加入0.1mL重水振荡混匀。进行NMR测试,记录血清样本1H NMR谱,实验温度 为25℃,预饱和法压制水峰,饱和时间为2 s。采用Carr-Purcell—Meiboom-Gill(CPMG)脉冲序列抑制血清中 较宽的蛋白质信号。采用2.1 s的弛豫延迟时间、8000.0 Hz的谱宽和1.000 s的采集时间。总回波时间100 ms, 累加次数128次。 【数据分析方案】 1. 1H NMR 图
对谱图进行傅立叶变换,相位调整,基线 校正、去偏置等处理,结合化合物归属表及相 关数据库,鉴定实验所检测到的代谢产物。
3
1
H NMR 图
3. PCA 分析 为了分析各个样品之间的关系,将校正后的数据做 PCA 分析(。PCA 是可以将分散在 n 维变量上的代谢 指纹图谱信息集中到某几个综合指标(主成分)上的统计分析方法。
PBS 0.010
t[2]
LC
C
0.015 0.01 0.005 0 -0.005
PBS LC C
0.005 t[2] 0.000 -0.005
-0.01
-0.010 -0.02 -0.01 0.00 t[1] 0.01 0.02
-0.015 -0.02 -0.01 0 t[1] 0.01
0.02
PCA scores plot
4. 组间差异代谢物统计 对组间变化显著的代谢产物进行统计分析,计算相关系数。实例如下表所示: Metabolites β-Glucose: 3.19(ddc), 3.34(t), 3.40(m), 3.43(t), 3.84(dd), 4.48(d) Valine: 0.91(d), 0.98(d) NAG: N-acetyl glycoprotein signals: 1.98(s) …
组间差异代谢物列表
ra PBS-LCb 0.867 -0.844 … PBS-C 0.852 0.712 … LC-C -0.919 0.770 0.867 …
5. PLS-DA 析 在分析得到差异代谢物的基础上,用偏最小二乘法-判别分 析(PLS-DA)识别的算法寻找一组代谢物作为生物标志物,建立 最优的判别模型,使达到最好的区分疾病和正常样品的效果。
t[2] 20 0 -20
PBS
LC
-60
-30
0 t[1]
30
60
PLS-DA 分析图
6. OPLS-DA 分析 对 PLS-DA 模型进行正交矫正处理(OPLS-DA),使用 SIMCA 软件进行正交偏最小二乘法-判别分析 (OPLS-DA),最大化地凸显模型内部不同组别之间的差异。OPLS-DA 使用自适换算(unit variance scaling) 的数据标度换算方式。
4
PBS 40 20 t[2]O 0 -20 -40 -40 -20 0 t[1]P 20
LC
40
R2X=67.7%, Q2=0.676
OPLS-DA 分析图
loading plots
7. 器官内部和不同器官之间显著变化代谢物之间的关联分析
计算器官内部及不同器官之间显著变 化代谢物之间的相关系数,利用 matlab 构 建像素图。
关联分析像素图
5
代谢组学的研究方法和研究流程分子微生物学112300003林兵 随着人类基因组计划等重大科学项目的实施,基因组学、转录组学及蛋白质组学在研究人类生命科学的过程中发挥了重要的作用,与此同时, 代谢组学(metabolomics)在20世纪90年代中期产生并迅速地发展起来,与基因组学、转录组学、蛋白质组学共同组成系统生物学。基因组学、转录组学、蛋白质组学和代谢组学等各种组学0在生命科学领域中发挥了重要的作用,它们分别从调控生命过程的不同层面进行研究, 使人们能够从分子水平研究生命现象, 探讨生命的本质, 逐步系统地认识生命发展的规律.这些组学手段加上生物信息学, 成为系统生物学的重要组成部分。 代谢组学的出现和发展是必要的, 同时也是必须的。对于基因组学和蛋白质组学在生命科学研究中的缺点和不足, 代谢组学正好可以进行弥补。代谢组学研究的是生命个体对外源性物质(药物或毒物)的刺激、环境变化或遗传修饰所做出的所有代谢应答, 并且检测这种应答的全貌及其动态变化。代谢组学方法为生命科学的发展提供了有力的现代化实验技术手段, 同时也为新药临床前安全性评价与实践提供了新的技术支持与保障. 1 代谢组学的概念及发展 代谢组学最初是由英国帝国理工大学Jeremy N icholson教授提出的,他认为代谢组学是将人体作为一个完整的系统,机体的生理病理过程作为一个动态的系统来研究, 并且将代谢组学定义为生物体对病理生理或基因修饰等刺激产生的代谢物质动态应答的定量测定。2000年,德国马普所的Fiehn等提出了代谢组学的概念,但是与N ichols on提出的代谢组学不同, 他是将代谢组学定位为一个静态的过程,也可以称为/代谢物组学, 即对限定条件下的特定生物样品中所有代谢产物的定性定量分析。同时Fiehn还将代谢组学按照研究目的的不同分为4类: 代谢物靶标分析,代谢轮廓(谱)分析, 代谢组学,代谢指纹分析。现在代谢组学在国内外的研究都在迅速地发展, 科学家们对代谢组学这一概念也进行了完善, 作出了科学的定义: 代谢组学是对一个生物系统的细胞在给定时间和条件下所有小分子代谢物质的定性定量分析,从而定量描述生物内源性代谢物质的整体及其对内因和外因变化应答规律的科学。 与基因组学、转录组学、蛋白质组学相同, 代谢组学的主要研究思想是全局观点。与传统的代谢研究相比, 代谢组学融合了物理学、生物学及分析化学等多学科知识, 利用现代化的先进的仪器联用分析技术对机体在特定的条件下整个代谢产物谱的变化进行检测,并通过特殊的多元统计分析方法研究整体的生物学功能状况。由于代谢组学的研究对象是人体或动物体的所有代谢产物, 而这些代谢产物的产生都是由机体的内源性物质发生反应生成的,因此,代谢产物的变化也就揭示了内源性物质或是基因水平的变化,这使研究对象从微观的基因变为宏观的代谢物,宏观代谢表型的研究使得科学研究的对象范围缩小而且更加直观,易于理解, 这点也是代谢组学研究的优势之一. 代谢组学的优势主要包括:对机体损伤小,所得到的信息量大,相对于基因组学和蛋白质组学检测更加容易。由于代谢组学发展的时间较短, 并且由于代谢组学的分析对象是无偏向性的样品中所有的小分子物质,因此对分析手段的要求比较高, 在数据处理和模式识别上也不成熟,存在一些不足之处。同时生物体代谢物组变化快, 稳定性较难控制,当机体的生理和药理效应超敏时,受试物即使没有相关毒性,也可能引起明显的代谢变化,导致假阳性结果。 代谢组学应用领域大致可以分为以下7个方面:
代谢组学的数据分析技术 摘要:代谢组学是效仿基因组学和蛋白质组学的研究思想,对生物体内所有代谢物进行定量分析,并寻找代谢物与生理病理变化的相对关系的研究方式,是系统生物学的组成部分。其研究对象大都是相对分子质量1000以内的小分子物质。先进分析检测技术结合模式识别和专家系统等计算分析方法是代谢组学研究的基本方法。文章主要综述了将代谢组学中的图谱、数据信息转换为相应的参数所采用的分析方法。 关键词:代谢组学;数据分析方法 代谢组学是以代谢物分析的整体方法来研究功能蛋白如何产生能量和处理体内物质,评价细胞和体液内源性和外源性代谢物浓度及功能关系的新兴学科,是系统生物学的重要组成部分,其相应的研究能反映基因组、转录组和蛋白组受内外环境影响后相互协调作用的最终结果,更接近反映细胞或生物的表型,因此被越来越广泛地应用。而代谢组学的数据分析包括预处理和统计分析方法,多元统计分析方法主要分为两大类:非监督和监督方法,非监督方法包括主成分分析PCA;聚类分析CA等;监督方法包括显著性分析、偏最小二乘法等,本文就是主要综述代谢组学图谱信息转化为参数信息所采用的数据分析方法。 1预处理 数据的预处理过程包括以下:谱图的处理;生成原始的数据矩阵;数据的归一化以及标准化处理过程。针对实验性质、条件以及样品等因素采用不同的预处理方法。在实际应用过程中,预处理可以通过实验系统自带的软件如XCMS软件。进行,因此一般较容易获得所需的数据形式。 2数据分析方法 2.1 主成分分析PCA是多元统计中最常用的一种方法,它是在最大程度上提取原始信息的同时对数据进行降维处理的过程,其目的是将分散的信息集中到几个综合指标即主成分上,有助于简化分析和多维数据的可视化,进而通过主成分来描述机体代谢变化的情况。PCA 的具体过程是通过一种空间转换,形成新的样本集,按照贡献率的大小进行排序,贡献率最大的称为第一主成分,依次类推。经验指出,当累计贡献率大于85%时所提取的主成分就能代表原始数据的绝大多数信息,可停止提取主成分。在代谢组数据处理中,PCA是最早且广泛使用的多变量模式识别方法之一。,具有不损失样品基本信息、对原始数据进行降维处理的同时避免原始数据的共线性问题等优点,但在实际应用过程中,PCA存在着自身的缺点[1]:离群样本点的存在严重影响其生物标志物的寻找;非保守性的代谢组分扰乱正确的分类以及尺度的差异影响小浓度组分的表现等,其他的问题之前也有讨论[2]。针对PCA 的缺陷采用了不同的改进措施,与此同时,为了简化计算,侯咏佳等[3]。提出了一种主成分分析算法的FPGA实现方案,通过Givens算法和CORD IC算法的矢量旋转,用简单的移位和加法操作来实现协方差矩阵的特征分析,只需计算上三角元素,因此计算复杂度小、迭代收敛速度快。 2.2 聚类分析CA是用多元统计技术进行分类的一种方法。其主要原理是:利用同类样本应彼此相似,相类似的样本在多维空间里的彼此距离应较小,而不同类的样本在多维空间里的
代谢组学在医药领域的应用与进展 一、学习指导 1.学习代谢组学的概念及内涵,掌握代谢组学的研究对象与分析方法。 2.熟悉代谢组学数据分析技术手段 3.了解代谢组学优势特点 4.了解代谢组学在医药领域的应用 5.了解代谢组学发展趋势 二、正文 基因组功能解析是后基因组时代生命科学研究的热点之一,由于基因功能的复杂性和生物系统的完整性,必然要从“整体”层面上来理解构成生物体系的各个模块功能。随着新的测量技术、高通量的分析方法、先进的信息科学和系统科学新理论的发展,加上生物学研究的深入和生物信息的大量积累,使得在系统水平上研究由分子生物学发现的组件所构成的生命体系成为可能[1]。系统生物学家们认为,将生命科学上升为“综合”科学的时机已经成熟,生命科学再次回到整合性研究的新高度,逐步由分子生物学时代进入到系统生物学时代[2]。系统生物学不同以往的实验生物学仅关注个别基因和蛋白质,它要研究所有基因、蛋白质,代谢物等组分间的所有相互关系,通过整合各组成成分的信息,以数学方法建立模型描述系统结构[3,4]。 (一)代谢组学的概念及内涵 代谢组学是继基因组学、转录组学和蛋白质组学之后,系统生物学的重要组成部分,也是目前组学领域研究的热点之一。代谢组学术语在国际上有两个英文名,即metabolomics 和metabonomics。Metabolomics是由德国的植物学家Fiehn等通过对植物代谢物研究提出来的,认为代谢组学(metabolomics)是定性和定量分析单个细胞或单一类型细胞的代谢调控和代谢流中所有低分子量代谢产物,从而监测机体或活细胞中化学变化的一门科学[5]。英国Nicholson研究小组从毒理学角度分析大鼠尿液成份时提出了代谢组学(Metabonomics)的概念,认为代谢组学是通过考察生物体系受扰动或刺激后(如某个特定基因变异或环境变化后),其代谢产物的变化或代谢产物随时间的变化来研究生物体系的代谢途径的一种技术[6]。国内的代谢组学研究小组基本用metabonomics一词来表示“代谢组学”。严格地说,代谢组学所研究的对象应该包括生物系统中所有的代谢产物。但由于实际分析手段的局限性,只对各种代谢路径底物和产物的小分子物质(MW<1Kd)进行测定和分析。 (二)代谢组学优势特点 代谢组学作为系统生物学的一个重要组成部分,代谢组可以更好地反映体系表型生物机体是一个动态的、多因素综合调控的复杂体系,在从基因到性状的生物信息传递链中,机体需通过不断调节自身复杂的代谢网络来维持系统内部以及与外界环境的正常动态平衡[7]。
基因组学的研究内容 结构基因组学: 基因定位;基因组作图;测定核苷酸序列 功能基因组学:又称后基因组学(postgenomics基因的识别、鉴定、克隆;基因结构、功能及其相互关系;基因表达调控的研究 蛋白质组学: 鉴定蛋白质的产生过程、结构、功能和相互作用方式 遗传图谱 (genetic map)采用遗传分析的方法将基因或其它dNA序列标定在染色体上构建连锁图。 遗传标记: 有可以识别的标记,才能确定目标的方位及彼此之间的相对位置。 构建遗传图谱 就是寻找基因组不同位置上的特征标记。包括: 形态标记; 细胞学标记; 生化标记;DNA 分子标记 所有的标记都必须具有多态性!所有多态性都是基因突变的结果! 形态标记: 形态性状:株高、颜色、白化症等,又称表型标记。 数量少,很多突变是致死的,受环境、生育期等因素的影响 控制性状的其实是基因,所以形态标记实质上就是基因标记。
细胞学标记 明确显示遗传多态性的染色体结构特征和数量特征 :染色体的核型、染色体的带型、染色 体的结构变异、染色体的数目变异。优点:不受环境影响。缺点:数量少、费力、费时、对生物体的生长发育不利 生化标记 又称蛋白质标记 就是利用蛋白质的多态性作为遗传标记。 如:同工酶、贮藏蛋白 优点: 数量较多,受环境影响小 ?
缺点: 受发育时间的影响、有组织特异性、只反映基因编码区的信息 DNA 分子标记: 简称分子标记以 DNA 序列的多态性作为遗传标记 优点: ? 不受时间和环境的限制 ? 遍布整个基因组,数量无限 ?
不影响性状表达 ? 自然存在的变异丰富,多态性好 ? 共显性,能鉴别纯合体和杂合体 限制性片段长度多态性(restriction fragment length polymorphism , RFLP ) DNA 序列能或不能被某一酶酶切,
基因组测序基础知识 ㈠De Novo测序也叫从头测序,是首次对一个物种的基因组进行测序,用生物信息学的分析方法对测序所得序列进行组装,从而获得该物种的基因组序列图谱。 目前国际上通用的基因组De Novo测序方法有三种: 1. 用Illumina Solexa GA IIx 测序仪直接测序; 2. 用Roche GS FLX Titanium直接完成全基因组测序; 3. 用ABI 3730 或Roche GS FLX Titanium测序,搭建骨架,再用Illumina Solexa GA IIx 进行深度测序,完成基因组拼接。 采用De Novo测序有助于研究者了解未知物种的个体全基因组序列、鉴定新基因组中全部的结构和功能元件,并且将这些信息在基因组水平上进行集成和展示、可以预测新的功能基因及进行比较基因组学研究,为后续的相关研究奠定基础。 实验流程: 公司服务内容 1.基本服务:DNA样品检测;测序文库构建;高通量测序;数据基本分析(Base calling,去接头, 去污染);序列组装达到精细图标准 2.定制服务:基因组注释及功能注释;比较基因组及分子进化分析,数据库搭建;基因组信息展 示平台搭建 1.基因组De Novo测序对DNA样品有什么要求?
(1) 对于细菌真菌,样品来源一定要单一菌落无污染,否则会严重影响测序结果的质量。基因组完整无降解(23 kb以上), OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;每次样品制备需要10 μg样品,如果需要多次制备样品,则需要样品总量=制备样品次数*10 μg。 (2) 对于植物,样品来源要求是黑暗无菌条件下培养的黄化苗或组培样品,最好为纯合或单倍体。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (3) 对于动物,样品来源应选用肌肉,血等脂肪含量少的部位,同一个体取样,最好为纯合。基因组完整无降解(23 kb以上),OD值在1.8~2.0 之间;样品浓度大于30 ng/μl;样品总量不小于500 μg,详细要求参见项目合同附件。 (4) 基因组De Novo组装完毕后需要构建BAC或Fosmid文库进行测序验证,用于BAC 或Fosmid文库构建的样品需要保证跟De Novo测序样本同一来源。 2. De Novo有几种测序方式 目前3种测序技术 Roche 454,Solexa和ABI SOLID均有单端测序和双端测序两种方式。在基因组De Novo测序过程中,Roche 454的单端测序读长可以达到400 bp,经常用于基因组骨架的组装,而Solexa和ABI SOLID双端测序可以用于组装scaffolds和填补gap。下面以solexa 为例,对单端测序(Single-read)和双端测序(Paired-end和Mate-pair)进行介绍。Single-read、Paired-end和Mate-pair主要区别在测序文库的构建方法上。 单端测序(Single-read)首先将DNA样本进行片段化处理形成200-500bp的片段,引物序列连接到DNA片段的一端,然后末端加上接头,将片段固定在flow cell上生成DNA簇,上机测序单端读取序列(图1)。 Paired-end方法是指在构建待测DNA文库时在两端的接头上都加上测序引物结合位点,在第一轮测序完成后,去除第一轮测序的模板链,用对读测序模块(Paired-End Module)引导互补链在原位置再生和扩增,以达到第二轮测序所用的模板量,进行第二轮互补链的合成测序(图2)。 图1 Single-read文库构建方法图2 Paired-end文库构建方法
2016-02,37(1)中国烟草科学 Chinese Tobacco Science 89 代谢组学技术在烟草研究中的应用进展 王小莉,付博,赵铭钦*,贺凡,王鹏泽,刘鹏飞 (河南农业大学烟草学院,国家烟草栽培生理生化研究基地,郑州 450002) 摘要:简述了作为研究植物生理生化和基因功能新方法的代谢组学在烟草研究中的主要技术流程及其应用现状,归纳了不同生态环境和不同组织中烟草代谢物差异及产生原因,总结了生物和非生物胁迫及化学诱导处理等条件下的烟草生理生化变化及相关基因功能。最后提出了目前烟草代谢组学研究所面临的问题,并指出与其他组学整合应用是代谢组学在烟草研究领域的发展趋势。 关键词:烟草;代谢组学;胁迫;化学诱导;基因功能 中图分类号:S572.01 文章编号:1007-5119(2016)01-0089-08 DOI:10.13496/j.issn.1007-5119.2016.01.016 Research of Metabolomics in Tobacco WANG Xiaoli, FU Bo, ZHAO Mingqin*, HE Fan, WANG Pengze, LIU Pengfei (College of Tobacco Science, Henan Agricultural University, National Tobacco Physiology and Biochemistry Research Center, Zhengzhou 450002, China) Abstract: Metabolomics has been considered one of the most effective means of investigating physiological and biochemical processes and gene function of plants. Here we review the main process of metabolomics and its application status in tobacco research, the regulation mechanisms of physiological and biochemical reactions when tobacco responds to different environmental, biotic and abiotic stresses, chemically induced processes and genetic modifications. Finally, issues of critical significance to current tobacco metabolomics research are discussed and it is noted that integration with other omics is the trend of metabolomics research in tobacco. Keywords: tobacco; metabolomics; stress; chemical induction; gene function 代谢组学与基因组学、转录组学和蛋白质组学分别从不同层面研究生物体对环境或基因改变的响应,它们都是系统生物学的重要组成部分。植物代谢组学是21世纪初产生的一门新学科,主要通过研究植物的次生代谢物受环境或基因扰动前后差异来研究植物代谢网络和基因功能[1-2]。与微生物和动物相比,植物的独特性在于它拥有复杂的代谢途径,目前发现的次生代谢产物达20万种以上[3]。代谢物差异是植物对基因或环境改变的最终响应[4],因此,对代谢物进行全面解析,探索相关代谢网络和基因调控机制,是从分子层面深入认识植物生命活动规律的一个重要环节[5-7]。 烟草不仅是重要的经济作物,同时还是一种重要的模式植物,作为生物反应器在研究植物遗传、发育、防御反应和转基因等领域中具有重要意义[8-10]。烟草代谢物非常丰富,目前从烟叶中已鉴定出3000多种[11],且代谢物理化性质和含量差异较大,给烟草化学及代谢规律研究带来挑战。传统的烟草化学主要集中于研究某一类化学成分或某几种重要物质,如萜类[12]、生物碱类[13]、多酚类等[14],这很难全面地系统地阐述烟草代谢网络。随着系统生物学的发展,烟草越来越广泛地被用于基因组学、转录组学、蛋白质组学和代谢组学的研究中,例如采用系统生物学的方法找出 基金项目:中国烟草总公司浓香型特色优质烟叶开发(110201101001 TS-01);上海烟草集团责任有限公司“浓香型特色优质烟叶风格定位研究及样品检测”(szbcw201201150) 作者简介:王小莉(1983-),女,博士研究生,主要从事烟草生理生化研究。E-mail:xiaoliwang325@https://www.doczj.com/doc/f54116301.html, *通信作者,E-mail:zhaomingqin@https://www.doczj.com/doc/f54116301.html, 收稿日期:2015-09-09 修回日期:2015-11-19
代谢组学分析系统 1.工作条件: 1.1 电压:220V(±10%)单相,50Hz(±1)。 1.2 环境温度:19-22o C 1.3 相对湿度:<70% * 2.设备用途和基本组成 2.1 仪器用途:所提供仪器为高分辨率,高灵敏度、高通量的分析系统,配以 专业的数据分析处理软件构成代谢组学专用分析系统,从而快速 寻找标记物。 2.2 仪器组成 2.2.1 仪器由超效液相色谱-四极杆/二级碰撞室/飞行时间质谱组成的系统,和 专用代谢组学分析软件以及代谢物分析软件构成,具有先进的中医药代 谢组学研究分析功能。 * 2.2.2 质谱主机要求配置同一厂家生产的液相色谱仪,具有良好的兼容性。 * 2.2.3 具备准确质量测定功能 准确质量测定的内标必须有独立于实测样品的通道进入离子源,内标不得 干扰实际样品的数据结果,并且质量准度<2ppm。 2.2.4 真空系统 要求完全被保护的多级真空系统,具有自动断电保护功能,采用分子涡轮 泵。离子源和质谱间有隔断阀。便于源清洗和日常维护。 * 2.2.5 碰撞室具有两级碰撞功能。分为以下部分: 捕获富集单元:具有离子传输富集、碰撞室两种功能 传输单元:具有离子传输、碰撞室两种功能 * 2.2.6 检测器 检测器由单个微通道板离子计数检测,可检测正负离子和采集MS和 MS/MS的数据, TDC转换速率>4.0 GHz。 * 2.2.7 数据采集和处理系统 工作站用于仪器控制和采集, 1024MB RAM, 200GB硬盘,DVD-ROM,
刻录光盘驱动器,1.44MB 3.5英寸软驱。 软件基于Windows XP 操作系统的应用软件包括集成化的仪器控制、数据处理等软件,代谢组学分析软件以及代谢物分析软件等。 3 仪器的详细技术指标 3.1 液相色谱仪 * 液相色谱仪必须是能够耐超高压(1000bar)的超高效液相色谱仪(UPLC)。3.1.1 可编程二元梯度泵。 溶剂数量:4 流速范围:0.010 - 2mL/min,步进0.001mL/min, 流速精度:< 0.075% RSD,流速准确度:±1%, 泵耐压:0 - 15000psi(1000bar) 梯度设定范围:0 - 100% *系统延迟体积:< 120uL 3.1.2 二极管阵列检测器 波长范围:190-700nm. *测量范围:0.0001~4.0000AUFS *采样速率:40点/秒 流通池:500nl低扩散 3.1.3 自动进样器系统 样品数量:96孔板、384孔板、24x4ml瓶、48x2ml瓶 进样范围:0.1- 50 μL, “针内针”样品探针。 温度范围:4-40摄氏度 3.1.4 在线脱气系统 真空脱气:六通道在线脱气机 3.1.5 柱加热系统 控温范围:室温+5---65摄氏度 3.1.6 专用色谱柱; * 1.7μ, 2.1 mm x 50 mm Column
代谢组学研究中数据处理新方法的应用 李 晶1 ,吴晓健1 ,刘昌孝 1,23 ,元英进 1 (1.天津大学化工学院制药工程系,天津300072; 2.天津药物研究院药物动力学与药效动力学省部共建国家重点实验室,天津300193) 摘要:目的 探索代谢组学研究中数据处理的新方法。方法 本文提出了在代谢组学数据预处理中,用稳健 PCA 的方法进行离群样品点的诊断,用变量的类内差异和类间差异的比较来判断非保守性代谢组分,用尺度同一化 的方法进行数据预处理来消除数据的尺度差异。并以A rabidopsis thaliana 属的四个基因型的植株代谢组学的数据为例,用以上的方法进行数据预处理后再用PC A 的方法分析。结果与结论 研究表明这三种数据预处理方法的应用会明显的改善代谢组学生物信息学分析中聚类分析的结果和生物标志物识别的准确性及全面性。 关键词:代谢组学;离群样本点诊断;非保守性代谢组分;数据尺度同一化;主成分分析法中图分类号:R969.1 文献标识码:A 文章编号:0513-4870(2006)01-0047-07 收稿日期:2005203220. 基金项目:科技部国家重点基础研究发展计划(973计划)资助 项目(2004CB518902);国家高技术研究发展计划(863计划)资助项目(2003AA2Z347D ). 3 通讯作者 Tel:86-22-23006863,Fax:86-22-23006860, E 2mail:liuchangxiao@https://www.doczj.com/doc/f54116301.html, Appli cati on of new method for dat a processi n g i n met abono m i c studi es L I J ing 1 ,WU Xiao 2jian 1 ,L I U Chang 2xiao 1,23 ,Y UAN Ying 2jin 1 (1.D epart m ent of Phar m aceutical Engineering,Institute of Che m ical Engineering,Tianjin U niversity,Tianjin 300072,China;2.S tate Key L aboratory of Phar m acokinetics and Phar m acodynam ics,T ianjin Institute of Phar m aceutical R esearch,Tianjin 300193,China ) Abstract:A i m T o search f or and app licati on of ne w method for data p r ocessing in metabonom ic studies .M ethods The paper p r oposed that in the p r ocessing of metabonom ic data,r obust PCA method can be used t o diagnose outliers;and unstable variables judged by comparis on bet w een difference within class and difference a mong classes should be excluded bef ore data analysis;moreover,the data should be p r operly scaled before further p r ocessing .The p r oposed methods were used t o p rep r ocess metabol om ic data of four genoty pes of the A rabidopsis tha liana p lants .Results and Conclusi on The outcome de monstrated that the app licati on of these methods can obvi ously i m p r ove clustering and bi omarker identifying results . Key words:metabol om ics;outlier diagnosis;unstable metabolite;data p re 2scaling;p rinci p le component analysis 代谢物组学是以代谢物分析的整体方法来研究功能蛋白如何产生能量和处理体内物质,其代谢物则以生化活性直接体现作用的结果,也就是说代谢物组学是评价细胞和体液的内源性和外源代谢物浓度与功能关系的学科[1~4] 。代谢物组学的出现,特别在药物安全性研究中的应用,认为该新兴的学科分支会对药物安全性研究产生革命性的影响。它与 药物的药效和毒性筛选和评价研究、作用机制研究和合理治疗用药密切相关。代谢物组是反应机体状况的分子集合,所有对机体健康影响的因素均可反映在代谢物组中,基因、环境、营养、药物(外源物)和时间(年龄)最终通过代谢物组对表达施加影响。代谢物组是评价健康和治疗的合适的分子集合。因 此研究代谢物组学对药物治疗有直接意义[4~9] 。 代谢组学是定量分析生物系统对机体反应或基因改变所产生的动态的、多参数应答的一项新发展 的技术[10] 。它可有效地应用于生物系统的机制研究及生物系统的生产优化研究中,代谢组学与代谢工程方法的联合在生物工程中的应用已显示出巨大 的潜力[11,12] 。代谢组学通常以核磁共振光谱 ? 74?药学学报Acta Phar maceutica Sinica 2006,41(1):47-53
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
代谢组学综述 摘要:代谢组学是20世纪90年代中期发展起来的对某一生物或细胞所有低相对分子质量代谢产物进行定性和定量分析的一门新学科,由于其广泛的应用前景,目前已成为系统生物学的重要组成部分。现简要介绍了代谢组学的含义、代谢组学研究的历史沿革、当前代谢组学研究中的分析技术、数据解析方法,综述了代谢组学在药物毒理学研究、疾病诊断、植物和中药等领域的应用情况,并对当前代谢组学研究中存在的问题及发展趋势进行探讨。 关键词:代谢组学研究技术 随着人类基因组计划等重大科学项目的实施,基因组学、转录组学及蛋白质组学在研究人类生命科学的过程中发挥了重要的作用, 与此同时, 代谢组学(metabolomics)在20世纪90年代中期产生并迅速地发展起来, 与基因组学、转录组学、蛋白质组学共同组成系统生物学。基因组学、转录组学、蛋白质组学和代谢组学等各种组学0在生命科学领域中发挥了重要的作用, 它们分别从调控生命过程的不同层面进行研究, 使人们能够从分子水平研究生命现象, 探讨生命的本质, 逐步系统地认识生命发展的规律。这些组学手段加上生物信息学, 成为系统生物学的重要组成部分。 代谢组学的出现和发展是必要的, 同时也是必须的。对于基因组学和蛋白质组学在生命科学研究中的缺点和不足, 代谢组学正好可以进行弥补。代谢组学研究的是生命个体对外源性物质(药物或毒物)的刺激、环境变化或遗传修饰所做出的所有代谢应答, 并且检测这种应答的全貌及其动态变化。代谢组学方法为生命科学的发展提供了有力的现代化实验技术手段, 同时也为新药临床前安全性评价与实践提供了新的技术支持与保障。 1 代谢组学的概念及发展 代谢组学最初是由英国帝国理工大学Jeremy N icholson教授提出的, 他认为代谢组学是将人体作为一个完整的系统, 机体的生理病理过程作为一个动态的系统来研究, 并且将代谢组学定义为生物体对病理生理或基因修饰等刺激产生的代谢物质动态应答的定量测定。2000年, 德国马普所的Fiehn等提出了代谢组学的概念, 但是与N icholson提出的代谢组学不同, 他是将代谢组学定位为一个静态的过程, 也可以称为/代谢物组学, 即对限定条件下的特定生物样品中所有代
代谢组学是一门对某一生物或细胞所有低分子质量代谢产物(以相对分子质量<1000的有机和无机的代谢物为研究核心区)进行分析的新兴学科。生物样本通过NMR、GC-MS、LC-MS等高通量仪器分析检测后,能产生大量的数据,这些数据具有高维,少样本、高噪声等复杂特征,同时代谢物多且代谢物之间联系密切,因此从复杂的代谢组学数据中确定与所研究的现象有关的代谢物,筛选出候选生物标记物成为代谢物组学研究的热点和难点。 代谢组学分析数据用于统计分析时,数据集通常为一个N ×K 的矩阵(X矩阵),N表示N个样本数,每一行代表一个样品,K表示K个变量,每一列代表一个变量,在代谢组学中变量通常是指代谢物含量。常用的分析方法如图1所示: 数据分析方法 单变量分析 多变量分析差异倍数分析 显著性检验 无监督分析 有监督分析 PLS-DA PCA OPLS-DA 图1 代谢组学常用的数据分析方法 单变量分析 单变量分析方法仅分别分析单个变量,不考虑多个变量的相互作用与内在联系。具有简单性、易应用性和可解释性。但是无法基于整
体数据对所测样品的优劣、差异进行综合评价和分析。 (1)差异倍数分析 差异倍数变化大小(Fold Change,FC)表示实验组与对照组的含量比值,可以快速考察各个代谢物在不同组别之间的含量变化大小。(2)显著性检验 p值即概率,反映某一事件发生的可能性大小,用于区分该变量是否具有统计显著性,通常认为p<0.05具有统计显著性。常用的检验方法有t-test、方差分析(Analysis of Variance,ANOVA),但是由于代谢组学的变量较多,必要时需要进行多重假设检验,对p值进行校正,减少Ⅰ类错误,降低假阳性。 多变量分析 多变量分析方法能同时处理数百或数千个变量,并且能处理变量之间的相互关系。利用变量之间的协方差或相关性,使原始数据在较低维空间上的投影能尽可能地捕获数据中的信息。但是如果存在大量无信息变量可能会妨碍多变量分析的能力,无信息变量的数量越多,减少真阳性数量的效果就越显著。 多变量分析分为无监督分析方法和有监督分析方法。在代谢组学分析中无监督学习有主成分分析(Principal Component Analysis,PCA),只需要数据集X,而有监督分析方法主要是偏小二乘判别分析(Partial Least Squares Discrimination Analysis, PLS-DA)和正交偏小二乘判别分析(Orthogonal Partial Least Squares
植物代谢组学的研究方法及其应用 近年来,随着生命科学研究的发展,尤其是在完成拟南芥(Arabidopsis thaliana) 和水稻(Oryza sativa) 等植物的基因组测序后,植物生物学发生了翻天覆地的变化。人们已经把目光从基因的测序转移到了基因的功能研究。在研究DNA 的基因组学、mRNA 的转录组学及蛋白质的蛋白组学后,接踵而来的是研究代谢物的代谢组学(Hall et al.,2002)。代谢组学的概念来源于代谢组,代谢组是指某一生物或细胞在一特定生理时期内所有的低分子量代谢产物,代谢组学则是对某一生物或细胞在一特定生理时期内所有低分子量代谢产物同时进行定性和定量分析的一门新学科(Goodacre,2004)。它是以组群指标分析为基础,以高通量检测和数据处理为手段,以信息建模与系统整合为目标的系统生物学的一个分支。 代谢物是细胞调控过程的终产物,它们的种类和数量变化被视为生物系统对基因或环境变化的最终响应(Fiehn,2002)。植物内源代谢物对植物的生长发育有重要作用(Pichersky and Gang,2000)。植物中代谢物超过20万种,有维持植物生命活动和生长发育所必需的初生代谢物;还有利用初生代谢物生成的与植物抗病和抗逆关系密切的次生代谢物,所以对植物代谢物进行分析是十分必要的。 但是,由于植物代谢物在时间和空间都具有高度的动态性(stitt and Fernie,2003)。尤其是次生代谢物种类繁多、结构迥异,且产生和分布通常有种属、器官、组织以及生长发育时期的特异性,难于进行分离分析,所以人们一直在寻找更为强大的检测分析工具。在代谢物分析领域,人们已经提出了目标分析、代谢产物指纹分析、代谢产物轮廓分析和代谢表型分析、代谢组学分析等概念。20世纪90年代初,Sauter 等(1991)首先将代谢组分析引入植物系统诊断,此后关于植物代谢组学的研究逐年增多。随着拟南芥等植物的基因组测序完成以及代谢物分析手段的改进和提高,今后几年进入此研究领域的科学家和研究机构将越来越多。 1研究方法 代谢组学分析流程包括样品制备、代谢物成分分析鉴定和数据分析与解释。由于植物中代谢物的种类繁多,而目前可用的成分检测和数据分析方法又多种多样,所以根据研究对象不同,采用的样品制备、分离鉴定手段及数据分析方法各不相同。 1.1样品制备 植物代谢物样品制备分为组织取样、匀浆、抽提、保存和样品预处理等步骤(Weckwerth and Fiehn,2002)。代谢产物通常用水或有机溶剂(如甲醇和己烷等)分别提取,获得水提取物和有机溶剂提取物,从而把非极性的亲脂相和极性相分开。分析之前,通常先用固相微萃取、固相萃取和亲和色谱等方法进行预处理(邱德有和黄璐琦,2004)。然而植物代谢物千差万别,其中很多物质稍受干扰结构就会发生改变,且对其分析鉴定所采用的设备也不同。目前还没有适合所有代谢物的抽提方法,通常只能根据所要分析的代谢物特性及使用的鉴定手段选择合适的提取方法。而抽提时间、温度、溶剂成分和质量及实验者的技巧等诸多因素也将影响样品制备的水平。 1.2成分分析鉴定
◇专论◇ 中国临床药理学与治疗学 中国药理学会主办 CN 3421206/R ,ISSN 100922501 E 2mail :ccpt96@https://www.doczj.com/doc/f54116301.html, 2010May ;15(5):481-489 2010203211收稿 2010204224修回E 2mail :ajiye333@hot https://www.doczj.com/doc/f54116301.html, 代谢组学数据处理方法———主成分分析 阿基业 中国药科大学药代动力学重点实验室&代谢组学研究室,南京210009,江苏 摘要 代谢组学在生命科学领域得到了越来越 广泛的应用并展现出良好的前景。代谢组学分析产生的含有大量变量的数据难以用常规方法进行分析,如何正确分析和解释代谢组学的数据是研究的关键。本文主要介绍了在代谢组学数据分析中占主导地位的主成分分析基本方法,旨在加强代谢组学数据分析的基础知识并规范数据分析的方法。关键词 代谢组学;主成分分析;偏最小二乘投影关联分析;偏最小二乘投影判别分析;正交偏最小二乘投影分析 中图分类号:R969.1文献标识码:A 文章编号:100922501(2010)0520481209代谢组学(metabolomics 或metabonomics )是“后基因组学”时期新兴的一门学科,“代谢组学”一词虽然常以“metabonomics ”或“metabolo 2mics ”出现,但多数学者通常并不对其加以严格区分,绝大多数情况下这两个词被认为包含了等同的意义。根据研究的内容、目的、偏重点和对代谢 组学的认识不同,代谢组学被赋予了不同的英文 名称,如代谢物分析(metabolite p rofiling )、代谢谱分析(metabolic profiling )、代谢指纹谱分析(metabolic fingerp rinting )等;根据分析的目标化合物不同,还可以分为目标化合物分析(targeting analysis )和(无设定目标的)全谱分析(non -tar 2geting analysis )。无论采用了哪种名称和说法,代谢组学的研究对象都是体内“代谢组” (metabo 2lome ,即生物样本、系统、组织或细胞中小分子化合物的总称)。不管采用哪种测定方法,应用代谢 组学技术都可以测定到许多内源性化合物的定性/定量信息。这些信息在输出的谱图上表现为许多信号峰,在色谱质谱图上表现为不同保留时间出现色谱峰,在核磁共振谱图上表现为不同化学位移处的色谱信号。每个信号峰都既包含了检测分子的定性/结构信息,也包含了定量信息。以气相色谱-质谱检测为例,样品中的分子经过数十米长的石英毛细管色谱柱色谱分离后在色谱图上的不同时间出现,输出的总离子流图上的每个色谱峰都包含该物质定性信息(质谱图)和定量信息(峰高或峰面积),图1。对于特定化合物来说,其定性信息就是该化合物的特征谱图和色谱保留时间,是化合物鉴定的基础;其定量信息就是该化合物的色谱响应强度,如峰高、峰面积,是定量比较不同样品或组别之间差异的基础 。 中国药科大学药物代谢动力学重点实验室&代谢组学研究室副研究员, 主要从事药物代谢动力学、代谢组学、代谢性疾病发病机制、与代谢相关的药效或药物毒性的分子机制等方向的研究。 ? 184?
代谢组学小常识 概念: ?代谢组:指一个细胞、组织或器官中所有代谢物的集合, 包含一系列不同类型的小分子(通常分子量<1000), 比如肽、碳水化合物、脂类、核酸等。 ?代谢组学:通过考察生物体系(细胞、组织或生物体)受刺激或扰动后,其代谢产物的变化或其随时间的变化,来研究生物体系的一门科学。 实验流程:(以液质联用为基础的代谢组学为例) ?样本前处理:在保证小分子代谢物完整的前提下,处理的步骤越简单越好,以保证操作容易重复,也为大批量样本的处理节约时间。 ?数据采集:依据实验目的有所不同。 o非目标代谢组学:选用高分辨质谱仪(TOF,Orbitrap等),有助于检测到尽可能多的化合物,另外高分辨的质核比数据也有助于数据库检索以及化合物的鉴定。 o目标代谢组学:通常使用三重四极其杆质谱,提高检测的灵敏度以及定量的准确性。 ?数据预处理:峰提取,排列,归一化。 o多数质谱商家都提供了配套的预处理软件,例如安捷伦公司的MassHunter,热电的Sieve,沃特世的MarkerLynx以及Progenisis QI。 o同时也有一些基于网络的可以免费获取的软件。建议使用配套的软件,因为不需要额外的数据转换,不需要上传数据,节省时间。 ?数据分析:多元统计分析包括主成份分析(PCA),偏最小二乘判别分析(PLS-DA),正交偏最小二乘判别分析(OPLS-DA),聚类分析(HCA)等。各个厂商也提供了相应的统计分析软件,比如安捷伦的MPP,热电的Sieve,沃特世的Ezinfor。目前常用的第三方软件是Simca-p,同时也有一些网络的开源软件可以使用。 ?化合物鉴定:数据库检索,标准品对比,二级质谱对比。 代谢组学文章中常见的统计图(一) 主成分分析(PCA) PCA得分图(score plot),用来看样本天然的分组情况,在分析时不加任何分组信息。图中每一个点代表一个样本,样本在空间中所处的位置由其中所含有的代谢物的差异决定。 PCA载荷图(loading plot),用来寻找差异变量。同种的每一个点代表样本中还有的一个代谢物物,距离原点越远的代谢物被认为对样本的分类贡献越大。 偏最小二乘判别分析(PLS-DA) 得分图和载荷图的解释同PCA。区别在于,PLS-DA在分析时提前赋予每个样本分组信息,简单说,就是在分