
中国化药注册分类(旧分类、新分类)Classification of Registration of Chemical Drugs in China (The Old Classification and the New Classification) 旧的注册分类是来源于2007年10月1日实施的《药品注册管理办法》(局令第28号)的附件2《化学药品注册分类及申报资料要求》(此注册分类实际在28号令之前就实施的,早期历史文件不再追溯)。
The old classification of registration dated from the Annex 2 “Registered Classification of Chemicals and Requirements of Application”of “Administrative Provision of Drug Registration” (Board Order No. 28), which implemented on October 1st, 2007 (Actually, this category had been implemented earlier than the date of the implementation of the 28 orders, and the early history file is no longer traceable).
旧的药品注册分类如下:/ The old classification of registration of drug is as follows:
进口化学药品申报,申请未在国内外获准上市销售的药品,按照注册分类1的规定报送资料;其他品种按照注册分类3的规定报送资料。也可以报送ICH 规定的CTD资料,但“综述资料”部分应按照化学药品《申报资料项目》要求报送。属于注册分类1的药物,应当至少是已在国外进入II期临床试验的药物。
For the application of import chemical drugs, apply the drugs which are not approved for marketing in domestic, submit the documentation in according to the provisions of Classified 1; others drugs should apply in according to the provisions of Classified 3. Also submit the CTD documentation in according to ICH is permitted, but, the part of “Documentation of Review” should be submitted in according to the requirements of “Projects of Application documentation”. The drugs of Classified 1 should be at least has entered Phase II clinical trials of the drug in a foreign country.
2016年3月4日国家食品药品监督管理总局(CFDA)正式发布《关于发布化学药品注册分类改革工作方案的公告(2016年第51号)》,并自公告发布之日起实施。
On March 4th, 2016, the State Food and Drug Administration (CFDA) was officially issued “Notice of reforming about registered classification of chemical drugs” (No. 51 of 2016)," and implemented from the issued date.
新的药品注册分类如下:/The new classification of registration of drug is as follows:
药品注册分类 The pony was revised in January 2021
化学药注册分类大变动,CFDA发布《化学药品注册分类改革工作方案》和《化学仿制药生物等效性试验备案管理规定》征求意见稿 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,昨日(11月6日),国家食药监总局官网发布了《化学药品注册分类改革工作方案(征求意见稿)》和《化学仿制药生物等效性试验备案管理规定(征求意见稿)》,面向社会公开征求意见。 《化学药品注册分类改革工作方案(征求意见稿)》 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,制定本工作方案。 一、工作原则 按照分类科学、标准严格、质量提高的原则,在原有化学药品注册分类的基础上,结合国务院改革意见中有关药品分类的调整原则,对原有化学药品注册分类进行调整和完善。首先,根据药品的安全风险程度,将药品分为新药和仿制药两大类;其次,根据药品原创性和新颖性的不同,将新药进一步分为创新药和改良型新药;第三,在仿制药中,根据被仿制药上市情况不同,进一步细分为对境外上市、境内未上市药品的仿制,对境内上市药品的仿制以及境外上市药品申请境内上市三类。 二、化学药品新注册分类及说明 新药是指未在中国境内外上市销售的药品,将境外上市境内未上市药品纳入仿制药。调整后,化学药品新注册分类共分为1-5类(表1),具体如下: (一)根据物质基础的原创性和新颖性不同,将新药分为创新药(注册分类1)和改良型新药(注册分类2)两类。其中,创新药是指含有新的结构明确的具有生理或药理作用的分子或离子,且具有临床价值的原料药及其制剂,包括用拆分或者合成等方法制得的已知活性成份的光学异构体及其制剂,但不包括对已知活性成份成酯、成盐(包括含有氢键或
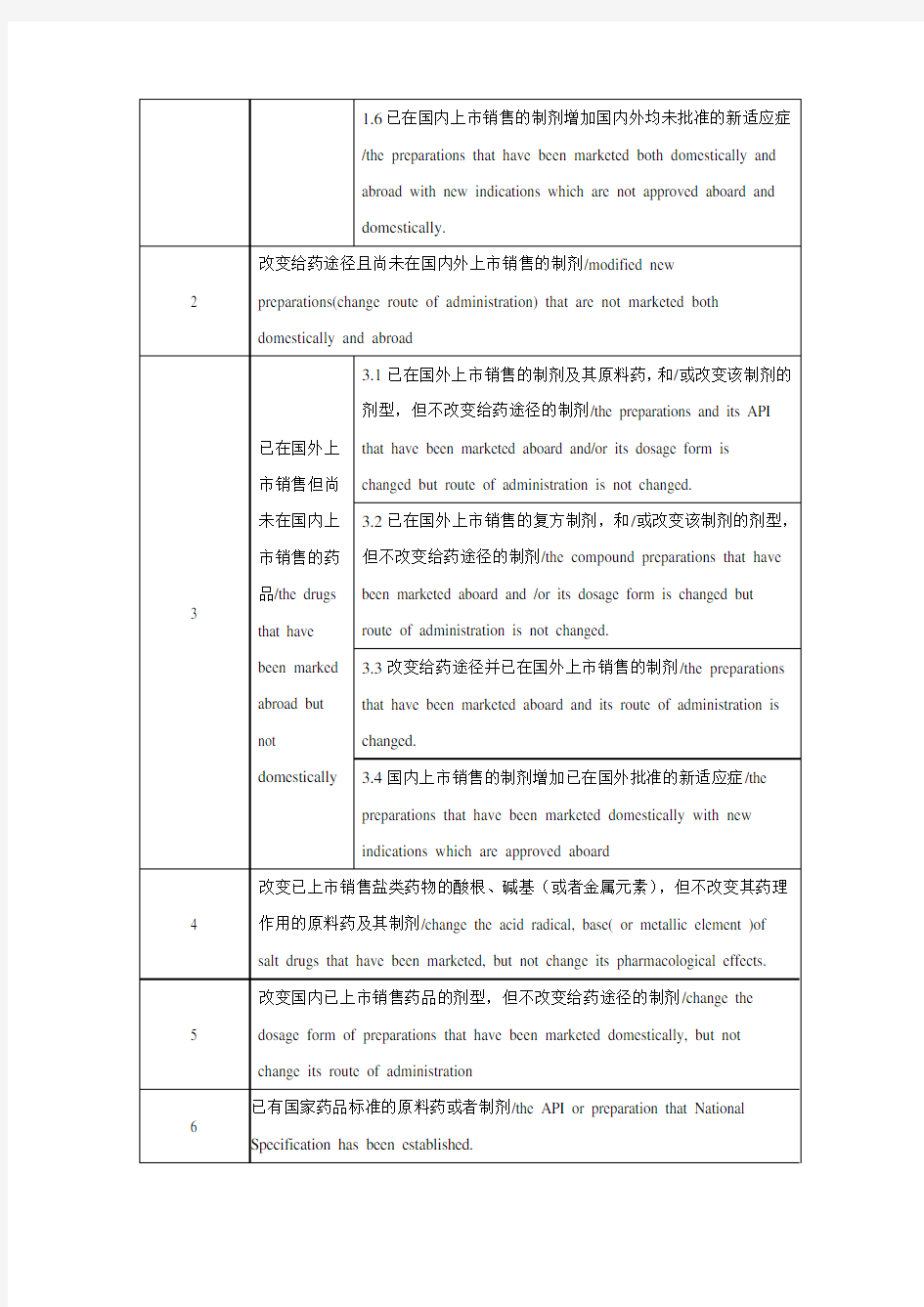
附件2:化学药品注册分类及申报资料要求 一、注册分类 1. 未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2. 改变给药途径且尚未在国内外上市销售的制剂。 3. 已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4. 改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原 料药及其制剂。 5. 改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6. 已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1. 药品名称。 2. 证明性文件。 3. 立题目的与依据。 4. 对主要研究结果的总结及评价。 5. 药品说明书、起草说明及相关参考文献。 6. 包装、标签设计样稿。 (二)药学研究资料 7. 药学研究资料综述。 8. 原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9. 确证化学结构或者组份的试验资料及文献资料。 10. 质量研究工作的试验资料及文献资料。 11. 药品标准及起草说明,并提供标准品或者对照品。 12. 样品的检验报告书。 13. 原料药、辅料的来源及质量标准、检验报告书。 14. 药物稳定性研究的试验资料及文献资料。 15. 直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16. 药理毒理研究资料综述。 17. 主要药效学试验资料及文献资料。
化学药注册分类大变动,CFDA发布《化学药品注册分类改革工作方案》和《化学仿制药生物等效性试验备案管理规定》征求意见稿为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,昨日(11月6日),国家食药监总局官网发布了《化学药品注册分类改革工作方案(征求意见稿)》和《化学仿制药生物等效性试验备案管理规定(征求意见稿)》,面向社会公开征求意见。 《化学药品注册分类改革工作方案(征求意见稿)》 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,制定本工作方案。 一、工作原则 按照分类科学、标准严格、质量提高的原则,在原有化学药品注册分类的基础上,结合国务院改革意见中有关药品分类的调整原则,对原有化学药品注册分类进行调整和完善。首先,根据药品的安全风险程度,将药品分为新药和仿制药两大类;其次,根据药品原创性和新颖性的不同,将新药进一步分为创新药和改良型新药;第三,在仿制药中,根据被仿制药上市情况不同,进一步细分为对境外上市、境内未上市药品的仿制,对境内上市药品的仿制以及境外上市药品申请境内上市三类。 二、化学药品新注册分类及说明
新药是指未在中国境内外上市销售的药品,将境外上市境内未上市药品纳入仿制药。调整后,化学药品新注册分类共分为1-5类(表1),具体如下: (一)根据物质基础的原创性和新颖性不同,将新药分为创新药(注册分类1)和改良型新药(注册分类2)两类。其中,创新药是指含有新的结构明确的具有生理或药理作用的分子或离子,且具有临床价值的原料药及其制剂,包括用拆分或者合成等方法制得的已知活性成份的光学异构体及其制剂,但不包括对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 改良型新药是在已知活性成份的基础上,对其结构、剂型、给药途径、适应症、用法用量、规格等进行优化,且具有明显临床优势的药品。结构优化是指对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 (二)被仿制的参比制剂来源不同,其上市情况存在差异,研制者和监管部门对其上市基础的认识也随之不同,为便于申报,将仿制药分为3-5类。其中,注册分类3是指仿境外上市、境内未上市药品;注册分类4是指仿制境内上市药品;注册分类5是指境外上市的药品申请在境内上市。 仿制药的基本要求是与参比制剂质量和疗效一致,参比制剂须为原研或国际公认的药品。原研药品指境外或境内首先批准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。国际公认的药品是指与原研药品质量和疗效一致的药品。 表1化学药品新注册分类、说明及包含的情形
许可事项化学药品注册分类 一、化学药品注册分类 1、未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物;(5)新的复方制剂。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的原料药及其制剂; (2)已在国外上市销售的复方制剂; (3)改变给药途径并已在国外上市销售的制剂。 4、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5、改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6、已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书样稿、起草说明及最新参考文献。 6、包装、标签设计样稿。
(二)药学研究资料 7、药学研究资料综述。 8、原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。 11、药品标准草案及起草说明,并提供标准品或者对照品。 12、样品的检验报告书。 13、辅料的来源及质量标准。 14、药物稳定性研究的试验资料及文献资料。 15、直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16、药理毒理研究资料综述。 17、主要药效学试验资料及文献资料。 18、一般药理研究的试验资料及文献资料。 19、急性毒性试验资料及文献资料。 20、长期毒性试验资料及文献资料。 21、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等主要与局部、全身给药相关的特殊安全性试验研究和文献资料。 22、复方制剂中多种成份药效、毒性、药代动力学相互影响的试验资料及文献资料。 23、致突变试验资料及文献资料。 24、生殖毒性试验资料及文献资料。 25、致癌试验资料及文献资料。
药品注册分类 一、中药、天然药物分类 (一)注册分类 1、未在国内上市销售的从中药、天然药物中提取的有效成份及其制剂。 2、未在国内上市销售的来源于植物、动物、矿物等药用物质制成的制剂。 3、中药材的代用品。 4、未在国内上市销售的中药材新的药用部位制成的制剂。 5、未在国内上市销售的从中药、天然药物中提取的有效部位制成的制剂。 6、未在国内上市销售的由中药、天然药物制成的复方制剂。 7、未在国内上市销售的由中药、天然药物制成的注射剂。 8、改变国内已上市销售药品给药途径的制剂。 9、改变国内已上市销售药品剂型的制剂。 10、改变国内已上市销售药品工艺的制剂。 11、已有国家标准的中成药和天然药物制剂。 (二)说明 1、“未在国内上市销售的中药、天然药物中提取的有效成份及其制剂”是指国家药品标准中未收载的从中药、天然药物中得到的未经过化学修饰的单一成份及其制剂。 2、“未在国内上市销售的来源于植物、动物、矿物等药用物质制成的制剂”是指未被国家药品标准或省、自治区、直辖市地方药材规范(以下简称“法定标准”)收载的中药材及天然药物制成的制剂。 3、“中药材的代用品”是指用来代替中药材某些功能的药用物质,包括: (1)已被法定标准收载的中药材; (2)未被法定标准收载的药用物质。 4、“未在国内上市销售的中药材新的药用部位制成的制剂”是指具有法定标准的中药材原动、植物新的药用部位制成的制剂。 5、“未在国内上市销售的中药、天然药物中提取的有效部位制成的制剂”是指从中药、天然药物中提取的一类或数类成份制成的制剂。
6、“未在国内上市销售的中药、天然药物制成的复方制剂”包括: (1)传统中药复方制剂; (2)现代中药复方制剂; (3)天然药物复方制剂。 7、“未在国内上市销售的中药、天然药物制成的注射剂”,其中包括水针、粉针、大输液之间的相互改变及其他剂型改成的注射剂。 8、“改变国内已上市销售药品给药途径的制剂”包括: (1)不同给药途径之间相互改变的制剂; (2)局部给药改为全身给药的制剂。 9、“改变国内已上市销售药品剂型的制剂”是指在给药途径不变的情况下改变剂型的制剂。 10、“改变国内已上市销售药品工艺的制剂”包括: (1)工艺有质的改变的制剂; (2)工艺无质的改变的制剂。 工艺有质的改变主要是指在生产过程中改变提取溶媒、纯化工艺或其他制备工艺条件等,使提取物的成份发生较大变化。 11、“已有国家标准的中成药或天然药物制剂”是指我国已批准上市销售的中药或天然药物制剂的注册申请。 二、化学药品注册分类 1、未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物;(5)新的复方制剂。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的原料药及其制剂; (2)已在国外上市销售的复方制剂;
一、化学药品注册分类 1、未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5、改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6、已有国家药品标准的原料药或者制剂。 二、中药、天然药物注册分类 中药是指在我国传统医药理论指导下使用的药用物质及其制剂。 天然药物是指在现代医药理论指导下使用的天然药用物质及其制剂。 1、未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。 2、新发现的药材及其制剂。 3、新的中药材代用品。 4、药材新的药用部位及其制剂。 5、未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。 6、未在国内上市销售的中药、天然药物复方制剂。 7、改变国内已上市销售中药、天然药物给药途径的制剂。 8、改变国内已上市销售中药、天然药物剂型的制剂。 9、仿制药。 三、治疗用生物制品注册分类 1、未在国内外上市销售的生物制品。 2、单克隆抗体。 3、基因治疗、体细胞治疗及其制品。 4、变态反应原制品。 5、由人的、动物的组织或者体液提取的,或者通过发酵制备的具有生物活性的多组份制品。 6、由已上市销售生物制品组成新的复方制品。 7、已在国外上市销售但尚未在国内上市销售的生物制品。 8、含未经批准菌种制备的微生态制品。 9、与已上市销售制品结构不完全相同且国内外均未上市销售的制品(包括氨基酸位点突变、缺失,因表达系统不同而产生、消除或者改变翻译后修饰,对产物进行化学修饰等)。 10、与已上市销售制品制备方法不同的制品(例如采用不同表达体系、宿主细胞等)。
化学药品注册分类改革工作方案解读 2016年03月16日发布食品药品监管总局组织制定了化学药品注册分类改革工作方案(以下简称方案),已于2016年3月4日正式发布实施。现就有关问题解读如下: 一、方案的出台背景。 2015年8月9日,国务院发布《关于改革药品医疗器械审评审批制度的意见》,明确调整药品注册分类。2015年11月4日,第十二届全国人民代表大会常务委员会第十七次会议审议通过《关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定》,同意国务院组织开展药品注册分类改革。为落实上述政策要求,满足实际工作需要,食品药品监管总局制定发布了本方案。 二、方案的适用范围。 本方案仅针对化学药品注册分类进行了调整,适用于方案发布实施后新受理的化学药品注册申请(包括临床、生产、进口注册申请)。 三、新注册分类中,关于新药的含义是什么? 新药指中国境内外均未上市的药品,分为创新药和改良型新药。 新注册分类1为创新药,强调含有新的结构明确的、具有药理作用的化合物;新注册分类2为改良型新药,在已知活性成份基础上进行优化,强调具有明显的临床优势。
四、创新药具体包括什么? 创新药指含有新的结构明确、具有药理作用的化合物,且具有临床价值的药品,不包括改良型新药中2.1类的药品。 五、新注册分类中,关于仿制药的含义是什么? 仿制药是指仿制已上市原研药品的药品,分为两类,一是仿制境外已上市境内未上市原研药品,二是仿制境内已上市原研药品。仿制药要求与原研药品质量和疗效一致。 如果已上市药品的原研药品无法追溯或者原研药品已经撤市的,建议不再申请仿制;如坚持提出仿制药申请,原则上不能以仿制药的技术要求予以批准,应按照新药的要求开展相关研究。 六、新注册分类中,关于第5类药品的含义是什么? 第5类药品是指境外上市的药品申请在中国境内上市,分为原研药品和非原研药品两类。对应原化学药品注册分类中的进口药品类别。 七、含有未知活性成份的新复方制剂按什么申报? 含有新的结构明确的、具有药理作用的化合物的新复方制剂,应按照新注册分类1进行申报。 八、仿制药是否要求处方工艺、规格、用法用量等均与原研保持一致? 仿制药要求与原研药品具有相同的活性成分、剂型、规格、适应症、给药途径和用法用
附件2: 化学药品注册分类及申报资料要求 一、注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1.药品名称。 2.证明性文件。 3.立题目的与依据。 4.对主要研究结果的总结及评价。 5.药品说明书、起草说明及相关参考文献。 6.包装、标签设计样稿。 (二)药学研究资料 7.药学研究资料综述。 8.原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9.确证化学结构或者组份的试验资料及文献资料。 10.质量研究工作的试验资料及文献资料。 11.药品标准及起草说明,并提供标准品或者对照品。 12.样品的检验报告书。 13.原料药、辅料的来源及质量标准、检验报告书。 14.药物稳定性研究的试验资料及文献资料。 15.直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16.药理毒理研究资料综述。
目录 1. 化学药品注册分类 2. 境内申请人新药申报流程 3. 化学药品申报资料要求 4. 化学药品临床试验要求
化学药品注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 以上,属注册分类1~5类按新药申报程序申请注册,6类按仿制药申请程序申请注册。
境内申请人,新药申报流程(以上注册分类中1~5类申报流程) 准备报临床申报资料(具体资料项目要求附后) 向省食品药品监督管理局报送申请资料 拿到受理号,相关进度, 便可以从SFDA网站上 查询https://www.doczj.com/doc/e618483598.html, 省局,受理,5工作日内组织对药物研制情况及原始资料进行现场核查;30工作日内完成现场核查,将初审意见,《药品注册研制现场核查报告》,申报资料送交 国家食品药品监督管理局药品审评中心(CDE) 审评进度,审评人员名 单及联系方式可以从 CDE网站查询。 https://www.doczj.com/doc/e618483598.html, CDE对申报资料进行技术审评(90工作日) 如果必要,CDE将要求申请人补充资料
化学药品注册分类对比(仅供参考) 2016年3月4日,食品药品监管总局发布了《化学药品注册分类改革工作方案》。现将《化学药品注册分类改革工作方案》与《药品注册管理办法》(2007版)相比,有以下这些不同之处:
相较于2015年11月6日《化学药品注册分类改革工作方案(征求意见稿)》中提到的2.5含有已知活性成分的新用法用量和新规格的制剂,在此方案中并未体现,可能只能走补充申请了。 相关注册管理要求 (一)对新药的审评审批,在物质基础原创性和新颖性基础上,强调临床价值的要求,其中改良型新药要求比改良前具有明显的临床优势。对仿制药的审评审批,强调与原研药品质量和疗效的一致。
(二)新注册分类1、2类别药品,按照《药品注册管理办法》中新药的程序申报;新注册分类3、4类别药品,按照《药品注册管理办法》中仿制药的程序申报;新注册分类5类别药品,按照《药品注册管理办法》中进口药品的程序申报。 新注册分类2类别的药品,同时符合多个情形要求的,须在申请表中一并予以列明。 (三)监测期有变动; (四)可以继续按照原规定进行审评审批,也可以申请按照新注册分类进行审评审批。如申请按照新注册分类进行审评审批,补交相关费用后,不再补交技术资料,国家食品药品监督管理总局药品审评中心要设立绿色通道,加快审评审批。符合要求的,批准上市;不符合要求的,不再要求补充资料,直接不予批准。 (五)新注册分类的注册申请所核发的药品批准文号(进口药品注册证/医药产品注册证)效力与原注册分类的注册申请核发的药品批准文号(进口药品注册证/医药产品注册证)效力等同。 (六)国家食品药品监督管理总局组织相关部门细化工作要求,做好受理、核查检查、技术审评及制定、修订相关国家药品标准等工作。 (七)《药品注册管理办法》与本方案不一致的,按照本方案要求执行。
目 录 1. 化学药品注册分类 2. 境内申请人新药申报 流程 3. 化学药品申报资料要求 4. 化学药品临床试验要求化学药品注册分类
1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂;(4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药 及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 以上,属注册分类1~5 类按新药申报程序申请注册, 6 类按仿制药申请程序申请注册。 境内申请人,新药申报流程(以上注册分类中1~5 类申报流程)
准备报临床申报资料(具体资料项目要求附后) 向省食品药品监督管理局报送申请资料 拿到受理号,相关进度, 便可以从SFDA 网站上 查询 https://www.doczj.com/doc/e618483598.html, 省局,受理,5工作日内组织对药物研制情况及原始资料进行现场核查;30工作日内完成现 场核查,将初审意见,《药品注册研制现场核查报告》,申报资料送交 国家食品药品监督管理局药品审评中心( CDE ) CDE 对申报资料进行技术审评(90工作日) 如果必要,CDE 将要求申请人补充资料 (补充资料准备时间 4个月,审评30工作日,一般最多要求补充 CDE 完成审评,提出技术审评意见,送国家食品药品监督管理局( SFDA )审批 符合规定,SFDA30工作日内作出审批,发给 《药物临床试验批件》 临床试验备案(SFDA,申报省局,临床试验基地所在省局 3处备案) 进行药物临床试验 临床试验完成后,准备报生产申报资料 审评进度,审评人员名 单及联系方式可以从 CDE 网站查询。 https://www.doczj.com/doc/e618483598.html, 2次)
附件1 化学药品注册申报资料指南(试行)(第一部分注册分类1、2、3、5.1类) 可编辑范本
目录 一、适用范围 (3) 二、基本要求 (3) (一)申请表的整理 (3) (二)申报资料的整理 (4) 三、申请表 (8) (一)《药品注册申请表》 (8) (二)《小型微型企业收费优惠申请表》 (14) 四、申报资料 (15) (一)申报资料项目 (15) (二)申报资料要求 (18) 五、其他 (24) 附件:1.申报资料袋封面格式 (25) 2.申报资料项目封面格式 (27) 3.申报资料项目目录 (28) 4.化学药品1、2、3、5.1类注册申报资料自查表 (29) 可编辑范本
化学药品注册申报资料指南(试行) 第一部分注册分类1、2、3、5.1类 一、适用范围 化学药品注册分类1、2、5.1类临床试验/新药生产(含新药证书)/上市申请;化学药品注册分类3类仿制药申请。 二、基本要求 (一)申请表的整理 1.种类与份数要求 药品注册申请表、申报资料情况自查表、小型微型企业收费优惠申请表(如适用)各四份,一份为原件;药品研制情况申报表(如适用)、药品注册生产现场检查申请表(如适用)各四份,三份为原件。 2.依据《关于启用新版药品注册申请表报盘程序的公告》,申请表的填报须采用国家食品药品监督管理总局统一发布的填报软件,提交由新版《药品注册申请表报盘程序》生成的电子及纸质文件。(确认所用版本为最新版[以最新发布的公告为准],所生成的电子文件的格式应为RVT文件。各页的数据核对码必须一致,并须与提交的电子申请表一致,申请表及自查表各页边缘应加盖所有申请人或注册代理机构骑缝章。) 3.填写应当准确、完整、规范,不得手写或涂改,并应符合可编辑范本
一、注册分类 1、未在国内外上市销售的药品: (1 )通过合成或者半合成的方法制得的原料药及其制剂; (2 )天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3 )用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; ( 4 )由已上市销售的多组份药物制备为较少组份的药物;( 5 )新的复方制剂。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1 )已在国外上市销售的原料药及其制剂; (2 )已在国外上市销售的复方制剂; (3 )改变给药途径并已在国外上市销售的制剂。 4 、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5、改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6、已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书样稿、起草说明及最新参考文献。 6、包装、标签设计样稿。
(二)药学研究资料 7、药学研究资料综述。 8、原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。 11、药品标准草案及起草说明,并提供标准品或者对照品。 12、样品的检验报告书。 13、辅料的来源及质量标准。 14、药物稳定性研究的试验资料及文献资料。 15 、直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16、药理毒理研究资料综述。 17、主要药效学试验资料及文献资料。 18、一般药理研究的试验资料及文献资料。 19、急性毒性试验资料及文献资料。 20、长期毒性试验资料及文献资料。 21 、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等主要与局部、全身给药相关的特殊安全性试验研究和文献资料。 22、复方制剂中多种成份药效、毒性、药代动力学相互影响的试验资料及文献资料。 23、致突变试验资料及文献资料。 24、生殖毒性试验资料及文献资料。 25、致癌试验资料及文献资料。 26、依赖性试验资料及文献资料。 27、动物药代动力学试验资料及文献资料。
— 1 — 附件 化学药品注册分类及申报资料要求 一、化学药品注册分类 基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 2.1含有用拆分或者合成等方法制得的已知活性成份的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金属元素,或者形成其他非共价键衍生物(如络合物、 螯合物或包合物),且具有明显临床优势的药品。 2.2含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的药品。 2.3 2.4
3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与参比制剂的质量和疗效一致。(境外原研药的国内首仿) 4类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与参比制剂的质量和疗效一致。 5类:境外上市的药品申请在境内上市。(——进口药) 5.1境外上市的原研药品和改良型药品申请在境内上市。改良型药品应具有明显临床优势。 5.2境外上市的仿制药申请在境内上市。 原研药品是指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。 评估确认的仿制药研制使用的对照药品。参比制剂的遴选与公布按照国家药品监管部门相关规定执行。 二、相关注册管理要求 (一)化学药品1类为创新药,应含有新的结构明确的、具有药理作用的化合物,且具有临床价值,不包括改良型新药中2.1类的药品。含有新的结构明确的、具有药理作用的化合物的新复方制剂,应按照化学药品1类申报。 (二)化学药品2类为改良型新药,在已知活性成份基础上进行优化,应比改良前具有明显临床优势。已知活性成份指境内或境外已上市药品的活性成份。该类药品同时符合多个情形要求 —2 —
化学药品注册分类 一、注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 中药、天然药物注册分类 本附件中的中药是指在我国传统医药理论指导下使用的药用物质及其制剂。 本附件中的天然药物是指在现代医药理论指导下使用的天然药用物质及其制剂。 一、注册分类及说明 (一)注册分类 1.未在国内上市销售的从植物、动物、矿物等物质中提取的有效成份及其制剂。 2.新发现的药材及其制剂。 3.新的中药材代用品。 4.药材新的药用部位及其制剂。 5.未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂。 6.未在国内上市销售的中药、天然药物复方制剂。 7.改变国内已上市销售中药、天然药物给药途径的制剂。 8.改变国内已上市销售中药、天然药物剂型的制剂。 9.仿制药。 (二)说明
化药注册分类新旧整合版 咸达数据 | 2016-03-05 09:05 公告发布日期:2016年3月4日 公告名字:总局关于发布化学药品注册分类改革工作方案的公告(2016年第51号) 公告背景:根据2015年11月4日第十二届全国人民代表大会常务委员会第十七次会议审议通过的《关于授权国务院在部分地方开展药品上市许可持有人制度试点和有关问题的决定》,国家食品药品监督管理总局制定了化学药品注册分类工作改革方案,已经国务院同意,现予以公告,并自公告发布之日起实施。 一、化学药品注册分类类别调整分析 公告对化学药品注册分类类别进行调整,化学药品新注册分类共分为5个类别。总体看来,这次化药注册分类改革的总体思路是:一创二改三抢四仿五进口,还给原研药正了名。 a)1类药:一创 定义:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。 分类说明:境内外均未上市的创新药。 包含的情形:含有新的结构明确的、具有药理作用的化合物,且具有临床价值的原料药及其制剂。 对应2007年版《药品注册管理办法》中附件2《化学药品注册分类及申报资料要求》旧分类(下文简称“对应旧分类”,只是笔者个人预估,一切以国家局颁布的标准为准):1.1类药(通过合成或者半合成的方法制得的原料药及其制剂)和1.2类药(天然物质中提取或者通过发酵提取的新的有效单体及其制剂)。 监测期:5年。 原监测期:5年。 申报程序:按照《药品注册管理办法》中新药的程序申报。 评点:不再纠结化合物的获得方法,更注重的是化合物的结构、药理作用和临床价值。
无论合成还是提取的,只要是「新的结构明确的、具有药理作用的化合物」,不仅有5 年监测期,还享有优先审评的特权。 b)2类药:二改 定义:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。分类说明:境内外均未上市的改良型新药。 包含的情形: 2.1类药含有用拆分或者合成等方法制得的已知活性成份(“已知活性成份”指“已上市药品的活性成份”,下同)的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金属元素,或者形成其他非共价键衍生物(如络合物、螯合物或包合物),且具有明显临床优势的原料药及其制剂。 2.1类药对应旧分类:1.3类药(用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂)和4类药(改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂)。 监测期:3年。 原监测期:1.3类药5年;4类药3年。 2.2类药含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的制剂。 2.2类药对应旧分类:2类药(改变给药途径且尚未在国内外上市销售的制剂)和5类药(改变国内已上市销售药品的剂型,但不改变给药途径的制剂)。 监测期:4年。 原监测期:2类药4年;5类药3年。 2.3类药含有已知活性成份的新复方制剂,且具有明显临床优势。(注意: 不包括“含有未知活性成份的新复方制剂”)。 2.3类药对应旧分类:1.5类药(新的复方制剂)。 监测期:4年。 原监测期:1.5类药4年。 2.4类药含有已知活性成份的新适应症的制剂。
中国药品注册的分类说明: 化学药品新注册分类共分为5个类别,具体如下: 1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。 2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。 4类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。 5类:境外上市的药品申请在境内上市。 涉及到本次双方的合作项目的进口注册类型为5.2类别:境外上市的非原研药品(包括原料药及其制剂)申请在境内上市。 The classification instruction of drug registration in China There are 5 categories of chemical drug new registrations. They are as follows. Category 1: Innovative drugs that are not marketed both domestically and abroad. These drugs contain new compounds with clear structures and pharmacological effects and they have clinical value. Category 2: Modified new drugs that are not marketed both domestically or abroad. With known active components, the drug’s structure, phase, prescription manufacturing process, administration route and indication are optimized and it has obvious clinical advantage. Category 3: The drugs that are imitated by domestic applicants to original drugs that have been marketed abroad but not domestically.This kind of drugs are supposed to have the same quality and effects with original drugs.Original drugs are the foremost drugs that are approved to be marketed domestically and /or abroad with complete and full safety and validity data as marketing evidence. Category 4: The drugs that are imitated by domestic applicants to original drugs that have been marketed domestically. This kind of drugs are supposed to have the same quality and effects with original drugs. Category 5: The drugs that have been marketed abroad are applied to be marketed domestically. The category of the imported registration involved in our collaboration program is category 5.2: non-original drugs( including API and its preparation) that have been marketed abroad are applied to be marketed domestically.
药品注册分类
化学药注册分类大变动,CFDA发布《化学药品注册分类改革工作方案》和《化学仿制药生物等效性试验备案管理规定》征求意见稿 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,昨日(11月6日),国家食药监总局官网发布了《化学药品注册分类改革工作方案(征求意见稿)》和《化学仿制药生物等效性试验备案管理规定(征求意见稿)》,面向社会公开征求意见。 《化学药品注册分类改革工作方案(征求意见稿)》 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,制定本工作方案。 一、工作原则 按照分类科学、标准严格、质量提高的原则,在原有化学药品注册分类的基础上,结合国务院改革意见中有关药品分类的调整原则,对原有化学药品注册分类进行调整和完善。首先,根据药品的安全风险程度,将药品分为新药和仿制药两大类;其次,根据药品原创性和新颖性的不同,将新药进一步分为创新药和改良型新药;第三,在仿制药中,根据被仿制药上市情况不同,进一步细分为对境外上市、境内未上市药品的仿制,对境内上市药品的仿制以及境外上市药品申请境内上市三类。
二、化学药品新注册分类及说明 新药是指未在中国境内外上市销售的药品,将境外上市境内未上市药品纳入仿制药。调整后,化学药品新注册分类共分为1-5类(表1),具体如下: (一)根据物质基础的原创性和新颖性不同,将新药分为创新药(注册分类1)和改良型新药(注册分类2)两类。其中,创新药是指含有新的结构明确的具有生理或药理作用的分子或离子,且具有临床价值的原料药及其制剂,包括用拆分或者合成等方法制得的已知活性成份的光学异构体及其制剂,但不包括对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 改良型新药是在已知活性成份的基础上,对其结构、剂型、给药途径、适应症、用法用量、规格等进行优化,且具有明显临床优势的药品。结构优化是指对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 (二)被仿制的参比制剂来源不同,其上市情况存在差异,研制者和监管部门对其上市基础的认识也随之不同,为便于申报,将仿制药分为3-5类。其中,注册分类3是指仿境外上市、境内未上市药品;注册分类4是指仿制境内上市药品;注册分类5是指境外上市的药品申请在境内上市。