
β2环糊精修饰胶束电动毛细管色谱法测定
大黄中有效成分
尚小玉 袁倬斌
3
(中国科学技术大学研究生院,北京100039)
摘 要 采用β2环糊精修饰的胶束电动毛细管色谱法分离测定了大黄中5种有效成分。缓冲溶液为磷酸盐
(pH 10~10.38)含20mm ol ΠL S DS 和10mm ol ΠL β2C D ,10%~15%(V ΠV )的甲醇或异丙醇。对生大黄和蒙古大
黄样品进行了测定,结果令人满意。
关键词 大黄,β2环糊精修饰胶束电动毛细管色谱
2001208206收稿;2002201211接受
本文系国家自然科学基金资助项目(N o.29675022,29875027)和国家“九五”科技攻关项目(N o.962A23201206)
1 引 言
大黄,古代亦称黄良和将军,是我国常用中药。大黄具有泻下、抗菌、抗病毒、止血、止痛、抗肿瘤及
抗突变等作用[1,2]
。大黄的主要成分是蒽醌类及其衍生物,包括大黄素、大黄酚、大黄酸、芦荟2大黄素、大黄素甲醚和它们的葡萄糖甙等。对其活性成分分离测定的常用方法是T LC 和HP LC [3,4]
等,但这些方
法存在着或者分辨率低或者预处理烦琐、溶剂消耗量大的缺点。近几年来,胶束电动毛细管色谱法[5]
、
微乳胶毛细管色谱法[6]及毛细管电色谱[7]
等方法应用到大黄的分离测定中。本文首次把β2环糊精(C D )修饰的胶束电动毛细管色谱法(C D 2MEK C )用于分离测定大黄中5种有效成分,并对两种大黄样品进行了测定。
2 实验部分
2.1 仪器与试剂
实验在北京新技术所1229型高效毛细管电泳仪上进行。未涂层的熔融石英毛细管,购于河北永年光纤厂,内径为50μm ,有效长度50cm 。柱上紫外检测(254nm ),恒压14kV ,电进样5s 。
磷酸盐缓冲溶液由Na 2HPO 4和K H 2PO 4(M Π30)配制。十二烷基硫酸钠(S DS )购自军事医学科学院药材供应站,β2C D 购自华北地区特种化学试剂开发中心。邻苯二甲酸(纯度为99.8%)购自北京化工厂。生大黄和蒙古大黄购于河北安国药材市场。无水乙醇、甲醇和异丙醇均为分析纯试剂。2.2 实验方法
0.5g 大黄样品置于索氏提取器中,加入15m L 2m ol ΠL H 2S O 4和70m L 氯仿,水浴中加热提取,直至氯仿液变成无色。取出氯仿层,水浴中加热蒸干,残余物用无水乙醇溶解,加入30μL 邻苯二甲酸内标
溶液后,加缓冲溶液稀释至5m L ,用0.45μm 滤膜(Φ25mm ×50张,北京建达实业总公司)过滤,供测定
用。
实验前,用0.1m ol ΠL NaOH 冲洗管壁10min ,然后用二次水冲洗10min ,最后用缓冲溶液冲洗5min 。
3 结果与讨论
3.1 pH 对分离的影响
考察了含10mm ol ΠL S DS 和10mm ol ΠL β2C D 的磷酸盐缓冲溶液在不同pH 值下的分离情况。并考察了在7.94~10.38范围内pH 值对迁移时间的影响。结果如图1所示。由图可以看出,5种成分的迁移时间随pH 值增大而降低。5种成分是弱酸性物质,在此pH 范围内,不同程度的带上负电荷。pH 值增
第30卷
2002年7月 分析化学(FE NXI H UAX UE ) 研究简报Chinese Journal of Analytical Chemistry
第7期
853~856
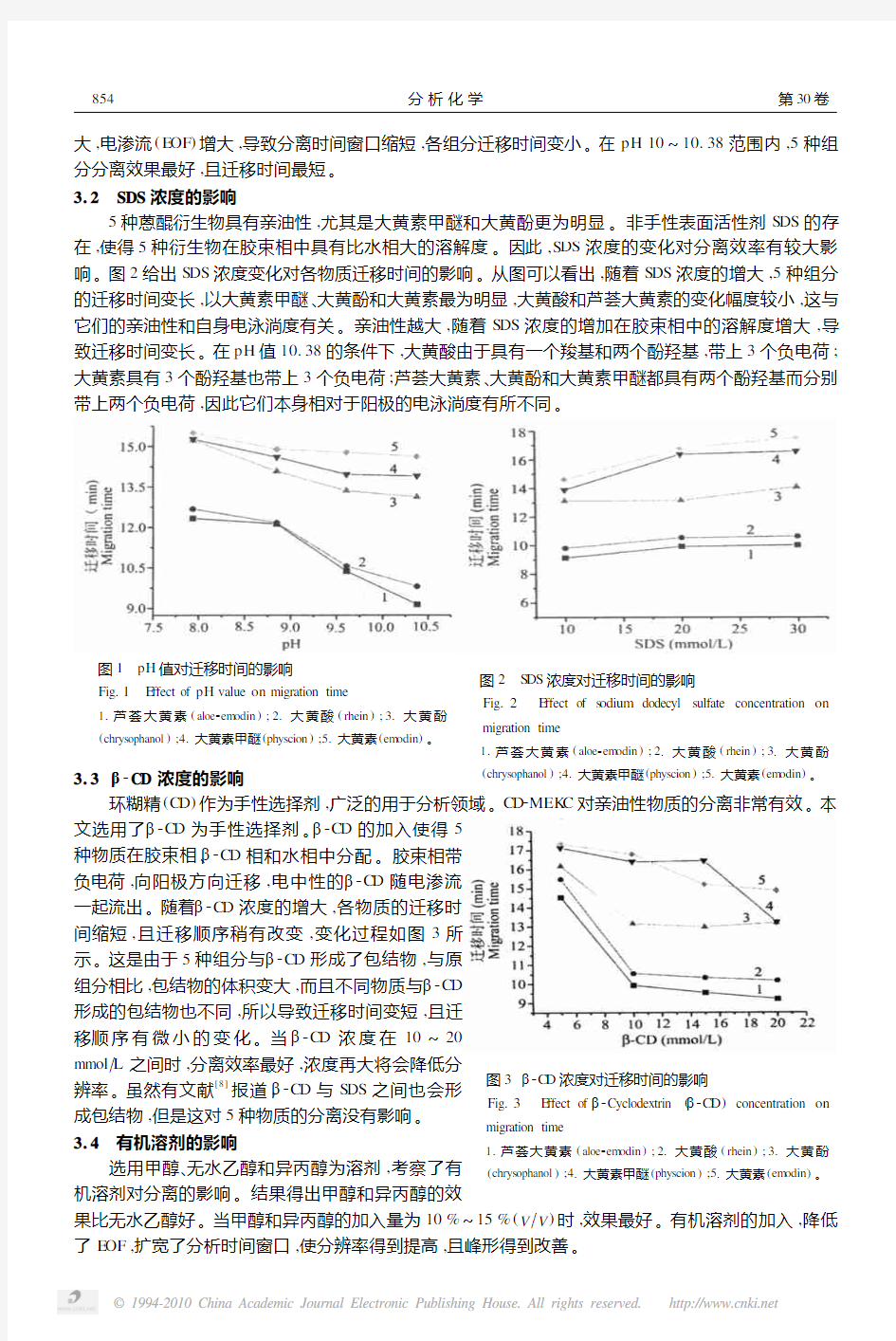
大,电渗流(E OF )增大,导致分离时间窗口缩短,各组分迁移时间变小。在pH 10~10.38范围内,5种组分分离效果最好,且迁移时间最短。3.2 SDS 浓度的影响
5种蒽醌衍生物具有亲油性,尤其是大黄素甲醚和大黄酚更为明显。非手性表面活性剂S DS 的存在,使得5种衍生物在胶束相中具有比水相大的溶解度。因此,S DS 浓度的变化对分离效率有较大影响。图2给出S DS 浓度变化对各物质迁移时间的影响。从图可以看出,随着S DS 浓度的增大,5种组分的迁移时间变长,以大黄素甲醚、大黄酚和大黄素最为明显,大黄酸和芦荟大黄素的变化幅度较小,这与它们的亲油性和自身电泳淌度有关。亲油性越大,随着S DS 浓度的增加在胶束相中的溶解度增大,导致迁移时间变长。在pH 值10.38的条件下,大黄酸由于具有一个羧基和两个酚羟基,带上3个负电荷;大黄素具有3个酚羟基也带上3个负电荷;芦荟大黄素、大黄酚和大黄素甲醚都具有两个酚羟基而分别带上两个负电荷,因此它们本身相对于阳极的电泳淌度有所不同
。
图1 pH 值对迁移时间的影响 Fig.1 E ffect of pH value on migration time
1.芦荟大黄素(aloe 2em odin );21大黄酸(rhein );31大黄酚(chrys ophanol );41大黄素甲醚(physcion );51大黄素(em odin )
。
图2 S DS 浓度对迁移时间的影响
Fig.2 E ffect of s odium dodecyl sulfate concentration on
migration time
1.芦荟大黄素(aloe 2em odin );21大黄酸(rhein );31大黄酚
(chrys ophanol );41大黄素甲醚(physcion );51大黄素(em odin )。3.3 β2CD 浓度的影响
环糊精(C D )作为手性选择剂,广泛的用于分析领域。C D 2MEK C 对亲油性物质的分离非常有效。本
图3 β2C D 浓度对迁移时间的影响
Fig.3 E ffect of β2Cyclodextrin (β2C D )concentration on
migration time
1.芦荟大黄素(aloe 2em odin );21大黄酸(rhein );31大黄酚(chrys ophanol );41大黄素甲醚(physcion );51大黄素(em odin )。文选用了β2C D 为手性选择剂。β2C D 的加入使得5
种物质在胶束相、β2C D 相和水相中分配。胶束相带
负电荷,向阳极方向迁移,电中性的β2C D 随电渗流
一起流出。随着β
2C D 浓度的增大,各物质的迁移时间缩短,且迁移顺序稍有改变,变化过程如图3所
示。这是由于5种组分与β2C D 形成了包结物,与原组分相比,包结物的体积变大,而且不同物质与β2C D 形成的包结物也不同,所以导致迁移时间变短,且迁移顺序有微小的变化。当β2C D 浓度在10~20mm ol ΠL 之间时,分离效率最好,浓度再大将会降低分
辨率。虽然有文献[8]
报道,β2C D 与S DS 之间也会形
成包结物,但是这对5种物质的分离没有影响。
3.4 有机溶剂的影响选用甲醇、无水乙醇和异丙醇为溶剂,考察了有机溶剂对分离的影响。结果得出甲醇和异丙醇的效
果比无水乙醇好。当甲醇和异丙醇的加入量为10%~15%(V ΠV )时,效果最好。有机溶剂的加入,降低了E OF ,扩宽了分析时间窗口,使分辨率得到提高,且峰形得到改善。
458 分析化学第30卷
3.5 标准曲线的绘制
在大黄素4.6~56.6mg ΠL 、芦荟大黄素3.86~32mg ΠL 和大黄酸4~36.6mg ΠL 的浓度范围内,精密
配制标准溶液。在上述3种不同浓度样品中分别加入10μL 邻苯二甲酸内标溶液。测得各组分的峰面积值,3.6 加样回收率实验
由对照品与内标面积比值(Y )和对照品重量与内标重量之比(X ),计算线性方程和相关系数,结果如下:芦荟大黄素:Y =0.9210X -0.3201,r =0.9998;大黄酸:Y =1.0535X -0.0028,r =0.9996;大黄素:Y =0.7613X -0.0089,r =0.9998。
图4 对照品和大黄样品的色谱图
Fig.4 Chromatograms of standard sam ples and rhubarb sam ples 1.芦荟大黄素(aloe 2em odin );21大黄酸(rhein );31大黄酚
(chrys ophanol );41大黄素甲醚(physcion );51大黄素(em odin )。
精密量取对照品溶液不同量,分别加到已知含量的生大黄和蒙古大黄样品中,按实验方法进行提取、测定,计算回收率。结果如下:芦荟大黄素为95112%,大黄酸为96.38%,大黄素为100167%。3.7 样品的测定
用此法对5种对照品和大黄样品进行了分离,结果见图4和表1。对生大黄和蒙古大黄进行了测定,平行3次,计算结果见表2。由于大黄酚和大黄素甲醚的重现性差,所以只对其它3种组分进行了测定。
表1 迁移时间的重现性T able 1 Reproducibility of migration time
化合物
C om pound
迁移时间M igration time
平均时间
M ean (m in )
RS D (%)
邻苯二甲酸o 2Phthalic acid 9.51 1.91芦荟大黄素Aloe 2em odin 13.61 1.96大黄酸Rhein
16.42 2.01大黄酚Chrys ophanol 18.39 2.11大黄素甲醚Physcion 20.68 2.15大黄素Em odin
22.93
1.93
表2 生大黄和蒙古大黄样品中蒽醌衍生物的含量
T able 2 C ontents of anthraquinone derivatives in Raw Rhubarb and M ong olian Rhubarb
样品Sam ple
含量C ontent (mg Πg )芦荟大黄素Aloe 2em odin
大黄酸Rhein
大黄素Em odin
生大黄Raw Rhubarb
0.9751 1.12100.8754蒙古大黄M ong olian Rhubarb
0.5213
0.8131
1.0428
4 结 论
采用环糊精修饰的胶束电动毛细管色谱方法分离测定了大黄中的5种有效成分,在23min 内5种物质全部分离。与文献报道的MEK C 方法比较,该法具有较高的分离选择性和分辨率,而且所用缓冲溶液基本上对环境友好。通过对各参数的筛选,得出了最佳分离条件:磷酸盐缓冲溶液(pH 10.38)含20mm ol ΠL S DS 和10mm ol ΠL β2C D ,10%~15%(V Π
V )的甲醇或异丙醇。从含量测定看,两种大黄样品中5种蒽醌衍生物的含量差异较大,因此,有必要对大黄的质量进行控制。R eferences
1 Wu X iuying (武秀英),Wu Qingtai (武庆泰).Acta Chinese Medicine and Pharmacology (中医药学报),1995,2:54~56
5
58第7期尚小玉等:β2环糊精修饰胶束电动毛细管色谱法测定大黄中有效成分
658 分析化学第30卷
2 T ao Y uzhen(陶玉珍),Liu Y i(刘 毅),G uo Qiming(郭启明).Carcinogenesis Teratogenesis and Mutagenesis(癌变?畸变?突变),1998,10(4):241~243
3 He Liyi(何丽一),Luo Shurong(罗淑荣).Acta Pharmaceutica Sin.(药学学报),1980,X V(9):555~562
4 Liu C L,Zhu P L,Liu M C.J.Chromatogr.A,1999,857:167~174
5 Sheu S J,Chen H R.Analytica Chimica Acta,1995,309:361~367
6 Li G B,Chen X G,Liu M C,Hu Z D.Analyst,1998,123:1501~1505
7 Li Y,Liu H W,Li J L.Electrophoresis,2000,21:3109~3115
8 Lu Runhua(鲁润华),Shi Shuo(石 硕),Wang Hanqing(汪汉卿).China Sur factant Detergent&Cosmetics(日用化学工业), 1999,4:28~29
Determination of E ffective Constituents in Rhubarb by β2Cyclodextrin Modified Micellar E lectrokinetic Chromatography
Shang X iaoyu,Y uan Zhuobin3
(Institute o f Applied Chemistry,Graduate School o f the Univer sity o f Science and Technology o f China,Beijing100039)
Abstract β2Cyclodextrin(β2C D)m odified micellar electrokinetic chromatographic method was adopted for the determination of five effective constituents in rhubarb.Under the optimized conditions:phosphate buffer(pH 10138)containing20mm olΠL s odium dodecyl sulfate(S DS),10mm olΠLβ2C D and10%~15%(VΠV)methanol or is opropanol,the five com ponents were separated within23min.T w o commercial Rhubarb sam ples were analyzed by this method,and the results obtained were satis factory.
K eyw ords Rhubarb,β2cyclodextrin m odified micellar electrokinetic chromatography
(Received6August2001;accepted11January2002)
国家自然科学基金委员会
重点学术期刊专项基金2001年度批准资助期刊名单
刊 名刊 期资助金额刊 名刊 期资助金额1数学学报(英)季刊8万13中华外科杂志月刊12万
2力学学报(英)季刊8万14作物学报双月刊10万
3物理学报月刊12万15中国农业科学双月刊10万
4中国物理快报(英)月刊12万16遗传学报双月刊10万
5化学学报月刊12万17大气科学进展(英)季刊8万
6中国化学快报月刊12万18地球物理学报双月刊10万
7分析化学月刊12万19材料科学技术学报(英)双月刊10万
8高等学校化学学报月刊12万20稀有金属材料与工程双月刊10万
9生物化学与生物物理学报双月刊10万21金属学报月刊12万
10植物学报月刊12万22硅酸盐学报双月刊10万
11中国药理学报(英)月刊12万23光学学报月刊12万
12中华医学杂志(英)月刊12万24电子学报月刊12万
毛细管胶束电动色谱分离原理 概述 在电泳缓冲液中加入离子表面活性剂,当溶液中表面活性剂浓度超过临界胶束 浓度时,表面活性剂的单体就结合在一起,形成一个球体,称为胶束。胶束一般是由10--50个碳原子单位的长链分子组成,具有头部(或外层)亲水、尾部(或内层)疏水的特性。在溶液中,头部露在外面,尾部包在胶束中。这种胶束的形成是疏水效应的结果,能使系统的自由能减少。胶束可分为正相胶束和反相胶束两类,以前者应用较多。反相胶束则是指在有机溶剂中形成的胶束,尚未作系统研究。 用来作为表面活性剂的化合物很多,大体有四类:阴离子、阳离子、两性离子和 非离子表面活性剂,其典型化合物及主要性质如表4.1所示。 应用最多的是前两类,且尤以十二烷基硫酸钠(SDS)等使用最为普遍。图4.1是SDS胶束的结构示意, 里面有一个疏水内核,外面布满了.S03ˉ离子。
在MECC系统中,实际上存在着类似于色谱的两相,一是流动的水相,另一是起到固定相作用的胶束相,溶质在这两相之间分配,由其在胶束中不同的保留能力而产生不同的保留值。与毛细管区带电泳一样,由于缓冲液在靠近管壁处形成的正电,使其显示出强烈的电渗流而向阴极移动。对于SDS胶束来说,由于其外壳带很大的负电荷,本应以较大的淌度朝阳极迁移,但由于在一般情况下电渗流的速度大于胶束的迁移速度,这就迫使胶束最终以较低的速度向阴极移动,如图4.2所示。由此可见,毛细管胶束电动色谱有别于普通色谱的一个重要特性为它的“固定相”是移动的,这种移动的“固定相”又被称之为“准固定相’。 在毛细管胶束电动色谱中,中性粒子由于本身疏水性的不同而得以分离.具有不同疏水性的粒子与胶束的相互作用不同,疏水性强的作用力
毛细管柱应用范围及使用温度 一、SPB-1型非极性柱(键合,聚二甲基硅氧烷) 对照品牌:HP-1,DB-1,BP-1,CP-SIL 5CB,UItra-1,007-1,RTx-1,AT-1 类似固定相:SE-30,SP-2100,OV-1,OV-101 使用温度:-60℃-320℃ 应用范围:烷烃,芳烃,多环芳烃,醇,酚,酮,酯,醛,胺,卤代烃,吡啶,糖衍生物,氨基酸衍生物,维生素衍生物,镇痛药,农药,溶剂,胆固醇,香料,咖啡,食品添加剂等 二、SPB-5型弱极性柱(键合,5%苯基,95%甲基聚硅氧烷) 对照品牌:HP-5,DB-5,BP-5,CP-SIL 8CB,UItra-2,007-2,RTx-5,AT-5 类似固定相:SE-54,SE-52,OV-73 使用温度:-60℃-320℃ 应用范围:烷基苯,多环芳烃,醇,酚,酮,脂肪酸酯,苯二甲酸酯,硝基芳烃,芳胺,烷基胺,联苯胺,卤代烃,多氯联苯,糖类衍生物,维生素衍生物,有机酸,镇痛药,农药,抗组胺药,溶剂,生物碱,防腐剂,香料等
三、SUPELCOWAX 10型极性柱(键合,聚乙二醇二万) 对照品牌:HP-Wax,DB- Wax,BP-20,CP- Wax 52CB,HP-INNO Wax,AT- Wax 类似固定相:PEG-20M,CARBOWAX-20M 使用温度:35℃-280℃ 应用范围:低沸点芳烃,醇,酮,酯,醛,醚,乙二醇,丙二醇,甘油,吡啶,胺,亚硝胺,卤代烃,胆汁酸衍生物,冰片,薄荷,精油,香料,酒,苯乙烯,茶,溶剂等 四、SPB-50型中等极性柱(键合,50%二苯基,50%二甲基聚硅氧烷) 对照品牌:HP-50,HP-17,DB-17,007-17,RTx-50,AT-50 类似固定相:OV-17,SP-2250 使用温度:30℃-310℃ 应用范围:烷烃,低沸点芳烃,多环芳烃,醇,甘油三酸酯,喹啉,卤素化合物,精油,香料,农药,酯,镇痛药,除草剂等 五SPB-1701型中等极性柱(键合,14%氰丙基,86%二甲基聚硅氧烷) 对照品牌:HP-1701,DB-1701,007-1701,RTx-1701,AT-1701,BP-10,CP-Sil 19CB 类似固定相:OV-1701,SP-2250 使用温度:室温-280℃ 应用范围:醇,卤素化合物,有机氯农药,酸性药物,有机磷,除草剂等
气相色谱分析法 一、选择题 ⒈对某一组分来说,在一定的柱长下,色谱峰的宽或窄主要决定于组分在色谱 柱中的( )。 A.保留值 B. 扩散速度 C.分配比 D. 理论塔板数 ⒉载体填充的均匀程度主要影响 A.涡流扩散相 B. 分子扩散 C.气相传质阻力 D. 液相传质阻力 ⒊在气相色谱检测器中通用型检测器是( )。 A.氢火焰离子化检测器 B.热导池检测器 C.示差折光检测器 D.火焰光度检测器 ⒋在气液色谱中,色谱柱的使用上限温度取决于: A.样品中沸点最高组分的沸点 B.样品中各组分沸点的平均值 C.固定液的沸点 D.固定液的最高使用温度 二、填空题 1. 在气液色谱中,被分离组分分子与固定液分子的性质越相近,则它们之间的作用力越,该组分在柱中停留的时间越,流出越。 2. 气液色谱法即流动相是,固定相是的色谱法。样品与固定相间的作用机理是。 3. 气固色谱法即流动相是,固定相是的色谱法。样品与固定相的作用机理是。 4. 气相色谱仪中气化室的作用是保证样品气化。气化室温度一般要比柱温度高℃,但不能太高,否则会引起样品。 5. 气相色谱分析的基本程序是从进样,气化了的样品在分离,分离后的各组分依次流经,它将各组分物理或化学性质的变化转换成电量变化,输给记录仪,描绘成色谱图。 6. 色谱柱的分离效率用α表示。α越大,则在色谱上两峰的距离,表明这两个组分分离,通常当α大于时,即可在填充柱上获得满意的分离。 7. 分配系数K用固定液和载气中的溶质浓度之比表示。待分离组分的K值越大,则它在色谱柱中停留的时间,其保留值。各组分的K值相差越大,则它们分离。 8. 一般地说,为了获得较高的柱效率,在制备色谱柱时,固定液用量宜,
基本原理 毛细管电色谱(Capillary electrochromatography, 简称 CEC)是在毛细管中填充或在管壁涂布、键合液相色谱的固定相,然后在毛细管的两端施加高压直流电,在电场作用下产生电渗流(Electroosmotic flow ,简称EOF),流动相在电渗流的驱动下通过色谱柱。对中性化合物,其分离过程和HPLC类似,即通过溶质在固定相和流动相之间的分配差异而获得分离;当被分析的物质在流动相中带电荷时,除了和中性化合物一样的分配机理外,自身电泳淌度的差异对物质的分离也起相当的作用。 毛细管电色谱(capillary electro chromatography,CEC)以内含色谱固定相的毛细管为分离柱,兼具毛细管电泳及高效液相色谱的双重分离机理,既可分离带电物质也可分离中性物质。毛细管电色谱法是用电渗流或电渗流结合压力流来推动流动相的一种液相色谱法。 因此,毛细管电色谱法可以说是HPLC和HPCE 的有机结合,它不仅克服了HPLC 中压力流本身流速不均匀引起的峰扩展,而且柱内无压降,使峰扩展只与溶质扩散系数有关,从而获得了接近于HPCE 水平的高柱效,同时还具备了HPLC 的选择性。 HPLC是用压力驱动流动相。流速是随填充微粒的大小和柱长而变化的。流速在管中呈抛物线轮廓,因而造成了色谱峰谱带的展宽,降低了柱效。而CEC是采用电场推动流动相。其线速度是与柱的直径和填微粒的大小无关的,因而在毛细管中几乎没有流速梯度。谱带展宽效应相应的就十分小。这点是CEC与HPLC的本质差别,也是CEC效率高于HPLC 的根本。 依靠电渗流(EOF)和电渗流结合压力流推动流动相,使中性和带电荷的样品分子根据它们在色谱固定相和流动相间吸附、分配平衡常数的不同和电泳速率不同而达到分离分析。 仪器设备: 毛细管电色谱的早期研究是在改装的CE商品仪器上进行的,随着研究的深入和对研究前景的良好预期,现在已有商品仪器既可进行电泳模式也可方便地进行电色谱研究。目前,主要是Beckman公司的 P/ACE 系列和HP公司的HP3D系列。检测器根据分析样品性质的不同,可选UV 检测器( 包括DAD ) 、电化学检测器、LIF及CE-MS等。 类型:在毛细管电色谱(CEC)中,色谱柱是电色谱的心脏,按照固定相的装填方式不同可以分为[7]:填充毛细管电色谱(PCCEC),开管毛细管电色谱(OTCEC),整体式毛细管电色谱(MCEC)。PCCEC是将固定相装填在毛细管中,OTCEC是将固定相涂渍或键合在毛细管内壁上,MCEC是通过在毛细管内原位聚合或固化的方法,制成的具有多孔结构的整体式固定相。根据分离过程中驱动力的不同可以将毛细管电色谱分
第六章毛细管柱气相色谱法 第一节毛细管气相色谱仪 现代的实验室用的气相色谱仪大都既可用作填充柱气相色谱又可用作毛细管色谱仪。毛细管色谱仪应用范围广,可用于分析复杂有机物,如石油成分,天然产物,环境污染,农药残留等。图6-1是毛细管气相色谱仪示意图,与填充柱色谱仪比,毛细管色谱仪在柱前多一个分流-不分流进样器,柱后加一个尾吹气路。由于毛细管柱体积很小,柱容量很小,出峰快,所以死体积一定要小,要求瞬间注入极小量样品,因此柱前要分流。对进样技术要求高,对操作条件要求严。尾吹的目的是减小死体积和柱末端效应。毛细管柱对固定液的要求不苛刻,一般2-3根不同极性的柱子可解决大部分的分析问题。毛细管柱一般配有响应快,灵敏度高的质量型检测器。 高分辨率毛细管气相色谱仪的三要素是:要选择好的毛细管柱及最佳分析条件;按样品选择合适的毛细管进样系统;选择高性能的毛细管气相色谱仪。 图6-1 毛细管气相色谱仪示意图 第二节毛细管色谱柱 1957年,美国科学家Golay提出毛细管柱的气相色谱法。Golay称毛细管色谱柱为开管柱。因这种色谱柱中心是空的。毛细管柱是内径为Φ0.1-0.5mm左右、长度为10-300m的毛细柱,虽然每米理论板数约为2000-5000,与填充柱相当,但由于柱子很长,总柱效可高达106。 一、毛细管色谱柱组成 通常来说,一根毛细管色谱柱由管身和固定相两部分组成。管身采用熔融二氧化硅(熔融石英),通常在其表面涂上一层聚酰亚胺保护层。涂层后的熔融石英毛细管呈褐色:但是涂层后的毛细管之间的颜色却不尽相同。色谱柱的颜色对于其色谱性能没有什么影响。经过持续的较高温度处理后.聚酰亚胺涂层管的的温度会变得比以前更深:标准的聚酰亚胺涂层管熔融石英管的温度上限为360℃,高温聚酰亚胺涂层管的温度上限为400℃。固定相种类很多,大部分的固定相是热稳定性好的聚合物,常用的有聚硅氧烷和聚乙二醇。另外还有一类是小的多孔粒子组成的聚合物或沸石(例如氧化铝、分子筛等)。 熔融石英管的内表面会用一些化学方法进行处理,尽量的减小样品和管壁之间可能存在的相互作用。所用的试剂和处理方法一般是依据将要涂在内壁上的固定相种类来确定的。硅烷化处理则是最为常用的处理方式,使用硅烷类的试剂和管壁内表面上的硅基醇基团进行反应,使其变为甲基硅烷基或苯甲基甲基硅烷基。 当实验要求更高的使用温度时,我们可以来用不锈钢毛细柱来代替熔融石英毛细柱。不锈钢毛细柱在使用温度(耐高温)及日常维护(不易折断等)的性能和指标上都优于熔融石英毛细柱。但是不锈钢材质的惰性没有熔融石英好,它可以和许多的化合物相互作用,产生反应。所以通常可以用化学方法对其进行处理,或者是在它的内壁再涂上薄薄的一层熔融石英,以增加不锈钢管的隋性:经过适当处理后,不锈钢毛细柱的惰性与熔融石英毛细柱的不相上下。 二、毛细管色谱柱固定相 (一)气-液色谱固定相 1.聚硅氧烷
毛细管气相色谱法条件及定量分析 指导老师:李建国 实验人:王壮 同组实验:陆潇、戈畅 实验时间:2016.4.18 一、实验目的 1.熟悉色谱分析的原理及色谱工作站的使用方法; 2、掌握气相色谱仪操作方法与氢火焰离子化检测器的原理; 3.用保留时间定性;用归一化法定量;用分离度对实验数据进行评价。 二、实验原理 不同组分在同一分离色谱柱上,在相同实验条件下有不同的保留行为,其保留时间的差异可以用来定性分析,每一组分的质量与相应色谱峰的积分面积成正比,因此可以公式计算,用归一化方法测定每一组分的质量百分含量。 1122100A is i i A A A s s ns n f A w f A f A f A =?++???+% 本实验是用气相色谱测定乙酸乙酯、乙酸丁酯及其混合试样,检测器用FID 。用色谱软件进行谱图处理和定量计算,让学生掌握用已知物对照定性、用归一化法测定混合物组分定量的实验。 混和试样的成功分离是气相色谱法定量分析的前提和基础,衡量一对色谱峰分 离的程度可用分离度:12121()2 R R t t R W W -=?+,式中1R t 、2R t 和1W 、2W 分别指两组分的保留时间和峰底宽度,R=1.5时两组分完全分离,实际中R=1.0(分离度98%)即可满足要求。 三、仪器与试剂 仪器:GC7890F 型气相色谱仪、氢火焰离子化检测器(FID )、氮气钢瓶、空气钢瓶、氢气发生器,微量注射器、3mm x 200cm 的10% SE-54不锈钢分离柱。GC5400型气相色谱仪、空气发生器、氮气发生器、氢气发生器,微量注射器、15m 毛细管分离柱。 试剂:乙酸乙酯、乙酸丁酯标准试样及其未知混合试样。 四、实验内容 1.按操作说明书使色谱仪正常运行,并调节至如下条件: 柱温:110C ? 检测器温度:120C ? 气化温度:120C ? 载气、氢气和空气流量分别为30、50和200mL/min 。 2.分别改变柱温至80、90、100、110、120C ?。每改变一次柱温,注入0.5L μ混合
毛细管气相色谱柱的选择方法 【关闭本页】【返回首页】【发布时间2004-1-14】 一、固定相的选择 1.如果不知道使用何种固定相,可以从非极性柱或弱极性柱如SPB-1或SPB-5开始试用,如效果不好,再按极性渐强的顺序选用中等极性直至高极性柱逐一尝试,直到有较令人满意的分析结果即可确定适用的柱极性。 2.低流失(“ms”)色谱柱通常更为惰性,有更高的温度上限,适用于MS检测器。3.使用能够提供满意的分离度和分析时间的极性最小的固定相,非极性固定相比极性固定相具有更长的寿命。 4.要使用和被分析物极性相近的固定相,使用这一选择方法常常是有效的,但是使用这一方法并不总是能找到最好的固定相。 5.如果被分离混合物具有不同的偶极或氢键力,改变使用具有不同偶极或氢键力(不一定要更大)的固定相后,会出现其他共流出物,所以新的固定相不一定提供更好的总分离度。 6.如果可能,要避免使用含有能使选择性检测器产生高响应值功能团的固定相,例如含有氰丙基的固定相,用NPD会产生不成比例地增大基线高度(由于柱流失)的现象。7.SPB-1或SPB-5,SPB-50,SPB-1701,和SUPELCOWAX 10以最少数量的色谱柱能覆盖最大范围的选择性。 8.PLOT柱用于在高于室温的柱温下来分析气体样品。 二、色谱柱直径的选择 1. 当需要有高柱效的色谱柱时应使用0.18-0.25mm的色谱柱。0.18mm的色谱柱很适合于泵容量低的GC/MS系统。小内径柱的容量最小而且需要最高的柱头压力。 2. 当需要样品容量大时就要使用0.32mm内径的色谱柱。与0.25mm内径柱相比, 0.32mm内径柱对不分流进样或大体积(>2μL)进样时早流出的色谱峰有较高的分离度。 3. 只有配备大口径直接进样口时,才使用0.53mm内径的色谱柱,它特别适合于高载气流速的应用,例如吹扫捕集,顶空进样。0.53mm内径色谱柱在恒定的液膜情况下具有最高的样品容量。 三、色谱柱柱长的选择 1. 当不知道最佳柱长时,尝试使用25-30m长的色谱柱。 2. 10-15m长的色谱柱适合于分离含有很容易分离的溶质混合物,或者分离为数不多的溶质混合物,较短的柱长用于直径很小的色谱柱,以便降低柱头压力。 3. 当使用其他方法(小内径柱,不同的固定液,改变柱温)不能达到分离度时,就使用50-60m长的色谱柱。它最适合于分离含有多组分的复杂混合物,长柱需要的分离时间长,费用也高。 四、色谱柱膜厚的选择 1. 0.18-0.32mm内径的色谱柱,其平均或标准膜厚在0.18-0.25μm,用于绝大多数的分析。 2. 0.45-0.53 mm内径的色谱柱,其平均或标准膜厚在0.18-1.5μm,用于绝大多数的分析。 3. 厚液膜色谱柱用于保留和分离挥发性物质(如轻溶剂,气体)。厚液膜色谱柱有更高的惰性,其柱容量也高;但厚液膜色谱柱具有较高流失性,使用温度上限也有所下降。 4. 薄液膜色谱柱用于降低高沸点物质和高分子量物质(如甾体,三甘油酸酯)的保留时间,并具有低流失性的特点;但薄液膜色谱柱的惰性较差,且柱容量较低。
毛细管气相色谱法条件及定量分析 指导老师:李建国 实验人:王壮 同组实验:陆潇、戈畅 实验时间:2016.4.18 一、实验目的 1.熟悉色谱分析的原理及色谱工作站的使用方法; 2、掌握气相色谱仪操作方法与氢火焰离子化检测器的原理; 3.用保留时间定性;用归一化法定量;用分离度对实验数据进行评价。 二、实验原理 不同组分在同一分离色谱柱上,在相同实验条件下有不同的保留行为,其保留时间的差异可以用来定性分析,每一组分的质量与相应色谱峰的积分面积成正比,因此可以公式计算,用归一化方法测定每一组分的质量百分含量。 1122100A is i i A A A s s ns n f A w f A f A f A =?++???+% 本实验是用气相色谱测定乙酸乙酯、乙酸丁酯及其混合试样,检测器用FID 。用色谱软件进行谱图处理和定量计算,让学生掌握用已知物对照定性、用归一化法测定混合物组分定量的实验。 混和试样的成功分离是气相色谱法定量分析的前提和基础,衡量一对色谱峰分 离的程度可用分离度:12121()2 R R t t R W W -=?+,式中1R t 、2R t 和1W 、2W 分别指两组分的保留时间和峰底宽度,R=1.5时两组分完全分离,实际中R=1.0(分离度98%)即可满足要求。 三、仪器与试剂 仪器:GC7890F 型气相色谱仪、氢火焰离子化检测器(FID )、氮气钢瓶、空气钢瓶、氢气发生器,微量注射器、3mm x 200cm 的10% SE-54不锈钢分离柱。GC5400型气相色谱仪、空气发生器、氮气发生器、氢气发生器,微量注射器、15m 毛细管分离柱。 试剂:乙酸乙酯、乙酸丁酯标准试样及其未知混合试样。 四、实验内容 1.按操作说明书使色谱仪正常运行,并调节至如下条件: 柱温:110C ? 检测器温度:120C ? 气化温度:120C ? 载气、氢气和空气流量分别为30、50和200mL/min 。 2.分别改变柱温至80、90、100、110、120C ?。每改变一次柱温,注入0.5L μ混合酯试样,记下保留时间,观察其出峰顺序和分离情况。
毛细管色谱法测定洛美沙星中的有机溶剂残留量 来源: 作者:王国成,陈莹,徐波 摘要:目的:建立毛细管气相色谱法测定洛美沙星中的有机溶剂残留量。方法: 用INNOWAX 毛细管气相色谱柱,FID检测器, 以22戊酮为内标进行测定。结果: 乙酸乙酯、四氢呋喃、乙醇、乙腈的线性范围分别为0~80μg/m l ( r =0.999 7)、0~11.52μg/ml( r=0.9996)、0~80μg/ml( r=0.9997) , 0~6.56 μg/ml( r= 0.9996);平均回收率分别为100.5%、100.1%、101.2%、100.1%; RSD 分别为1.30%、0.9%、1.18%和1.23% (n = 9)。结论: 本方法简单、准确、灵敏度高、重现性好, 适用于洛美沙星中有机溶剂残留量的测定。 关键词洛美沙星,毛细管气相色谱法,有机溶剂残留量 药物生产过程中残存的有机溶剂均有不同程度的毒性, 不仅对人体有害, 而且这些溶剂与药物的治疗作用无关, 原则上应愈少愈好。洛美沙星为第三代喹诺酮类广谱抗菌药, 是由日本北陆株式会社研制的第一代口服长效抗菌药。该药物在合成过程中采用了乙酸乙酯、四氢呋喃、乙醇、乙腈等有机溶剂, 故对此4种有机溶剂加以检测有利于药物质量控制。本试验采用毛细管色谱法测定洛美沙星原料药中有机溶剂的含量,方法简便,结果准确可靠。 1 仪器与试剂 Agilent6890 增强型气相色谱仪, Agilent6890 工作站。乙酸乙酯、四氢呋喃、乙醇、乙腈均为分析纯(上海化学试剂公司) , 22戊酮(内标物) 为色标试 剂(天津化学试剂一厂) , 1-甲基-2-吡咯烷酮, 溶解样品用溶剂为化学纯(上海化学试剂公司)。 2 方法与结果 2.1 色谱条件色谱柱: Agilent HP-NNOWAX (固定液为键合聚乙二醇, 30m×0.53mm,1.0μm) 毛细管柱; 气化室温度: 220℃; 程序升温: 起始温度为40℃, 保持10min, 然后以20℃/min 升温至220℃,保持4 min;载气为氮气;分流比:1∶1; 进样量2μl; 检测器温度: 氢焰离子化检测器(FID) , 240 ℃。 2.2 溶液及试样制备 2. 2. 1 内标溶液的制备精密量取色标试剂2-戊酮124.0μl (约相当于100mg) , 置100ml 量瓶中, 用1-甲基-2-吡咯烷酮稀释至刻度, 摇匀; 精密量取1m l, 置10ml容量瓶中, 用1-甲基-2-吡咯烷酮稀释至刻度, 摇匀, 作为内标溶液。 2. 2. 2 对照溶液的制备精密量取乙酸乙酯111. 1μl (约相当于100mg) , 乙醇126.6μl(约相当于100mg) 置100ml 量瓶中, 用1-甲基-2-吡咯烷酮稀释至刻度,摇匀,作为对照贮备液A; 精密量取四氢呋喃162.2μl(约相当于144m g) , 乙腈105.0μl (约相当于82mg) 置100ml量瓶中, 用1-甲基-2-吡咯烷酮稀释至刻度, 摇匀, 作为对照贮备液B。精密量取对照贮备液A 10ml 与对照贮备液B 1ml 置同一100ml 量瓶中, 用1-甲基-2-吡咯烷酮稀释至刻度, 摇匀, 作为对照贮备液。精密量取对照贮备液5ml, 置10ml 量瓶中,精密加入内标溶液 1ml, 加1-甲基-2-吡咯烷酮稀释至刻度, 摇匀, 即得对照溶液。 2. 2. 3 供试品溶液的配制取本品约0.1g, 精密称定, 置10ml 量瓶中, 精密加入内标溶液1ml, 加1-甲基-2-吡咯烷酮适量, 振摇使溶解并稀释至刻度, 摇匀,作为供试品溶液。 2.3 系统适用性试验精密量取对照品溶液2μl, 注入气相色谱仪, 记
?毛细管色谱柱的选择 毛细管色谱柱的选择和使用。 1 毛细管色谱柱柱型规格与操作条件的选择依据 目前, 市面上的商品毛细管柱品种、规格、牌号繁多, 这给使用毛细管柱的分析人员带来一定困难。其实各供给商产品相互可以替代, 不一定要追求某一特定的商品牌号。在选择毛细管柱时只需考虑以下四个因素: 固定相、内径、膜厚及柱长。 1.1 固定相 由于毛细管柱具有高分离能力, 在大多数应用在大多数应用方面, 有限数目的固定相就能替代几百种在填充柱中使用的固定相。目前可选择近200种固定液, 但是使用最广泛的仍是侧链为甲基或被其它基团取代的聚硅氧烷类, 如SE-30,OV-1,OV-101,DC-200等。固定相的选择依样品性质而定,一般有如下原则。①相似相容原则:从极性、化学官能团和主要差别来考虑。对非极性组分一般选非极性固定液,如麦氏常数(P)为0或+1;中等极性组分选P为+2或+3;强极性组分选P为+4或+5。官能团相同时相互作用力强,选择性高。另外,当组分主要差别为沸点时,选非极性固定液;当主要差别为极性时选极性固定液。②麦氏常数法:对照组分结构,比较固定液的相常数,值越大则对分离越有利。 通常, 固定相热稳定性随着其极性增加而降低,色谱工作者必须非凡注重固定相热稳定性极限。这些固定相在室温下粘度不同, 又会影响
它们的最低使用温度,例如:OV-1(胶状)的最低使用温度为50℃,OV-101(粘液)的最低使用温度为-50℃。因此,在室温分离低沸点化合物OV-101较OV-1要好。 1.2 内径选择 内径直接影响柱效、保留特性和样品容量。 ID 0.25mm: 适合于复杂多组份试样分析,高分离工作的常规柱,与大口径毛细管柱比,负荷量低,柱流失较小,必须采用分流或无分流进样, 适合与质谱等高灵敏检测器联用。 ID 0.32mm: 柱效稍低于ID 0.25mm 常规柱,负荷量大于常规柱的60%。可采用柱头进样,分流/不分流进样,有些情况下还可使用直接进样。 ID 0.53mm: 这种使用广泛的毛细管柱可以替代大部分填充柱,具有近似填充柱的负荷量, 与小孔径毛细管柱相比使用更简便,可采用直接进样技术。 1.3 固定液膜厚 液膜厚度影响柱子的保留特性和柱容量,随着膜厚的增加,对两个流出时间相近的化合物的分离越好,但是固定相的温度上限降低,柱流失增加。因此,毛细管柱液膜厚度(df)是一个重要的柱参数。 对于低挥发性高沸物或热敏化合物,往往选用薄液膜柱(df:0.25~0.5μm),主要用于在300℃以下流出的大部分样品。对于流出温度高于300℃的高分子量的化合物来说,选用df0.1μm 薄液膜柱是理想的,而且通常选择较短的柱子(10~15m)。对于质谱等高灵敏度检测