
生物物理学报第十七卷第一期二★★一年三月
ACTA BIOPHYSICA SINICA Vol.17No.1Mar.2001
生物信息学在基因芯片中的应用
孙啸,王晔,何农跃,赵雨杰,陆祖宏
(东南大学分子与生物分子电子学实验室,江苏南京210096)
摘要:生物信息学和基因芯片是生命科学研究领域中的两种新方法和新技术,生物信息学与基因芯片密切相关,生物信息学促进了基因芯片的研究与应用,而基因芯片则丰富了生物信息学的
研究内容。本论文探讨生物信息学在基因芯片中的应用,将生物信息学方法运用到高密度基因芯片
设计和芯片实验数据管理及分析。从信息学的角度提出基因芯片设计准则,提出寡核苷酸探针的优
化设计方法,将该方法运用于再测序型芯片和基因表达型芯片的设计,在此基础上研制出高密度基
因芯片设计软件系统和实验结果分析系统。
关键词:生物信息学;基因芯片;DNA阵列;探针设计
中图分类号:Q617文献标识码:A文章编号:1000-6737(2001)01-0027-08
1引言
生命科学与信息科学是目前发展最为迅速的两大领域,作为这两大学科交叉的产物之一,生物信息学在基因组学研究中已发挥巨大的作用,而凝结这两大学科研究成果的另一项崭新技术-基因芯片,已成为大规模提取和探索生物分子信息的强有力手段,将在后基因组研究中发挥突出的作用。
基因芯片(g ene chi p),又称DNA微阵列(microa rra y),是由大量DNA或寡核苷酸探针密集排列所形成的探针阵列,其基本原理是通过杂交检测信息。基因芯片是分子生物学和微电子学及信息学相互结合所形成的新型技术,利用基因芯片,可以实现基因信息的大规模检测[1-3]。这一技术的成熟和应用将在新世纪里给遗传研究、疾病诊断和治疗、新药发现和环境保护等生命科学相关领域带来一场革命。基因芯片应用主要分为两大类,一是用于研究基因型[4],一是用于分析基因表达[5,6]。从本质上来讲,前者实际上是利用基因芯片进行序列分析,其中包括识别DNA序列的突变和研究基因的多态性;而后者则是利用基因芯片研究基因的功能。
随着基因芯片需求和应用的不断增长,基因芯片及其相关的研究内容将会越来越丰富,基因芯片的应用已产生或即将产生大量的生物分子信息。生物信息学是分析处理生物分子信息、揭示生物分子信息内涵的一种技术[7,8],它在基因芯片研究与应用中起着重要的作用[9]。从确定基因芯片检测对象到基因芯片设计,从芯片检测结果分析到实验数据管理和信息挖掘,无不需要生物信息学的支持和帮助。通过合理的芯片设计和芯片优化,可以提高基因芯片获取生物
收稿日期:2000-04-07
基金项目:国家自然科学基金资助项目(69971010)
作者简介:孙啸,1962年出生,副教授,博士,电话:(025)3792245,E-mail:x sun@https://www.doczj.com/doc/e35730785.html,.
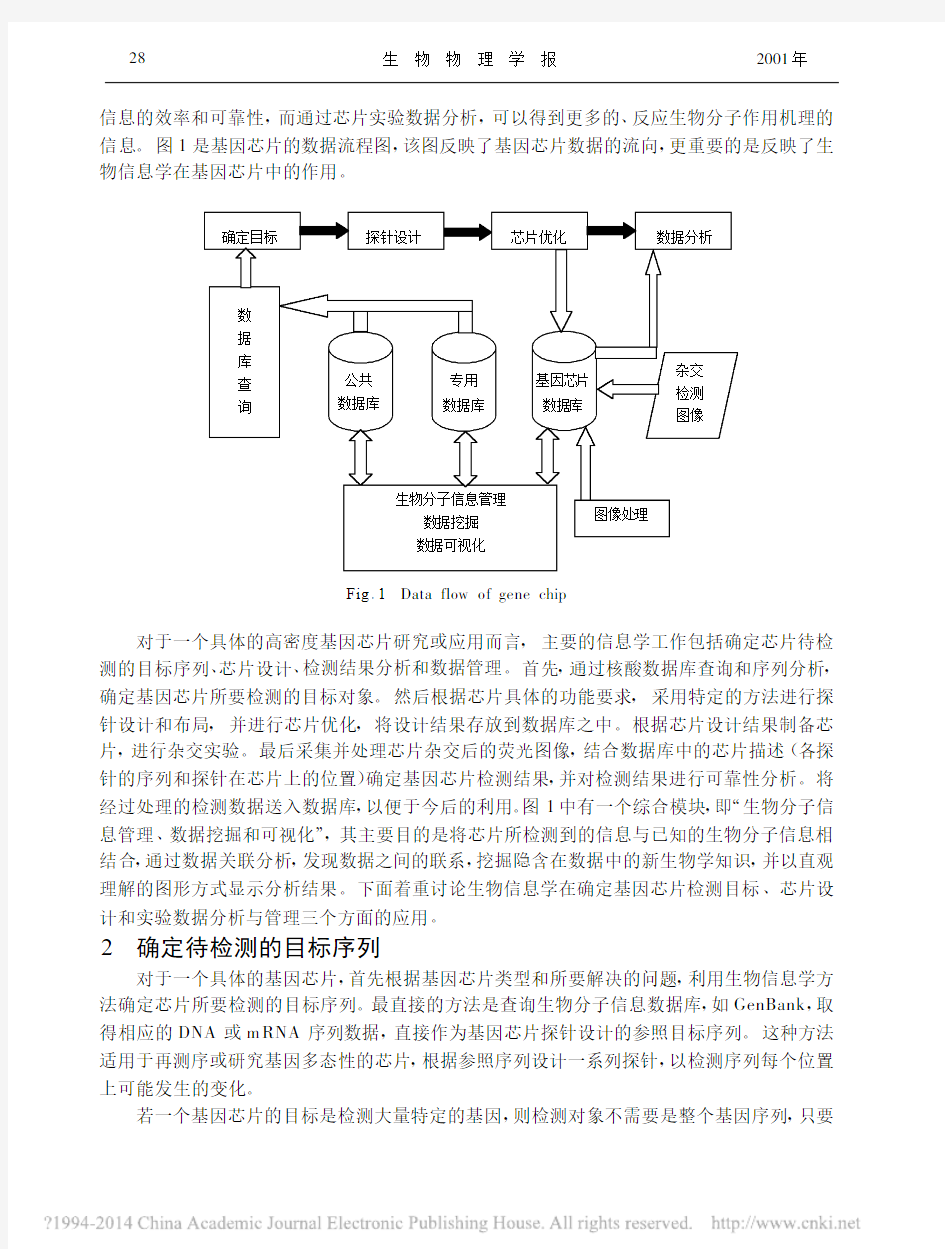
生物物理学报2001年28信息的效率和可靠性,而通过芯片实验数据分析,可以得到更多的、反应生物分子作用机理的信息。图1是基因芯片的数据流程图,该图反映了基因芯片数据的流向,更重要的是反映了生物信息学在基因芯片中的作用。
对于一个具体的高密度基因芯片研究或应用而言,主要的信息学工作包括确定芯片待检测的目标序列、芯片设计、检测结果分析和数据管理。首先,通过核酸数据库查询和序列分析,确定基因芯片所要检测的目标对象。然后根据芯片具体的功能要求,采用特定的方法进行探针设计和布局,并进行芯片优化,将设计结果存放到数据库之中。根据芯片设计结果制备芯片,进行杂交实验。最后采集并处理芯片杂交后的荧光图像,结合数据库中的芯片描述(各探针的序列和探针在芯片上的位置)确定基因芯片检测结果,并对检测结果进行可靠性分析。将经过处理的检测数据送入数据库,以便于今后的利用。图1中有一个综合模块,即“生物分子信息管理、数据挖掘和可视化”,其主要目的是将芯片所检测到的信息与已知的生物分子信息相结合,通过数据关联分析,发现数据之间的联系,挖掘隐含在数据中的新生物学知识,并以直观理解的图形方式显示分析结果。下面着重讨论生物信息学在确定基因芯片检测目标、芯片设计和实验数据分析与管理三个方面的应用。
2确定待检测的目标序列
对于一个具体的基因芯片,首先根据基因芯片类型和所要解决的问题,利用生物信息学方法确定芯片所要检测的目标序列。最直接的方法是查询生物分子信息数据库,如GenBank ,取得相应的DNA 或m RNA 序列数据,直接作为基因芯片探针设计的参照目标序列。这种方法适用于再测序或研究基因多态性的芯片,根据参照序列设计一系列探针,以检测序列每个位置上可能发生的变化。
若一个基因芯片的目标是检测大量特定的基因,则检测对象不需要是整个基因序列,只要
图像处理
专用数据库公共数据库杂交检测
图像
数据分析
确定目标探针设计数
据
库
查
询芯片优化基因芯片数据库生物分子信息管理
数据挖掘
数据可视化Fig .1Data flow of gene chip
29
生物信息学在基因芯片中的应用
第1期
检测能够代表该基因的一小段特征序列即可。所谓特征序列就是一段高度特异的序列,独一无二,它代表一个基因。从一个给定的序列中任选一段并不一定是特异的序列片段,因为序列之间可能存在着相似性,必须通过数据库的序列搜索比较,才能确定一段序列是否是特异的。在这种情况下,首先从核酸数据库中取得基因序列,然后通过序列比对分析,找出其特征序列,作为探针设计的参照序列。序列比对分析是生物信息学中最常用的方法,可以直接利用现有的软件。生物信息学中常用的核酸序列搜索比较算法是BLAS T。上述确定目标序列的方法多用于基因检测型芯片或基因表达型芯片。
提取特征片段需要用到序列的两两比较,与其相对应,在有些情况下需要利用多重序列比对技术[10]。以我们设计的检测乙型肝炎病毒(H BV)的基因芯片为例,由于致病HBV的基因组序列并不完全相同[11],临床上一般将其分为五种主要类型,各种亚型的基因组序列存在碱基插入、缺失和替换而导致的细微差别。利用多重序列比较可以发现HBV的S区相对保守,因此选择这段保守区作为芯片设计的参考序列。
在设计用于表达分析的基因芯片时,常用到表达序列标记EST。由于ES T是基因的子序列,首先必须知道众多的ES T序列究竟包含那些基因。这需要对ES T片段进行聚类分析,将相互重叠的序列归并成更长的序列[6],从而作为寡核苷酸探针设计的参考序列。
核酸序列数据库搜索及生物信息学公共服务器上的序列分析工具为确定基因芯片检测目标序列提供了帮助,通过数据库的搜索,还可以得到关于序列突变的信息及其它相关信息。
3高密度基因芯片设计
芯片设计的目的在于:通过设计,提取更多的生物分子信息,并提高信息的可靠性。高密度基因芯片设计包括寡核苷酸探针设计、探针布局和芯片优化。根据参照序列设计探针,尽可能使最终芯片的荧光检测图像中完全互补杂交信号突出,提高基因芯片检测的可靠性。芯片优化是指在设计后续阶段对芯片制备过程进行优化,如减少制备芯片所需要的掩膜板,精简探针合成环节。在芯片设计的不同阶段,都要用到信息学中的优化方法,如探针优化、布局优化及芯片优化。
各种基因芯片的功能不同,相应的芯片设计要求和设计方法也有所不同,必须根据具体的芯片功能采用不同的设计方法。根据我们的经验,从信息学角度提出以下设计准则:
(1)互补性:探针与待检测的目标序列片段互补;
(2)敏感性和特异性;既有较高的敏感性,也有较高的特异性,要求探针仅对特定目标序列片段敏感,而对其他序列不产生杂交信号;
(3)容错性:通过探针设计,提高基因芯片检测的容错性,常用的方法是使用冗余探针;
(4)可靠性:通过探针设计,提高基因芯片检测的可靠性;
(5)可控性:在基因芯片上设置质量监控探针,以便于监控基因芯片产品的质量;
(6)可读性:通过探针布局,使得最终的杂交检测图像便于观察理解,如将检测相关基因的探针放在芯片上相邻的区域。
在探针设计方面,为了提高芯片杂交错配辨别能力,我们提出一种优化设计方法。该方法的基本思想是通过动态调节各个探针的长度及探针之间的覆盖长度,使所设计的各个探针的解链温度T m最大程度地保持一致,从而有效地提高对碱基杂交错配的辨别能力,提高基因芯
生物物理学报2001年30片检测结果的可靠性。我们采用生物信息学中常用的动态规划算法进行优化,以各探针具有相近解链温度作为优化目标,筛选并优化组合各候选探针。在优化组合时要求各探针的长度和相邻探针之间的交叠长度满足给定的约束条件,经过优化组合以后得到一组覆盖目标序列的探针。
对于高密度基因芯片,往往需要同时检测多个目标序列。首先根据前面介绍的基本方法设计检测第一个目标序列的探针集合S 1,然后顺序处理其它目标序列,依此得到对应的探针集合S i 。在处理第i +1(i =1,2,…,n -1,n 是目标序列个数)个目标序列时,已经得到S 1、S 2、…、S i ,计算S 1、S 2、…和S i 中所有探针T m 的平均值T avg 。这样在对第i +1个目标序列进行探针优化设计时,以各探针的T m 值与T avg 的均方差作为动态规划算法的代价,经过优化设计以后得到探针集合S i +1。该过程一直进行到求出S n 为止。最后,S 1、S 2、…、S n 就是所要设计的探针集合,将各集合中的探针按一定规律分区排布在基因芯片上。
对于再测序或进行基因多态性研究的芯片,需要筛选可能的碱基变化。首先根据目标序列进行探针优化设计,得到一系列解链温度相近的野生型探针,这些探针用于检测正常的目标序列。计算这些探针T m 的平均值,以T av g 表示,作为设计其它检测单碱基变异(替换)探针的T m 参照值。然后从目标序列的前端开始,针对每个点,设计检测对应点A 、G 、C 、T 变异的四个探针,每个探针检测的变异点位于探针的中心,并使各探针的T m 值与T avg 接近。在探针布局时,将野生型探针按非连续方式排放在第一行,即Tar g et 行,然后将检测各点变异的探针依次连
A A T C G A C
T arget -lane
A -lane
G -lane
C -lane
T -lane Target
T C C G T T A G C T G A C T G C
(a )
Variation T ※G
(b )Tar g et
T C C G T T A G C T G A C T G C
Target -lane
A -lan e
G -lan e
C -lane
T -lane
Fi g .2(a )La y out of the chi p f or rese q uencin g .(b )H y bridization p at tern
31
生物信息学在基因芯片中的应用
第1期
续地放在A行、G行、C行和T行,如图2(a)所示。其实Targe t行的探针包含于A、G、C、T各行,但在示意图中列出Target行是为了说明用我们的方法所设计的野生型探针之间的覆盖长度为可变的,这类探针在芯片上对应于目标序列的排列是非连续的,而检测碱基变异的探针排列是连续的。图2(b)为基因芯片杂交后荧光检测图像模式示意图。假设在目标序列的第6位置上有一个T到G的变异,则在芯片的G行的第6个位置上有一个强的荧光信号(深黑色表示),而在第6列附近其他位置上由于存在一个碱基失配,故荧光信号相对较弱(浅灰色表示)。
上述方法仅用一个探针检测单碱基变异。若要可靠地检测目标序列上特定的位置是否有变异,则可以设计四组探针,每一组探针检测一种突变。用一组探针检测一种突变,可提高芯片检测的容错性。设已知目标序列中一个变异点位置k,以k为中心,在目标序列中取一定长度(25-39)的片段。对于这个片段,将其中心的核苷酸分别替换为A、G、C、T,由此得到四个不同的序列片段。将这四个片段看成是四个目标子序列,利用前面所提出的多目标优化探针设计方法,得到四组探针,所有各组探针的杂交解链温度相近。
除了单碱基替换之外,还可以设计检测碱基插入或缺失的探针。H acia在最近的一篇文献中综述了再测序型芯片的设计[12],他所在的研究小组还报道了用基因芯片对种属内和种属之间遗传变异进行快速比较序列分析的结果[13]。
对于表达分析型基因芯片,一般是设计多组探针以监控多个基因的表达水平,并且使同一块芯片所能监控的基因越多越好。要求探针之间相互独立,尽可能不重叠或少重叠,以提高探针的特异性[14]。设计的关键是探针冗余,用不同的探针检测同一个目标序列的不同区域,这提高了信号噪声比,同时也提高了定量检测目标序列的精确程度。另一种冗余来自于错配检测探针,所谓错配检测探针与正常探针基本相同,仅仅是探针的中心位置有一个碱基替换,利用这种探针用来辨别完全匹配与非完全匹配。
基因芯片设计的结果是形成芯片合成方案和步骤,产生制作掩膜板的方案。为提高基因芯片制作效率,需要对芯片设计结果进行优化。通过优化减少制作芯片所需的掩膜板个数,减少芯片上寡核苷酸探针的循环合成次数。
我们已经开发出一个高密度基因芯片设计的软件系统H GD V1.0,对于给定的参考序列,该系统可以根据要求采用不同的方法(包括优化方法)进行探针设计,并进行探针布局和芯片优化,产生芯片描述和芯片制备文件。
4基因芯片检测结果分析及数据管理
基因芯片与经过荧光标记的样品杂交以后,产生荧光图像。用图像扫描仪器捕获芯片上的荧光图像,并对图像进行处理,去除图像上的污点以及其它形式的噪声。由于芯片制备的原因,每个芯片单元的大小和位置可能会发生变化,这影响解释芯片图像。自动对准是芯片图像处理中的一个关键问题,需要用图像分割技术来解决该问题。
分析经过处理的基因芯片荧光图像,根据芯片的功能给出检测结果。如果芯片检测的目的是测定样本序列,则需要根据芯片上每个探针的杂交结果判断样本中是否含有对应的互补序列片段,并利用生物信息学中的片段组装算法连接各个片段,形成更长的目标序列。片段组装算法有基于片段覆盖图的贪婪算法和非循环子图方法[15]。如果芯片检测的目的是进行序列变
32
生物物理学报2001年
异分析,则要根据全匹配探针以及错配探针在基因芯片对应位置上的荧光信号强度,给出序列变化的位点,并指明发生什么变化。如果芯片检测的目的是进行基因表达分析,则需要给出芯片上各个基因的表达图谱,定量描述基因的表达水平。进一步的分析还包括根据基因表达模式进行聚类,寻找基因之间的相关性,发现协同工作的基因。
我们目前已经研制出一个基因芯片荧光图像处理和实验结果分析系统DAGC V1.0,该系统能够较好地去除荧光图像上的噪声,准确地进行芯片单元定位,能够完成序列片段组装,检测样品序列中出现的变异,并用层次式聚类方法和自组织映射神经网络实现对基因表达模式的聚类分析。
基因芯片是一个非常复杂的系统,包括许多环节,由于目前技术上的限制,在基因芯片制备、杂交及检测等方面都可能出现误差,芯片检测结果并非100%可靠。因此,必须对芯片检测结果作出可靠性的评价。可靠性分析可以从两个方面进行,一是根据实验统计误差(如探针合成的错误率、全匹配探针与错配探针的误识率等),分析基因芯片最终实验结果的可靠性。二是对基因芯片与样本序列杂交过程进行分子动力学研究,建立芯片杂交过程的计算机仿真实验模型,以便在制作芯片之前分析所设计芯片的性能,预测芯片实验结果的可靠性。
基因芯片实验将产生大量的数据,有效地管理这些数据是生物信息学所面临的一个挑战,数据管理的目的是为了更好地利用和共享数据。基因芯片产生的数据主要是基因表达数据,目前在国际互连网上有许多公共基因表达数据库,如欧洲生物信息学研究所EBI建立的A r-ra y Ex p ress数据库。基因组信息是相互关联的,不仅要建立基因芯片数据库,还与其它生物分子数据库、分析工具集成在一起,建立交叉索引,使基因芯片数据成为更有价值的生物学资源。在此基础上,引入数据挖掘技术,进行深层次的数据分析,从大量的基因芯片实验数据及其他相关实验数据中提取隐含的生物学信息,并上升为生物学知识。
5基因表达数据分析的最新进展
基因表达数据分析是目前生物信息学研究的热点和重点。在以往生物信息学数据分析处理中,一次数据处理的对象往往是单个或几个生物分子,而现在一块基因芯片就可以产生上千个基因的表达数据,数据处理量大幅度增加,数据之间的关系也格外复杂。对基因表达数据,在大规模数据集上进行分析、归纳,可以了解基因表达的时空规律,探索基因表达的代谢控制,了解基因的功能,理解遗传网络,提供疾病发病机理的信息[16]。研究基因表达数据的处理和分析方法已成为生物信息学发展的一个重要方向。
目前对基因表达数据的处理主要是进行聚类分析,将表达规律相似的基因聚为一类,在此基础上寻找相关基因,分析基因的功能。所用方法有相关分析方法,模式识别技术中的层次式聚类方法,人工智能中的自组织映射神经网络[17-19]。此外还有主元分析方法,利用主元分析可以在多维数据集合中确定关键变量的特点,分析在不同条件下基因响应的规律和特征[20]。进一步的分析还可以探索基因的转录调节网络[21],发现基因在环境或药物作用下表达模式的变化,阐明一些基因对另一些基因的调节作用[22]。利用聚类分析的结果可以研究基因的启动子,分析表达模式相同的一类基因的启动子组成特性,通过多重序列比对操作,在各个基因序列的上游区域寻找共同的启动子。虽然聚类方法是基因表达数据分析的基础,但是目前这类方法只能找出基因之间简单的、线性的关系[23],需要发展新的分析方法以发现基因之间复杂的、非
33
生物信息学在基因芯片中的应用
第1期
线性的关系。
最近国际上在基因调控网络分析方面出现了许多有意义的工作,建立起一些基因调控网络的数学模型,如布尔网络模型、线性关系网络模型、微分方程模型、互信息相关网络模型等,在此基础研究基因调控网络的动力学性质。最近几届太平洋地区生物计算会议上有许多这方面的文章,可在其电子版论文集中查看(http://psb.stanfo https://www.doczj.com/doc/e35730785.html,/)。
生物信息学在基因芯片研究和产业化中起着至关重要的作用,从更广的范围来看,基因芯片的应用和发展应该紧密地与信息技术相结合。我们需要加强研究和开发基因芯片设计软件及相关数据分析软件,并根据研究和应用需要,设计出有自主知识产权的、有特色的和市场认可的基因芯片。
参考文献:
[1]Ram say G.DN A chips:S tate-of-the art[J].Natu re B iotechno logy,1998,16:40-44.
[2]Ch ee M,Yang R,H ubbel,et al.Accessing g enetic in forma tion w ith h ig h-d ensity D NA arrays[J].
Science,1996,274:610-613.
[3]Marshall A,Hod g so n J.DNA ch i p s:A n arra y o f p ossibilities[J].Natu re B iotechno lo gy,1998,16:27-31.
[4]Lindblad-Toh K,et https://www.doczj.com/doc/e35730785.html,rge-sca le dis covery a nd g enotyping o f sing le-nucleotide
poly morp hisms in the mou s e[J].Nature Genetics,2000,24:381-386.
[5]Duggan DJ,et al.E xp res sion p rof iling using cD NA microarrays[J].Nature Genetics supp lement,
1999,21:10-14.
[6]Bow tell DDL.O p tions ava ilable-f rom start to f in is h-f or o btain in g ex p ression d ata b y m icroarra y[J].
Natu re Genetics sup plement,1999,21:25-32.
[7]陈润生.生物信息学[J].生物物理学报,1999,15(1):5-12.
[8]孙啸.生物信息学-揭示生物分子数据的内涵[J].电子科技导报,1998,11,5-9.
[9]Dou g las EB,M ichael BE,Mask SB.Gene ex p ression in f orm atics-it's all in y ou r mine[J].
Natu re Genetics su p p lement,1999,21:51-55.
[10]Hertz GZ,S to rm o GD.Iden tifying DN A an d p rotein p atterns w ith statistica lly
sig nif ica nt alig n men ts of m ultip le s equences[J].Bioin forma tics,1999,15:563-577.
[11]姚桢.分子乙型肝炎病毒相关病学.北京:中国医药科技出版社,1998,1-33.
[12]Hacia JG.Rese q uencin g an d mu ta tiona l an al y sis u sin g oli g onu cleotide m icro a rra y s[J].
Natu re g enetics su p p lem en t,1999,21:42-47.
[13]Hacia JG,et al.Determina tion of a ncestral alleles for h u man sin glen ucleotid e polymorphisms
using high-d en sity oligonu cleotide arrays[J].Na tu re Genetics,1999,22:164-167.
[14]Lipshu tz R,Fo dor S,Gingeras T,et al.Hig h density s yn thetic oligon ucleotide arrays[J].
Natu re Genetics su p p lement,1999,21:20-24.
[15]Dietz D.I ntrod uction to com p u ta tiona l mo lecular biolo https://www.doczj.com/doc/e35730785.html, A:PWS P ublishin g C o m p a n y,1997,105-138.
[16]Derisi JL,Iyer V R,B row n PO.E xp lorin g the Metab o lic an d Genetic Contro l of Gene
E xpression on a Genomic S ca le[J].S cience,1997,278:680-686.
[17]T am ay o P,et al.In terp retin g pa ttern s o f gene expression with s elf-organ izing map s:
Methods a nd a pp lica tion to hem ato p oietic d i ff erentia tion.In:Lan der ES.Proc.Natl.Aca d.
Sci.,https://www.doczj.com/doc/e35730785.html,A:Na tional A cadem y o f Sciences,1999,2907-2912.
[18]Mich ael BE,et al.Cluster ana lysis and d isp lay of genome-w id e expression p atterns.In:
B otstein D.P ro c.Natl.A cad.S ci.,https://www.doczj.com/doc/e35730785.html, A:Na tiona l Academy o f Sciences,1998,14863-4868.
34
生物物理学报2001年
[19]Hill A,et al.Genom ic An alysis of Gene E xp ression in C.E leg ans[J].S cience,2000,290:809-812.
[20]Raychaudh uri S,Stuart JM,Altman RB.Princi p a l Com p onen ts A na l y sis to S um ma rize
Microarra y E x p erim en ts:A pp lication to S p oru la tion Time S eries.In:Paci f ic S y m p osium
on B iocom p u tin g,2000,5:452-463.
[21]T avazoie S,et al.System atic determ ination of genetic network arch itecture[J].
Na ture Genetics,1999,22:281-285.
[22]Coller HA,et al.Ex p res sion an al y sis w ith oli g on ucleotid e m icroarra y s revea ls tha t MY C
re g ulates g enes in volved in g row th,cell c y cle,si g n alin g,a nd adhesion.In:Eis enm an RN.
P roc.Natl.A cad.Sci.,https://www.doczj.com/doc/e35730785.html,A:Nation al Academ y o f S cien ces,2000,3260-3265.
[23]Bittner M,M eltzer P,T rent J.Da ta ana lysis a nd integration:of step s a nd arrows[J].
Na ture Genetics,1999,22:213-215.
APPLICATION OF BIOINFORMATICS TO GENE CHIP
SUN Xiao,WANG Ye,HE Non g-y ao,ZHAO Yu-j ie,LU Zu-hon g
(Lab.of Molecular and Biomolecular Electronics,Southeast U niversity,Nanjing,210096,China)
Abstract:Bioinform atics and gene chip are tw o new techno logies in the field of Life S cience.Bioinfo rmatics is ver y im p ortant to the research and the a pp lication of g ene chi p.It p la y s a ke y role in g ene chi p desi g n,ex p erime nt data anal y sis and data m anagement.The applicatio ns of Bioinform atics to g ene chip are discussed in this pa-p e r in g reat detail.Some rules fo r desi g nin g g ene chi p are p ro p osed.A new a pp roach for g ene chi p desi g n,b y w hich an o p tim al set of oli g onucleo tide p robes can be g ener-ated using dy nam ic prog ramming,is also proposed.T he probes have nearly the same melting tem perature Tm,and are of va riable leng th and variable o verlap.T he metho d is a pp lied to desi g nin g g ene chi p s for DNA rese q uencin g,mutatio nal anal y sis and g ene ex p ression anal y sis.Tw o softw are s y stem s,o ne fo r g ene chi p desi g n and o ne for data analy sis have been developed.
Ke y Words:Bioinfo rm atics;Gene chi p;DNA arra y;Probe desi g n
生物芯片研究进展 摘要 生物芯片是切采用生物技术制备或应用于生物技术的微处理器是便携式生物化学分析器的核心技术。通过对微加工获得的微米结构作生物化学处理能使成千上万个与生命相关的信息集成在一块厘米见方的芯片上。生物芯片发展的最终目标是将从样品制备、化学反应到检测的整个生化分析过程集成化以获得所谓的微型全分析系统或称缩微芯片实验室。生物芯片技术的出现将会给生命科学、医学、化学、新药开发、生物武器战争、司法鉴定、食品和环境卫生监督等领域带来一场革命。本文主要阐述了生物芯片技术种类和应用方面的近期研究进展。 关键词 生物芯片,疾病诊断,研究运用,基因表达 基因芯片的种类 基因芯片产生的基础则是分子生物学、微电子技术、高分子化学合成技术、激光技术和计算机科学的发展及其有机结合。根据基因芯片制造过程中主要技术的区别,下面主要介绍四类基因芯片。 一、光引导原位合成技术生产寡聚核苷酸微阵列 开发并掌握这一技术的是Affymetrix公司,Affymetrix采用了照相平板印刷技术技术结合光引导原位寡聚核苷酸合成技术制作DNA芯片,生产过程同电子芯片的生产过程十分相似。采用这种技术生产的基因芯片可以达到1×106/cm2的微探针排列密度,能够在一片1厘米多见方的片基上排列几百万个寡聚核苷酸探针。 原位合成法主要为光引导聚合技术(Light-directed synthesis),它不仅可用于寡聚核苷酸的合成,也可用于合成寡肽分子。光引导聚合技术是照相平板印刷技术(photolithography)与传统的核酸、多肽固相合成技术相结合的产物。半导体技术中曾使用照相平板技术法在半导体硅片上制作微型电子线路。固相合成技术是当前多肽、核酸人工合成中普遍使用的方法,技术成熟且已实现自动化。二者的结合为合成高密度核酸探针及短肽列阵提供了一条快捷的途径。 Affymetrix公司已有诊断用基因芯片成品上市,根据用途可以分为三大类,分别为基因表达芯片、基因多态性分析芯片和疾病诊断芯片,基因表达分析芯片和基因多态性分析芯片主要用于研究机构和生物制药公司,可以用来寻找新基因、基因测序、疾病基因研究、基因制药研究、新药筛选等许多领域,Affymetrix公司主要生产通用寡聚核苷酸芯片;疾病诊断芯片则主要用于医学临床诊断,包括各种遗传病和肿瘤等,目前Affymetrix公司生产
2012年第35期生物信息学是利用计算机为工具,用数学及信息科学的理论和方法研究生命现象,对生物信息进行收集、加工、存储、检索和分析的科学。生物信息学的核心是基因组信息学,基因组学是研究生物基因组和如何利用基因的一门学问,该学科提供基因组信息以及相关数据系统,试图解决生物、医学和工业领域的重大问题。对于基因组学研究所产生的大量数据必须借助于先进的计算机技术收集和分析处理这些生物学信息,因此计算机科学为生物信息学的研究和应用提供了非常好的支撑。 1.序列比对 序列比对其意义是从核酸、氨基酸的层次来比较两个或两个以上符号序列的相似性或不相似性,进而推测其结构功能及进化上的联系。研究序列相似性的目的是通过相似的序列得到相似的结构或功能,也可以通过序列的相似性判别序列之间的同源性,推测序列之间的进化关系。序列比对是生物信息学的基础,非常重要。 序列比对中最基础的是双序列比对,双序列比较又分为全局序列比较和局部序列比较,这两种比较均可用动态程序设计方法有效解决。在实际应用中,某些在生物学上有重要意义的相似性不是仅仅分析单条序列,只能通过将多个序列对比排列起来才能识别。比如当面对许多不同生物但蛋白质功能相似时,我们可能想知道序列的哪些部分是相似的,哪些部分是不同的,进而分析蛋白质的结构和功能。为获得这些信息,我们需要对这些序列进行多序列比对。多重序列比对算法有动态规划算法、星形比对算法、树形比对算法、遗传算法、模拟退火算法、隐马尔可夫模型等,这些算法都可以通过计算机得以解决。 2.数据库搜索 随着人类基因组计划的实施,实验数据急剧增加,数据的标准化和检验成为信息处理的第一步工作,并在此基础上建立数据库,存储和管理基因组信息。这就需要借助计算机存储大量的生物学实验数据,通过对这些数据按一定功能分类整理,形成了数以百计的生物信息数据库,并要求有高效的程序对这些数据库进行查询,以此来满足生物学工作者的需要。数据库包括一级数据库和二级数据库,一级数据库直接来源于实验获得的原始数据,只经过简单的归类整理和注释;二级数据库是对基本数据进行分析、提炼加工后提取的有用信息。 分子生物学的三大核心数据库是GenBank 核酸序列数据库,SWISS-PROT 蛋白质序列数据库和PDB 生物大分子结构数据库,这三大数据库为全世界分子生物学和医学研究人员了解生物分子信息的组织和结构,破译基因组信息提供了必要的支撑。但是用传统的手工分析方法来处理数据显然已经无法跟上新时代的步伐,对于大量的实验结果必须利用计算机进行自动分析,以此来寻找数据之间存在的密切关系,并且用来解决实际中的问题。 3.基因组序列分析 基因组学研究的首要目标是获得人的整套遗传密码,要得到人的全部遗传密码就要把人的基因组打碎,测完每个小的序列后再把它们重新拼接起来。所以目前生物信息学的大量工作是针对基因组DNA 序列的,建立快速而又准确的DNA 序列分析方法对研究基因的结构和功能有非常重要的意义。对于基因组序列,人们比较关心的是从序 列中找到基因及其表达调控信息,比如对于未知基因,我们就可以通过把它与已知的基因序列进行比较,从而了解该基因相关的生理功能或者提供疾病发病机理的信息,从而为研发新药或对疾病的治疗提供一定的依据,使我们更全面地了解基因的结构,认识基因的功能。因此,如何让计算机有效地管理和运行海量的数据也是一个重要问题。 4.蛋白质结构预测 蛋白质是组成生物体的基本物质,几乎一切生命活动都要通过蛋白质的结构与功能体现出来,因此分析处理蛋白质数据也是相当重要的,蛋白质的生物功能由蛋白质的结构所决定,因此根据蛋白质序列预测蛋白质结构是很重要的问题,这就需要分析大量的数据,从中找出蛋白质序列和结构之间存在的关系与规律。 蛋白质结构预测分为二级结构预测和空间结构预测,在二级结构预测方面主要有以下几种不同的方法:①基于统计信息;②基于物理化学性质;③基于序列模式;④基于多层神经网络;⑤基于图论;⑥基于多元统计;⑦基于机器学习的专家规则;⑧最邻近算法。目前大多数二级结构预测的算法都是由序列比对算法BLAST 、FASTA 、CLUSTALW 产生的经过比对的序列进行二级结构预测。虽然二级结构的预测方法其准确率已经可以达到80%以上,但二级结构预测的准确性还有待提高。 在实际进行蛋白质二级结构预测时,往往会把结构实验结果、序列比对结果、蛋白质结构预测结果,还有各种预测方法结合起来,比较常用的是同时使用多个软件进行预测,把各个软件预测结果分析后得出比较接近实际的蛋白质二级结构。将序列比对与二级结构预测相结合也是一种常见的综合分析方法。 蛋白质二级结构指蛋白质多肽链本身的折叠和盘绕的方式。二级结构主要有α-螺旋、β-折叠、β-转角等几种形式,它们是构成蛋白质高级结构的基本要素,常见的二级结构有α-螺旋和β-折叠。三级结构是在二级结构的基础上进一步盘绕,折叠形成的。研究蛋白质空间结构的目标是为了了解蛋白质与三维结构的关系,预测蛋白质的二级结构预测只是预测蛋白质三维形状的第一步,蛋白质折叠问题是非常复杂的,这就导致了蛋白质的空间结构预测的复杂性。蛋白质三维结构预测方法有:同源模型化方法、线索化方法和从头预测的方法但是无论用哪一种方法,结果都是预测,采用不同的算法,可能产生不同的结果,因此还需要研究新的理论计算方法来预测蛋白质的三维结构。 图4.1蛋白质结构(下转第100页) 计算机在生物信息学中的应用 王帆刘帅 (长春工程学院计算机基础教学中心吉林 长春 130012) 【摘要】生物信息学是一门新兴的、正在迅速发展的交叉学科,它不仅对认识生物体的起源与进化研究有重要意义,而且还可以为人类诊断疾病及物种的改良提供一定的理论依据。生物研究过程中产生的海量数据又需要具有数据处理和分析能力的大容量、高性能的超级计算机的支持,因此计算机技术在生物信息学的研究中显得尤为重要,本文就简单介绍了计算机在生物信息学研究中的哪些方面起到了不可忽略的作用。 【关键词】生物信息学;计算机科学;基因组学 作者简介:王帆(1980—),男,长春人,毕业于长春理工大学,本科学历,信息与计算科学专业。 刘帅(1979—),女,长春人,东北师范大学硕士研究生,主要研究方向为计算机软件与理论 。 ◇高教论述◇
常用分子生物学软件简 介 公司内部编号:(GOOD-TMMT-MMUT-UUPTY-UUYY-DTTI-
常用分子生物学软件 一、基因芯片: 1、基因芯片综合分析软件。 ArrayVision 一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。Arraypro Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。 phoretix? Array Nonlinear Dynamics公司的基因片综合分析软件。 J-express 挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境后后,才能运行。 2、基因芯片阅读图像分析软件 ScanAlyze ,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。输出为分隔的文本格式,可很容易地转化为任何数据库。 3、基因芯片数据分析软件 Cluster
斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。 SAM Significance Analysis of Microarrays 的缩写,微矩阵显着性分析软件,EXCEL软件的插件,由Stanford大学编制。 4.基因芯片聚类图形显示 TreeView 斯坦福开发的用来显示Cluster软件分析的图形化结果。现已和Cluster成为了基因芯片处理的标准软件。 FreeView 是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。 5.基因芯片引物设计 Array Designer DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具 二、RNA二级结构。 RNA Structure RNA Sturcture 根据最小自由能原理,将Zuker的根据RNA一级序列预测RNA二级结构的算法在软件上实现。预测所用的热力学数据是最近由Turner实验室获得。提供了一些模块以扩展Zuker算法的能力,使之为一个界面友好的RNA折叠程序。允许你同时打开多个数据处理窗口。主窗口的工具条提供一些基本功能:打开文件、导入文件、关闭文件、设置程序参数、重排窗口、以及即时帮助和退
——古A.名词解释 1. 生物信息学:广义是指从事对基因组研究相关的生物信息的获取,加工,储存,分配,分析和解释。狭义是指综合应用信息科学,数学理论,方法和技术,管理、分析和利用生物分子数据的科学。 2. 基因芯片:将大量已知或未知序列的DNA片段点在固相载体上,通过物理吸附达到固定化(cDNA芯片),也可以在固相表面直接化学合成,得到寡聚核苷酸芯片。再将待研究的样品与芯片杂交,经过计算机扫描和数据处理,进行定性定量的分析。可以反映大量基因在不同组织或同一组织不同发育时期或不同生理条件下的表达调控情况。 3. NCBI:National Center for Biotechnology Information.是隶属于美国国立医学图书馆(NLM)的综合性数据库,提供生物信息学方面的研究和服务。 4. EMBL:European Molecular Biology Laboratory.EBI为其一部分,是综合性数据库,提供生物信息学方面的研究和服务。 5. 简并引物:PCR引物的某一碱基位置有多种可能的多种引物的混合体。 6. 序列比对:为确定两个或多个序列之间的相似性以至于同源性,而将它们按照一定的规律排列。
7. BLAST:Basic Local Alignment Search Tool.是通过比对(alignment)在数据库中寻找和查询序列(query)相似度很高的序列的工具。 8. ORF:Open Reading Frame.由起始密码子开始,到终止密码子结束可以翻译成蛋白质的核酸序列,一个未知的基因,理论上具有6个ORF。 9. 启动子:是RNA聚合酶识别、结合并开始转录所必须的一段DNA序列。原核生物启动子由上游调控元件和核心启动子组成,核心启动子包括-35区(Sextama box)TTGACA,-10区(Pribnow Box)TATAAT,以及+1区。真核生物启动子包括远上游序列和启动子基本元件构成,启动子基本元件包括启动子上游元件(GC岛,CAAT盒),核心启动子(TATA Box,+1区帽子位点)组成。 10. motif:模体,基序,是序列中局部的保守区域,或者是一组序列中共有的一小段序列模式。 11. 分子进化树:通过比较生物大分子序列的差异的数值重建的进化树。 12. 相似性:序列比对过程中用来描述检测序列和目标序列之间相似DNA碱基或氨基酸残基序列所占的比例。 13. 同源性:两个基因或蛋白质序列具有共同祖先的结论。
浅谈生物信息学在生物方面的应用 生物信息学(bioinformaLics)是以核酸和蛋白质等生物大分子数据库及其相关的图书、文献、资料为主要对象,以数学、信息学、计算机科学为主要手段,对浩如烟海的原始数据和原始资料进行存储、管理、注释、加工,使之成为具有明确生物意义的生物信息。并通过对生物信息的查询、搜索、比较、分析,从中获得基因的编码、凋控、遗传、突变等知识;研究核酸和蛋白质等生物大分子的结构、功能及其相互关系;研究它们在生物体内的物质代谢、能量转移、信息传导等生命活动中的作用机制。 从生物信息学研究的具体内容上看,生物信息学可以用于序列分类、相似性搜索、DNA 序列编码区识别、分子结构与功能预测、进化过程的构建等方面的计算工具已成为变态反应研究工作的重要组成部分。针对核酸序列的分析就是在核酸序列中寻找过敏原基因,找出基因的位置和功能位点的位置,以及标记已知的序列模式等过程。针对蛋白质序列的分析,可以预测出蛋白质的许多物理特性,包括等电点分子量、酶切特性、疏水性、电荷分布等以及蛋白质二级结构预测,三维结构预测等。 生物信息学中的主要方法有:序列比对,结构比对,蛋白质结构的预测,构造分子进化树,聚类等。基因芯片是基因表达谱数据的重要来源。目前生物信息学在基因芯片中的应用主要体现在三个方面。 1、确定芯片检测目标。利用生物信息学方法,查询生物分子信息数据库,取得相应的序列数据,通过序列比对,找出特征序列,作为芯片设计的参照序列。 2、芯片设计。主要包括两个方面,即探针的设计和探针在芯片上的布局,必须根据具体的芯片功能、芯片制备技术采用不同的设计方法。 3、实验数据管理与分析。对基因芯片杂交图像处理,给出实验结果,并运用生物信息学方法对实验进行可靠性分析,得到基因序列变异结果或基因表达分析结果。尽可能将实验结果及分析结果存放在数据库中,将基因芯片数据与公共数据库进行链接,利用数据挖掘方法,揭示各种数据之间的关系。 生物信息学在人类基因组计划中也具有重要的作用。 大规模测序是基因组研究的最基本任务,它的每一个环节都与信息分析紧密相关。目前,从测序仪的光密度采样与分析、碱基读出、载体标识与去除、拼接与组装、填补序列间隙,到重复序列标识、读框预测和基因标注的每一步都是紧密依赖基因组信息学的软件和数据库的。特别是拼接和填补序列间隙更需要把实验设计和信息分析时刻联系在一起.拼接与组装中的难点是处理重复序列,这在含有约30%重复序列的人类基因组中显得尤其突出。 人类基因组的工作草图即将完成,因此发现新基因就成了当务之急。使用基因组信息学的方法通过超大规模计算是发现新基因的重要手段,可以说大部分新基因是靠理论方法预测出来的。比如啤酒酵母完整基因组(约1300万bp)所包含6千多个基因,大约60%是通过信息分析得到的。 当人类基因找到之后,自然要解决的问题是:不同人种间基因有什么差别;正常人和病人基因又有什么差别。”这就是通常所说的SNPs(单核苷酸多态性)。构建SNPs及其相关数据库是基因组研究走向应用的重要步骤。1998年国际已开展了以EST为主发现新Spps 的研究。在我国开展中华民族SNPs研究也是至重要的。总之,生物信息学不仅将赋予人们各种基础研究的重要成果,也会带来巨大的经济效益和社会效益。在未来的几年中DNA 序列数据将以意想不到的速度增长,这更离不开利用生物信息学进行各类数据的分析和解释,研制有效利用和管理数据新工具。生物信息学在功能基因组学同样具有重要的应用目前应用最多的是同源序列比较、模式识别以及蛋白结构预测。所谓同源序列,是指从某一共同祖先经趋异进化而形成的不同序列。利用数据库搜索找出未知核酸或蛋白的同源序列,是序列分析的基础[lol。如利用BLASTn和BLASTx两种软件分别进行核苷酸和氨基
[生工0902] BLOSUM矩阵及其在生物 信息学中的使用 生物信息学 齐阳,汪锴,袁理 2011/11/25 什么是BLOSUM矩阵?BLOSUM矩阵有什么使用?
BLOSUM矩阵及其在生物信息学中的使用 齐阳汪锴袁理 摘要BLOSUM矩阵是一种蛋白质序列对比的算法,在生物信息学领域中被广泛使用。本文综述了BLOSUM矩阵的由来、如何构建BLOSUM矩阵和其打分规则、使用以及现代算法。并指出了BLOSUM矩阵的发展前景。 关键词BLOSUM矩阵;生物信息学;使用 0 引言 序列比对是现代生物学最基本的研究方法之一, 最常见的比对是蛋白质序列之间或核酸序列之间的两两比对,通过比较两个序列之间的相似区域和保守性位点,寻找二者可能的分子进化关系,进而可以有效地分析和预测一些新发现基因的功能。目前各种蛋白质序列对比算法主要利用一种替代矩阵来计算序列间的相似性,过去所普遍使用的Dayhoff矩阵只能用来进行相似度85%以上的序列对比「1」,为了满足大量生命科学研究的需求,1992年Henikoff夫妇从蛋白质模块数据库BLOCKS中找出一组替代矩阵,即BLOSUM系列,很好的解决了序列的远距离相关的问题,此后十几年来BLOSUM及其衍生替代矩阵已经成为蛋白质多序列对比的常用方法。 1BLOSUM矩阵概况 序列比对是现代生物学最基本的研究方法之一,常见的比对是蛋白质序列之间或核酸序列之间的两两比对,通过比较两个序列之间的相似区域和保守性位点,寻找二者可能的分子进化关系,进而可以有效地分析和预测一些新发现基因的功能。在比对两个序列时,不仅要考虑完全匹配的字符,还要考虑一个序列中的空格或间隙(或者,相反地,要考虑另一个序列中的插入部分)和不匹配,这两个方面都可能意味着突变「2」。在序列比对中,需要找到最优的比对即将匹配的数量最大化,将空格和不匹配的数量最小化。为了确定最优的比对,必须为每个比对进行评估和打分,于是引入了打分函数「3」。
1.DNA:遗传物质(遗传信息的载体) 双螺旋结构,A,C,G,T四种基本字符的复杂文本 2.基因(Gene):具有遗传效应的DNA分子片段 3.基因组(Genome):包含细胞或生物体全套的遗传信息的全部遗传物质。人类包括细胞核基因组和线粒体基因组 OR一个物种中所有基因的整体组成 4.人类基因组:3.0×109bp模式生物 5.HGP的最初目标通过国际合作,用15年时间(1990~2005)至少投入30亿美元,构建详细的人类基因组遗传图和物理图,确定人类DNA的全部核苷酸序列,定位约10万基因,并对其它生物进行类似研究。 6.HGP的终极目标 阐明人类基因组全部DNA序列; 识别基因; 建立储存这些信息的数据库; 开发数据分析工具; 研究HGP实施所带来的伦理、法律和社会问题。 7.遗传图谱(genetic map)又称连锁图谱(linkage map),它是以具有遗传多态性(在一个遗传位点上具有一个以上的等位基因,在群体中的出现频率皆高于1%)的遗传标记为“路标”,以遗传学距离(在减数分裂事件中两个位点之间进行交换、重组的百分率,1%的重组率称为1cM)为图距的基因组图。 遗传图谱的建立为基因识别和完成基因定位创造了条件。 8.遗传连锁图:通过计算连锁的遗传标志之间的重组频率,确定它们的相对距离,一般用厘摩(cM,即每次减数分裂的重组频率为1%)表示。 9.物理图谱(physical map)是指有关构成基因组的全部基因的排列和间距的信息,它是通过对构成基因组的DNA分子进行测定而绘制的。绘制物理图谱的目的是把有关基因的遗传信息及其在每条染色体上的相对位置线性而系统地排列出来。 10.转录图谱是在识别基因组所包含的蛋白质编码序列的基础上绘制的结合有关基因序列、位置及表达模式等信息的图谱。 11.序列图谱:随着遗传图谱和物理图谱的完成,测序就成为重中之重的工作。 DNA序列分析技术是一个包括制备DNA片段化及碱基分析、DNA信息翻译的多阶段的过程。通过测序得到基因组的序列图谱 12.大规模测序基本策略 逐个克隆法:对连续克隆系中排定的BAC克隆逐个进行亚克隆测序并进行组装(公共领域测序计划) 全基因组鸟枪法:在一定作图信息基础上,绕过大片段连续克隆系的构建而直接将基因组分解成小片段随机测序,利用超级计算机进行组装(美国Celera公司) 13.基因识别(gene identification)是HGP的重要内容之一,其目的是识别全部人类的基因。 基因识别包括: 识别基因组编码区 识别基因结构 基因识别目前常采用的有二种方法: 从基因组序列中识别那些转录表达的DNA片段 从cDNA文库中挑取并克隆。 14.基因组多态性(Polymorphism):是指在一个生物群体中,同时和经常存在两种或多种不连续的变异型或基因型(genotype)或等位基因(allele),亦称遗传多态性(genetic
分子生物学考试重点 一、名词解释 1、分子生物学(molecular biology):分子生物学是研究核酸、蛋白质等所有生物大分子的形态、结构特征及其重要性、规律性和相互关系的科学。 2、C值(C value):一种生物单倍体基因组DNA的总量。在真核生物中,C值一般是随生物进化而增加的,高等生物的C值一般大于低等生物。 3、DNA多态性(DNA polymorphism):DNA多态性是指DNA序列中发生变异而导致的个体间核苷酸序列的差异。 4、端粒(telomere):端粒是真核生物线性基因组DNA末端的一种特殊结构,它是一段DNA序列和蛋白质形成的复合体。 5、半保留复制(semi-conservative replication):DNA在复制过程中碱基间的氢键首先断裂,双螺旋解旋并被分开,每条链分别作为模板合成新链,产生互补的两条链。这样形成的两个DNA分子与原来DNA分子的碱基顺序完全一样。一次,每个子代分子的一条链来自亲代DNA,另一条链则是新合成的,所以这种复制方式被称为DNA 的半保留复制。 6、复制子(replicon):复制子是指生物体的复制单位。一个复制子只含一个复制起点。 7、半不连续复制(semi-discontinuous replication):DNA复制过程中,一条链的合成是连续的,另一条链的合成是中断的、不连续的,因此
称为半不连续复制。 8、前导链(leading strand):与复制叉移动的方向一致,通过连续的5ˊ-3ˊ聚合合成的新的DNA链。 9、后随链(lagging strand):与复制叉移动的方向相反,通过不连续的5ˊ-3ˊ聚合合成的新的DNA链。 10、AP位点(AP site):所有细胞中都带有不同类型、能识别受损核酸位点的糖苷水解酶,它能特异性切除受损核苷酸上N-β糖苷键,在DNA链上形成去嘌呤或去嘧啶位点,统称为AP位点。 11、cDNA(complementary DNA):在体外以mRNA为模板,利用反转录酶和DNA聚合酶合成的一段双链DNA。 12、C值反常现象(C value paradox):也称C值谬误。指C值往往与种系的进化复杂性不一致的现象,即基因组大小与遗传复杂性之间没有必然的联系。 13、DNA甲基化(DNA methylation):CpG二核苷酸(CpG岛)通常成串出现在DNA上,在甲基转移酶的作用下,胞嘧啶(C)的第5位碳原子能被修饰加上甲基。这种现象称为DNA甲基化。 14、DNA聚合酶(DNA polymerase):一种催化由脱氧核糖核苷三磷酸合成DNA的酶。 15、DNA拓扑异构酶(DNA topoisomerase):能在闭环DNA分子中改变两条链的环绕次数的酶。 16、DNA重组技术(recombinant DNA technology):又称基因工程(genetic engineering),将不同的DNA片段按照预先的设计定向连接
分生考点 Copyright by 孙倩1.顺式作用元件(cis-acting elements): 存在于基因内外,与基因表达调控相关、能够被基因调控蛋白特异性识别和结合的特定的DNA序列称为顺式作用元件。 2.启动子(promoter):真核基因的启动子指的是RNA聚合酶识别、结合的基因转录调控区中启动基因转录的一段特异DNA序列,包含一组转录调控功能组件,其中每一个功能组件的DNA序列约7~20 bp。 3.典型的启动子核心序列(core sequences)是在转录起始位点上游25~35 bp处,有一保守的TATA序列,被称为TATA盒(TATA box),真核细胞的TATA盒多为TATAAAA序列。TA TA盒与原核细胞的启动子一样,对RNA聚合酶II的转录起始位点起定位作用。 4. 有一些编码蛋白质基因不含TA TA盒或起始子,多在起始位点上游约100bp内含有20~50个核苷酸的CG序列,被称做CpG岛(CpG island)。此种基因可有多个转录起始点,可产生含不同5’末端的mRNA。这些基因大多为低转录基因,编码中间代谢酶的管家基因。 5.启动子上游元件(promoter-proximal elements, 或upstream promoter elements)是一些位于TATA盒上游的DNA序列,与调节蛋白结合,调节通用转录因子与TATA盒的结合、RNA聚合酶与启动子的结合,以及转录起始复合物的形成,从而决定基因的转录效率与专一性。常见的序列是CAA T盒和GC盒。 6.一些真核细胞基因含有另一种启动子元件,称为起始子(initiator,Inr),决定启动子的强度。 7.增强子(enhancer):是能够结合特异基因调节蛋白,促进邻近或远隔特定基因表达的DNA 序列。在酵母中,被称为上游活化序列(upstream activator sequences, UASs)。增强子的作用通常与其所处的位置和方向无关。 8.沉默子(silencer)是指某些真核基因转录调控区中抑制或阻遏基因转录的一段(数百bp)DNA序列。沉默序列促进局部DNA的染色质形成致密结构,从而阻止转录激活因子结合DNA,是基因转录的负性调节因素。 9.能够帮助RNA聚合酶转录RNA的蛋白质统称转录因子(transcription factors,TF)。以反式作用方式调节基因转录的转录因子称为反式作用因子(trans-acting factor),以顺式作用方式调节基因转录的转录因子称为顺式作用蛋白(cis-acting protein)。 10.基本转录因子(general transcription factors)是RNA聚合酶结合启动子所必需的一组蛋白因子,决定三种RNA(mRNA、tRNA及rRNA)转录的类别。 11.特异转录因子(special transcription factors)为个别基因转录所必需,决定该基因的时空特异性表达。 12.常染色质(euchromatin)结构松弛,分散分布在核内的染色质,对DNase I敏感,DNA 可降解为约200 bp 或其倍数的片断;基因表达处于活性状态,故亦称为活性染色质。使用DNase I处理活性染色质时,常会出现一些高敏感位点(hypersensitive sites),通常位于转录基因的5’和3’侧翼转录调控区的蛋白结合位点附近的裸露DNA上。 13.异染色质(heterochromatin)结构高度致密,处于凝聚状态的染色质,对DNase I不敏感。基因表达处于阻遏状态。 14.染色质重塑(chromatin remodeling)通过改变基因的启动子和调节序列区域的染色质结构来调节基因的表达,称为染色质重塑,也称为核小体重塑(nucleosome remodeling)。主要包括:CpG岛甲基化和组蛋白共价修饰。 15.核小体重塑(nucleosome remodeling):ATP依赖性核小体重塑复合体参与的核小体的移位、替换和去组装改变等。核小体重塑过程: 基因活化蛋白结合;ATP依赖性酶蛋白复合体结合转录活性区;A TP依赖性酶水解A TP,提供能量;移去或替换核小体。 16.组蛋白修饰包括组蛋白的乙酰化、甲基化、磷酸化、泛素化和多聚ADP-核糖基化。这些
生物信息学在医学领域的应用前沿 摘要:生物信息学是有生命科学、信息学、数学、物理、化学等学科相互交融而形成的新兴学科。生物信息数据库几乎覆盖了生命科学的各个领域,截止至2010年,总数已达1230个。生物信息学已不断渗透到医学领域的研究中。生物信息学在医学领域中主要应用于医学基础研究、临床医学、药物研发和建立与医学有关的生物信息学数据库。 关键词:生物信息学;医学;基因;应用 生物信息学是20世纪80年代以来随着人类基因组生命科学与信息科学以及数学、物理、化学等学科相互交融而形成的新兴学科,是当今最具发展前途的学科之一。人类基因组计划的顺利推进产生了海量基因数据,这些数据中蕴藏着丰富的生物学内涵,如果能充分挖掘并加以利用,可能揭示出很多对人类有用的信息。生物信息学已经成为生物学、医学、农学、遗传学、细胞生物学等学科发展的强大推动力量。随着生物信息学研究的深入与发展,它已不断渗透到医学领域的研究中。近年来,伴随着对基因组的研究不断深入,部分应用领域取得了令人瞩目的突破,其潜在的经济利益更是吸引了众多国家、企业及大量科研人员投入到相关研究中,生物信息学得到了迅猛的发展。 一、主要数据库 数据库是生物信息学的主要内容,各种数据库几乎覆盖了生命科学的各个领域。截止至2010年,生物信息数据库总数已达1230个。生物信息数据可可分为一级数据库和二级数据库。一级数据库的数据都直接来源于实验获得的原始数据,只经过简单的归类整理和注释,如Genbank数据库、SWISS-PROT数据库;二级数据库是在一级数据库、实验数据和理论分析的基础上针对特定目标衍生而来,是对生物学知识和信息的进一步整理,如人类基因组图谱库GDB。 在医学领域中常用的生物信息数据库主要有:核酸类数据库,如NCBI核苷酸序列数据库(Gen Bank )、欧洲核苷酸序列数据库(EMBL)、日本DNA 数据库(DDB)等;蛋白相关数据库,如蛋白质数据库(SWISS-PROT)、蛋白质信息资源库(HR)、Entrez 的蛋白三维结构数据库(MMDB)、蛋白质交互作用数据库(DIP)等;疾病相关数据库,包括综合临床数据库,如NCBI疾病基因数据库、Gene Cards等;遗传性疾病数据库,如遗传性疾病数据库(GDB)、人类遗传性疾病数据库(Gene Dis)等;肿瘤相关数据库,如肿瘤基因组解剖工程(CGAP)等;心血管疾病相关数据库,如心血管疾病相关生物医学数据库(Cardio)、心脏疾病计划及临床决策支持系统(HDP &CDM)等;免疫性疾病数据库,如免疫功能分子数据库( HMM)、免疫缺陷资源库(IDR)等;药物相关数据库,如药物和疾病数据库(Drugs)、FDA药品评审与研究中心(CDER)等。 二、生物信息学在医学领域的应用 2.1 生物信息学在医学基础研究中的应用 2.1.1 新基因的发现与鉴定 疾病的发生发展与特异基因的改变有关,鉴定与疾病相关的基因是科学家在积极探索的一个方向,对治疗某些疑难杂症带来新的契机。发现新基因是当前国际上基因组研究的热点,使用生物信息学的方法是发现新基因的重要手段。现在很多疾病的致病基因已经发现,包括癌症、肥胖、哮喘、心脑血管病等,其中与癌症相关的原癌基因约有1000个,抑癌基因约有100个。 目前发现新基因的主要方法有以下3种:①通过多序列比对从基因组DNA序列中预测新基因,其本质是把基因组中编码蛋白质的区域和非编码蛋白质的区域区分开来。②基因的电子克隆,即以计算机和互联网为手段,通过发展新算法,对生物信息数据库中存储的表达序列标签进行修正、聚类、拼接和组装,获得完整的基因序列,以期发现新基因。③发现单核苷酸多态性。 例如,2010年我国学者通过生物信息学EST 拼接技术,RT-PCR等技术,克隆出30个人类未知功能的新基因,并通过生物信息学分析该基因
核酸序列的基本分析 运用DNAMAN软件分析核酸序列的分子质量、碱基组成和碱基分布。同时运用BioEdit(版本7.0.5.3)软件对基因做酶切谱分析。 碱基同源性分析 运用NCBI信息库的BLAST程序对基因进行碱基同源性分析(Translated query vs.protien database(blastx))网站如下:https://www.doczj.com/doc/e35730785.html,/BLAST/ 参数选择:Translated query-protein database [blastx];nr;stander1 开放性阅读框(ORF)分析 利用NCBI的ORF Finder程序对基因做开放性阅读框分析,网址如下: https://www.doczj.com/doc/e35730785.html,/projects/gorf/orfig.cgi 参数选择:Genetic Codes:1 Standard 对蛋白质序列的结构功能域分析 运用简单模块构架搜索工具(Simple Modular Architecture Research Tool,SMART)对基因的ORF出的蛋白质序列进行蛋白质结构功能域分析。该数据库由EMBL建立,其中集成了大部分目前已知的蛋白质结构功能域的数据。 网址如下:http://smart.embl-heidelberg.de/ 运用NCBI的BLAST程序再对此蛋白质序列进行rpsBlast分析 参数选择:Search Database:CDD v2.07-11937PSSM Expect:0.01 Filter:Low complexity Search mode:multiple hits 1-pass 同源物种分析 用DNAMAN软件将蛋白质序列相关基因序列比对,根据结果绘出系统进化树,并进行分析。 蛋白质一级序列的基本分析 运用BioEdit(版本7.0.5.3)软件对基因ORF翻译的蛋白的一些基本性质,对分子量、等电点、氨基酸组成等作出分析。 二级结构和功能分析 信号肽预测 利用丹麦科技大学(DTU)的CBS服务器蛋白质序列的信号肽(signal peptide)预测,进入Prediction Serves 页面。 网址如下:http://www.cbs.dtu.dk/services/SignalP/ 参数选择: Eukaryotes;Both;GIF (inline);Standard; 疏水性分析 利用瑞士生物信息学研究所(Swiss Institute of Bioinformatics,SIB)的ExPASy服务器上的ProtScale程序对ORF 翻译后的氨基酸序列做疏水性分析 网址如下: https://www.doczj.com/doc/e35730785.html,/cgi-bin/protscale.pl 参数选择:
分子生物学基础知识太仓生命信息研究所 2011-7
前言 本文仅适用于对非生物专业的员工进行基础知识普及。如有深入学习的要求,请选用正规权威教材。 本教材以蛋白质、DNA、RNA、复制、转录和翻译为主要讲解内容,目的是帮助员工理解在工作中会遇到的常见生物学概念及术语 目录 前言 (2) 目录 (2) 蛋白质 (3) 1. 什么是蛋白质 (3) 2. 蛋白质的3D结构 (5) DNA (7) 1. DNA的组成—4种碱基 (7) 2. DNA的复制 (8) 3. DNA转录为RNA (9) 4. mRNA翻译成氨基酸序列 (11)
蛋白质 1.什么是蛋白质 蛋白质是由20中基本氨基酸链接而成的,生物体的大部分是有蛋白质构成的。每种氨基酸由4部分组成:碳原子C,羧基coo-,氨基H3N和R group。 20中氨基酸按照不同的排列和不同的长度,就形成了蛋白质。不同的R group把氨基酸分为5类: 无极性脂肪类R Group:
芳香类R Group 有极性,无电荷R Group
正电荷R Group 负电荷R Group 2.蛋白质的3D结构 氨基酸链在三维空间里呈现出一定的结构。各个氨基酸分子于相邻的氨基酸之间有氢键连接。 一级结构:氨基酸的排列顺序,可以用氨基酸的缩写在书面上表达。 氨基和羧基之间的氢键使得单个的氨基酸分子能够链接起来。
二级结构:单条氨基酸链所形成的2D形态。常见的有Alpha helix Beta sheet。 Alpha helix:氨基酸分子按顺时针或逆时针的方向螺旋上升。 Beta sheet:多条氨基酸分子链并列在一起。 三级结构:氨基酸链在各个方向的形态综合在一起。
常用分子生物学软件 一、基因芯片: 1、基因芯片综合分析软件。 ArrayVision 7.0 一种功能强大的商业版基因芯片分析软件,不仅可以进行图像分析,还可以进行数据处理,方便protocol的管理功能强大,商业版正式版:6900美元。 Arraypro 4.0 Media Cybernetics公司的产品,该公司的gelpro, imagepro一直以精确成为同类产品中的佼佼者,相信arraypro也不会差。 phoretix?Array Nonlinear Dynamics公司的基因片综合分析软件。 J-express 挪威Bergen大学编写,是一个用JAVA语言写的应用程序,界面清晰漂亮,用来分析微矩阵(microarray)实验获得的基因表达数据,需要下载安装JAVA运行环境JRE1.2后(5.1M)后,才能运行。 2、基因芯片阅读图像分析软件 ScanAlyze 2.44 ,斯坦福的基因芯片基因芯片阅读软件,进行微矩阵荧光图像分析,包括半自动定义格栅与像素点分析。输出为分隔的文本格式,可很容易地转化为任何数据库。 3、基因芯片数据分析软件 Cluster 斯坦福的对大量微矩阵数据组进行各种簇(Cluster)分析与其它各种处理的软件。 SAM Significance Analysis of Microarrays 的缩写,微矩阵显著性分析软件,EXCEL软件的插件,由Stanford大学编制。 4.基因芯片聚类图形显示 TreeView 1.5 斯坦福开发的用来显示Cluster软件分析的图形化结果。现已和Cluster成为了基因芯片处理的标准软件。 FreeView 是基于JAVA语言的系统树生成软件,接收Cluster生成的数据,比Treeview增强了某些功能。 5.基因芯片引物设计 Array Designer 2.00 DNA微矩阵(microarray)软件,批量设计DNA和寡核苷酸引物工具 二、RNA二级结构。 RNA Structure 3.5 RNA Sturcture 根据最小自由能原理,将Zuker的根据RNA一级序列预测RNA二级结构的算法在软件上实现。预测所用的热力学数据是最近由T urner实验室获得。提供了一些模块以扩展Zuker算法的能力,使之为一个界面友好的RNA折叠程序。允许你同时打开多个数据处理窗口。主窗口的工具条提供一些基本功能:打开文件、导入文件、关闭文件、设置程序参数、重排窗口、以及即时帮助和退出程序。RNAdraw中一个非常非常重要的特征是鼠
生物信息学在医学领域的应用研究现状 摘要生物信息学是研究生物信息处理(采集、管理和分析应用),并从中提取生物学新知识的一门科学,它连接生物数据和医学科学研究。生物信息数据库几乎覆盖了生命科学的各个领域,截止至2010年,总数已达1230个。生物信息学已不断渗透到医学领域的研究中。生物信息学在医学领域中主要应用于医学基础研究、临床医学、药物研发和建立与医学有关的生物信息学数据库。 关键词生物信息学,医学,应用 前言据统计,生物学信息正以每14个月翻一倍的速度增长。随着基因组及蛋白质序列数据库的快速增长,以及从这些序列中获取最大信息的需求,生物信息学(bioinformatics)作为一门独立学科应运而生。简言之,生物信息学就是利用计算和分析工具去收集、解释生物学数据的学科。生物信息学是一门综合学科,是计算机科学、数学、物理、生物学的结合。它对于管理现代生物学和医学数据具有重大意义,其研究成果将对人类社会和经济产生巨大推动作用。生物信息学的基础是各种数据库的建立和分析工具的发展。 数据库 迄今为止,生物学数据库总数已达500个以上。归纳起来可分为4大类:即基因组数据库、核酸和蛋白质一级结构数据库、生物大分子三维空间结构数据库,以及以上述3类数据库和文献资料为基础构建的二级数据库。 生物信息学在临床医学上的应用 1.疾病相关基因的发现:很多疾病的发生与基因突变或基因多态性有关。发 现新基因是当前国际上基因组研究的热点,使用生物信息学的方法是发现新基因的重要手段。目前发现新基因的主要方法有多种:(1)基因的电脑克隆:所谓基因的“电脑克隆”, 就是以计算机和互联网为手段,发展新算法,对公用、商用或自有数据库中存储的表达序列标签(express sequence tags,EST)进行修正、聚类、拼接和组装, 获得完整的基因序列, 以期发现新基因。(2)通过多序列比对从基因组DNA 序列中预测新基因[1]:从基因组序列预测新基因,本质上是把基因组中编码蛋白质的区域和非编码蛋白质的区域区分开来。(3)发现单核苷酸多态性[2]:现在普遍认为SNPs研究是人类基因组计划走向应用的重要步骤。这主要是因为SNPs将提供一个强有力的工具,用于高危群体的发
摘要:生物信息学已成为整个生命科学发展的重要组成部分,成为生命科学研究的前沿。本文对生物信息学的产生背景及其研究现状等方面进行了综述,并展望生物信息学的发展前景。生物信息学的发展在国内、外基本上都处在起步阶段。因此,这是我国生物学赶超世界先进水平的一个百年一遇的极好机会。 关键字:生物信息学、产生背景、发展现状、前景 随着生物科学技术的迅猛发展,生物信息数据资源的增长呈现爆炸之势,同时计算机运算能力的提高和国际互联网络的发展使得对大规模数据的贮存、处理和传输成为可能,为了快捷方便地对已知生物学信息进行科学的组织、有效的管理和进一步分析利用,一门由生命科学和信息科学等多学科相结合特别是由分子生物学与计算机信息处理技术紧密结合而形成的交叉学科——生物信息学(Bioinformatics)应运而生,并大大推动了相关研究的开展, 被誉为“解读生命天书的慧眼”。 一、生物信息学产生的背景 生物信息学是80年代未随着人类基因组计划(Human genome project)的启动而兴起的一门新的交叉学科。它通过对生物学实验数据的获取、加工、存储、检索与分析,进而达到揭示数据所蕴含的生物学意义的目的。由于当前生物信息学发展的主要推动力来自分子生物学,生物信息学的研究主要集中于核苷酸和氨基酸序列的存储、分类、检索和分析等方面,所以目前生物信息学可以狭义地定义为:将计算机科学和数学应用于生物大分子信息的获取、加工、存储、分类、检索与分析,以达到理解这些生物大分子信息的生物学意义的交叉学科。事实上,它是一门理论概念与实践应用并重的学科。 生物信息学的产生发展仅有10年左右的时间---bioinformatics这一名词在1991年左右才在文献中出现,还只是出现在电子出版物的文本中。事实上,生物信息学的存在已有30多年,只不过最初常被称为基因组信息学。美国人类基因组计划中给基因组信息学的定义:它是一个学科领域,包含着基因组信息的获取、处理、存储、分配、分析和解释的所有方面。 自1990年美国启动人类基因组计划以来,人与模式生物基因组的测序工作进展极为迅速。迄今已完成了约40多种生物的全基因组测序工作,人基因组约3x109碱基对的测序工作也接近完成。至2000年6月26日,被誉为生命“阿波罗计划”的人类基因组计划终于完成了工作草图,预示着完成人类基因组计划已经指日可待。截止目前为止,仅登录在美国GenBank数据库中的DNA序列总量已超过70亿碱基对。此外,迄今为止,已有一万多种蛋白质的空间结构以不同的分辨率被测定。基于cDNA序列测序所建立起来的EST数据库其纪录已达数百万条。在这些数据基础上派生、整理出来的数据库已达500余个。这一切构成了一个生物学数据的海洋。这种科学数据的急速和海量积累,在人类的科学研究历史中是空前的。 数据并不等于信息和知识,但却是信息和知识的源泉,关键在于如何从中挖掘它们。与