
微生物学通报
NOV 20, 2011, 38(11): 1705?1714 Microbiology China
? 2011 by Institute of Microbiology, CAS
tongbao@https://www.doczj.com/doc/b315427538.html,
基金项目:中国科学院知识创新工程项目(No. KSCX2-EW-R-01-04) *通讯作者:耿佳宁
: Tel: 86-10-82995362;
: gengjianing@https://www.doczj.com/doc/b315427538.html, 胡松年: Tel: 86-10-82995362;
: husn@https://www.doczj.com/doc/b315427538.html,
?共同第一作者
收稿日期:2011-05-24; 接受日期:2011-09-13
专论与综述
基于第二代测序技术的细菌基因组与
转录组研究策略简介
刘万飞1,2? 王西亮1,2? 赵宇慧1,2 曾瀞瑶1,2 耿佳宁1* 胡松年1*
(1. 中国科学院北京基因组研究所基因组科学及信息重点实验室 北京 100029)
(2. 中国科学院研究生院 北京 100049)
摘 要: 随着基于第二代测序技术的细菌基因组与转录组研究越来越广泛, 选择合适的研究策略变得越来越重要。就基于第二代测序技术的细菌基因组和转录组研究策略进行综述, 并简要介绍细菌基因组和转录组研究中的机遇和挑战。综述细菌基因组与转录组研究的常规方法及步骤, 并简要地介绍存在的问题。细菌基因组和转录组研究策略为大多数细菌的研究提供了一个相对完整的研究路线, 同时也会促进其它领域的研究, 如生命形成、生物进化、基础代谢、疾病、药物等。
关键词: 细菌, 基因组, 转录组, 第二代测序技术
The brief introduction of research strategies for bacterial genome and transcriptome based on the next-generation
sequencing technologies
LIU Wan-Fei 1,2? WANG Xi-Liang 1,2? ZHAO Yu-Hui 1,2 ZENG Jing-Yao 1,2
GENG Jia-Ning 1* HU
Song-Nian 1* (1. Key Laboratory of Genome Science and Information , Beijing Institute of Genomics , Chinese Academy of Sciences ,
Beijing 100029, China )
(2. Graduate University of Chinese Academy of Sciences , Beijing 100049, China )
Abstract: Recently, many bacterial genome and transcriptome studies have been performed based on the next-generation sequencing technologies. Therefore, how to select the appropriate research strate-gies is becoming increasingly important. In this paper, we discussed the research strategies of bacterial genome and transcriptome based on the next-generation sequencing technologies, and stated the oppor-
1706
微生物学通报
2011, Vol.38, No.11
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
tunities and challenges in these fields briefly. We reviewed the conventional methods and procedures and presented the existing problems briefly for bacterial genome and transcriptome studies. The re-search strategies of bacterial genome and transcriptome provide a relatively complete pipeline for the majority of bacteria. Moreover, it will promote the research of other fields, such as the course of life, biological evolution, basal metabolism, disease and drugs.
Keywords: Bacteria, Genome, Transcriptome, The next-generation sequencing technologies 伴随着我国1%人类基因组计划的完成, 基因组学及转录组学在我国取得了快速发展。以454、Solexa 和SOLiD 为代表的第二代测序技术的出现, 更使大规模应用基因组及转录组方法解决科学问题成为可能。与此同时, 遗传学的研究对象由少量基因及其功能转变为生物体的全基因组结构、基因功能、表观修饰、细胞调控等, 遗传学研究进入了基因组和后基因组时代。其中, 通过细菌基因组和转录组研究来揭示生命基本过程, 如生命形成、生物进化、基础代谢、疾病发生、药物靶点等, 成为生物学研究的重要手段。目前, 以高通量低成本为特点的第二代测序技术使得单个实验室或者研究组独立进行细菌基因组及转录组研究成为可能。然而, 如何将海量测序结果组装成一个完整的细菌基因组, 以及如何将RNA 测序结果还原成细菌特定生理状态下的转录情况是科学家面临的主要问题。本文对现阶段基于第二代测序技术的细菌基因组及转录组研究策略进行综述, 旨在为细菌基因组及转录组
研究提供帮助。
1 细菌基因组学研究
1.1 细菌基因组学简介
细菌基因组学是研究细菌全基因组DNA 序列及其结构与功能的学科。1995年, 科学家获得了流感嗜血杆菌(Haemophilus influenzae Rd)的全基因组序列[1], 这是第一个完整的基因组序列, 也是第一个完成的细菌基因组序列。紧接着古细菌詹氏甲烷球菌(Methanococcus jannaschii )基因组[2]、大肠杆菌(Escherichia coli K-12)基因组[3]等也相继完成。细菌基因组研究不仅有利于研究细菌的基本生命过程, 同时也对高等真核生物的基因组学及后基因组学研究提供了参考和平台。到目前为止, NCBI 上记录了1 534个细菌基因组, 包括了103个古细菌和1 431个真细菌(2011-4-24)[4], 其中中国科学家完成了44个细菌基因组的测序工作。此外关于基因组学的研究报告也在逐年增加, 如表1[5]所示。
表1 PubMed 检索到的有关基因组学和转录组学的文献[5]
Table 1 The literature number retrieved in PubMed database using “Genomics” and “Transcriptomics” as keywords [5]
年份 Year 基因组学文章 Articles about Genomics
基因组学综述 Reviews about Genomics
转录组学文章
Articles about Transcriptomics
转录组学综述
Reviews about Transcriptomics
2000 1 543 383 2 0 2001 2 072 559 4 2 2002 2 689 826 18 7 2003 3 570 984 23 13 2004 4 810 1 238 56 23 2005 6 109 1 417 88 52 2006 6 905 1 559 105 49 2007 7 295 1 613 133 55 2008 8 119 1 603 162 59 2009 8 551 1 579 234 75 2010
8 986
1 487
296
70
刘万飞等: 基于第二代测序技术的细菌基因组与转录组研究策略简介
1707
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
1.2 细菌基因组研究策略
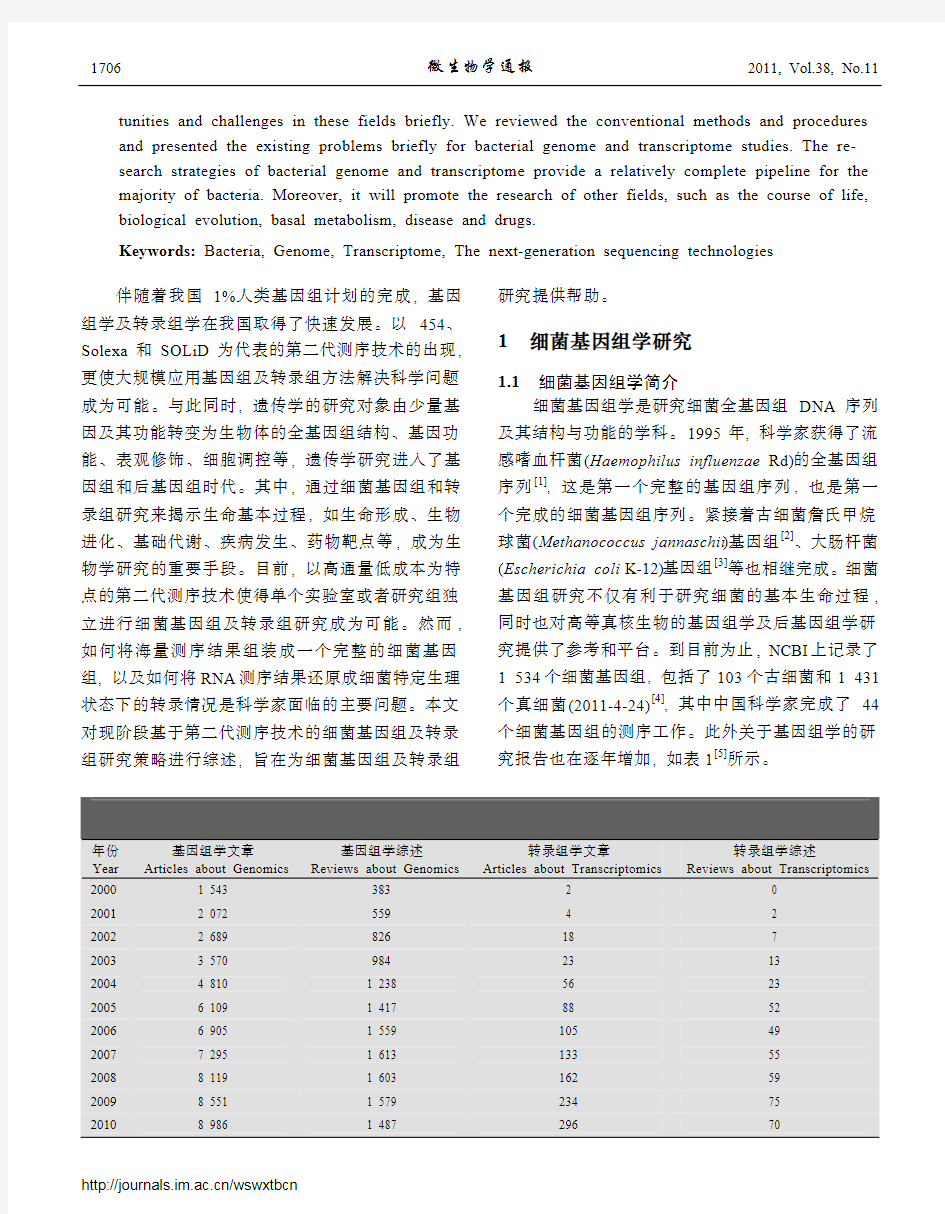
细菌基因组的研究策略主要分为DNA 的提取及测序、基因组组装、基因组完成(Genome finish-ing)、基因预测、基因注释和基因组比较分析六大部分, 如图1所示。
首先是DNA 的提取及测序。DNA 提取时要保证DNA 纯度, 同时要避免DNA 污染。目前主要应用的DNA 测序技术是以Roche 公司的454 (https://www.doczj.com/doc/b315427538.html,/)、Illumina 公司的Solexa (https://www.doczj.com/doc/b315427538.html,/)和ABI 公司的SOLiD (https://www.doczj.com/doc/b315427538.html,/)为代表的第二代测序技术[6?8]。与第一代测序技术相比, 第二代测序技术在测序成本和测序速度方面有了极大的提高。454采用焦磷酸测序法, 平均读长400 bp, 每个循环能产生400?600 Mb 序列, 耗时7.5 h 左右。经过最近的升级, 454测序长度已能达到1 kb, 是进行未知基因组测序(De novo genome sequencing)的理想平台。然而, 454在处理单碱基重复序列(Homopolymer)时会因为荧光信号强度判断错误而引入核苷酸缺失或插入。此外, 454因测序通量较小,使测序费用相对较高。Solexa 采用边合成边测序的方法, Solexa GAIIx 序列长度能达到100 bp, 每个反应能产生100 Gb 左右的碱基序列, 耗时约9 d 。升级版的HiSeq 2000在测序速度和测序通量上都有很大的提高,序列长度可达到120 bp, 每个反应能产生约600 Gb 的碱基序列。Solexa 的优势在于测序通量大,成本低,但因为序列相对较短, 会增加后继序列拼接组装等分析的难度和计算量。SOLiD 采用双碱基编码原理, 利用DNA 连接酶在连接过程中进行测序, 读长50 bp, SOLiD 4测序仪每个反应能产生100 Gb 左右的数据, 耗时6?7 d 。SOLiD 与Solexa 一样, 由于读长短, 后继分析相对复杂; 但是因为测序通量也较大, 测序费用相对较低。SOLiD 产生的序列是由0、1、2、3组成的Colorspace, 因此数据处理时需要特殊的处理。相较基因组测序而言,SOLiD 在转录组测序上具有很大的优势, 因为它可以获得RNA 的正负链信息, 这对于转录组研究, 尤其是Antisense RNA 分析, 具有重要意义。最近, 一
款新的DNA 测序方法——粒子流(Ion-torrent)半导体基因组测序方法问世(http://www.iontorrent. com/)[9]。此方法不依赖于光信号, 通过直接测知基于模板的DNA 聚合酶合成反应产生的离子来获得序列信息, 并且成本和通量的性价比较好。因此, 测序时应该根据各个测序平台的优缺点来选择合适的测序平台。比如细菌De novo 基因组测序,可以利用454或454与Solexa 相结合的测序方法, 即能降低测序成本, 又便于进行序列拼接组装。另外, 为了方便基因组组装, 我们可以适当的构建Pair-end 或Mate-pair 文库, 通过利用配对Reads 之间的关系来确定Contigs 之间的关系。
第二步, 基因组组装。常用的软件有Newbler [10]、AMOScmp [11]、Phred/Phrap/Consed [12?13]和Velvet [14]等, 可以根据自己的数据选择合适的组装软件, 也可以结合多种方法获得较好的组装结果。
第三步, 基因组完成, 即确定组装获得的Contigs 之间的连接顺序并修补Gaps 。可以按照以下几个步骤进行: 首先, 计算Contigs 和基因组的平均Reads 覆盖度, 通过Contigs 与基因组平均Reads 覆盖度的比较, 获得Unique contigs 和Repeat con-tigs 以及Repeat contigs 的重复次数。这一阶段, 可以过滤掉那些覆盖度明显低于基因组平均Reads 覆盖度的Contigs (可能来自污染DNA 序列)以及一些较短的Contigs (如长度小于500 bp 的Contigs, 这些Contigs 会在修补Gaps 时被填补回去)。其次, 根据Contigs 之间的Reads 连接数来确定Contigs 之间可能的连接顺序。这一步还可以通过一些间接的方法来定位Contigs 之间的连接关系: (1) 通过Contigs 与近缘物种基因组的比较(如MAUVE [15]和MUMmer [16]), 获得Contigs 之间的连接顺序; (2) 利用基因在基因组上排列顺序的保守性, 根据远缘物种基因组上基因的排列顺序来确定Contigs 之间的连接顺序(把这些基因定位到Contigs 上); (3) 将Contigs 与NCBI 的nr/nt 库进行序列比对, 根据已知序列定位Contigs 之间的连接顺序; (4) 进行随机PCR 扩增来确定Contigs 之间的连接顺序; (5) 利用
1708
微生物学通报
2011, Vol.38, No.11
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
Pair-end 或者Mate-pair 文库中的配对Reads 获得Contigs 之间的连接关系。最后, 根据Contigs 之间可能的连接顺序设计引物, 并进行PCR 扩增与测序来验证Contigs 之间的连接顺序和修补Gaps, 从而获得完整的基因组序列。在这一阶段, 可以先确定Unique contigs 之间的关系, 然后再把Repeat con-tigs 放回对应的位置。
第四步, 基因预测。常用的蛋白质编码基因预测软件有Glimmer [17]、GeneMarkS [18]和Prodigal [19], 通常可以任选其中一款软件进行预测, 也可以结合多个软件以获得较好的预测结果。此外, ZCURVE 是基于DNA 序列Z curve 理论的蛋白质编码基因识别软件, 具有较高的基因起始位点预测准确性[20]; GS-Finder 是不依赖于rRNA 序列的细菌基因组翻译起始位点识别软件, 能大大提高翻译起始位点预测的准确性[21]; OperonDB 是比较常用的操纵子预测软件, 可以用来预测共同转录的基因簇[22]。另外, 非蛋白质编码基因的预测也有较成熟的软件, 通常用RNAmmer [23]预测rRNA 、tRNAscan-SE [24]预测tRNA 以及Rfam [25]预测Small RNA 等(可以利用Splitter [26]将基因组分成较小序列, 然后用Rfam 寻找Small RNA; 或者下载并安装Rfam database, 在本地寻找Small RNA)。
第五步, 基因注释。这一步通常要整合多个数据库, 如NCBI 的nr 库、InterPro [27]、COG [28]和KEGG [29]等, 通过序列比对进行预测基因的注释。此外, 还可以利用一些特定功能的软件或者数据库进行相应的分析, 如用SignalP [30]预测信号肽、TMHMM [31]预测跨膜结构、ISfinder [32]预测插入序列、VFDB 预测毒力因子[33]、Islander 数据库查询基因组岛[34]、MobilomeFINDER [35]和IslandViewer [36]鉴定基因组岛、PAIDB 预测潜在的致病岛[37]、Repeat-match 预测基因组重复序列、Tandem repeat Finder [38]寻找串联重复序列、CRISPR finder [39]预测CRISPR 序列、Phage-finder [40]寻找噬菌体序列、TCDB [41]注释膜转运蛋白、Ori-Finder [42]寻找复制起始位点、ARDB [43]鉴定和注释抗菌素抗性基因、ACLAME [44]注释可变遗传因子(Mobile genetic ele-ments)和TADB [45]数据库搜索Type2 toxin-antitoxin 位点等。另外, 有些基因是生物体生存不可或缺的基因, 即必需基因, 它们是生命的基础。DEG [46]数据库收集了一些物种的必需基因, 也可以用于注释必需基因, 这些必需基因是很好的抗菌药物靶基因。注释结束后, 对基因注释结果进行检查, 比如基因之间是否有Overlap 、是否存在假基因等, 可以利用Mciobial Genome Submission Check [47]程序进行检查。
最后, 基因组比较分析。获得完整基因组及其注释后, 通常会进行相近物种之间或同一物种不同株之间的基因组比较分析。常用的细菌基因组比较分析软件和数据库有ACT [48]、Mauve [15]、MUMmer [16]、MicrobesOnline [49]、mGenomeSubtractor [50]和xBASE [51] 等。ACT (Artemis Comparison Tool), 是一款进行基因组及其注释之间比较的可视化软件, 支持多种输入格式(EMBL, GenBank, FASTA 和GFF 格式), 可以用来鉴定相似序列、插入、缺失、重排等。另外, 对于未注释序列, 可以寻找CDS 序列并进行相应的比较。目前, 有两款基于ACT 的网站, WebACT [52]和DoubleACT (http://www.hpa-bioinfotools. https://www.doczj.com/doc/b315427538.html,/pise/
double_act.html)。WebACT 提供了已知物种的基因组比较结果, 同时支持上传序列进行在线比较分析, 而DoubleACT 提供生成ACT 可读的基因组比较文件。Mauve 可以非常有效地构建多基因组比对结果, 而且可以容忍基因组重排和倒置, 同时Mauve 会根据比对结果绘制不同基因组之间的进化树。MUMmer 可以用来比较完整的或者不完整的基因组序列(如Contigs), 既可以在DNA 水平比较, 也可以在蛋白质水平比较, 而且还可用于高等真核生物基因组之间的比较。另外, ACT 可以利用MUMmer 的比较结果进行可视化操作。MicrobesOnline 是一个提供原核生物比较和功能基因组分析的数据库, 包含了1 000多个基因组和许多物种的芯片表达数据。MicrobesOnline 包含的模块有比较基因组浏览器、基因调控预测、系统发生搜索、代谢途径比较、操纵子预测、序列分析以及和其他微生物基因组资源的整合分析等。mGenomeSubtractor 是通过相近物种
刘万飞等: 基于第二代测序技术的细菌基因组与转录组研究策略简介
1709
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
基因组的在线比较分析得到保守的和特异性的基因组片段, 从而获得一个特定细菌的表型、环境适应和疾病相关等信息。xBASE 是一个细菌基因组比较注释软件, 只要提供完整或不完整的Fasta 格式基因组序列, 就能获得基因组注释结果。xBASE 特别适合那些有参考基因组的基因组序列。另外, xBASE 开发了针对第二代测序数据的xBASE-NG 模块, 可以进行SNP 分析、新序列挖掘和基因组注释等。
通过细菌基因组比较分析有助于阐明微生物的许多特性(如耐高温、耐盐、降解塑料和抗药性等), 许多研究成果可应用于工业生产(如发酵)、环境治理(如分解石油)以及医药(如抗生素)等方面。当前对致病菌的研究主要集中于毒力因子、致病岛、耐药基因、耐药机制以及与寄主的关系等, 通过比较可以发现或预测致病菌的致病相关基因, 为疾病的诊断、药靶的寻找和疫苗或抗生素的研制提供理论支
持。通过基因组比较还可以研究进化史、构建生物进化树。CVTree [53]是一个不基于序列比对的系统发生树分析工具。它是利用组成向量法(Composition vector)构建全基因组的系统发生树, 这样即避免了因为基因选择而带来的系统发生树的可变性, 又避免了不同长度和内容基因之间的序列比对。
2 细菌转录组学研究
2.1 细菌转录组学简介
细菌转录组学是从RNA 水平研究基因表达的情况。目前细菌转录组学主要应用于基因注释校正、基因表达、操纵子鉴定、转录起始位点(TSS)鉴定、新基因鉴定、Small RNA 分析等内容。此外, 通过细菌转录组比较分析, 研究细菌在不同环境、宿主等情况下的基因表达变化, 从而阐述宿主、环境对细菌的影响、细菌致病机制等。目前转录组学的研
图1 基于第二代测序技术的细菌基因组与转录组研究策略图
Fig. 1 The research strategies of bacteria genome and transcriptome based on the next-generation sequencing technologies
1710
微生物学通报
2011, Vol.38, No.11
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
究也在逐年增加, 如表1所示, 但是相对于基因组学来说, 细菌转录组学尚处于发展阶段。
2.2 细菌转录组研究策略
细菌转录组的研究策略主要包括总RNA 的提取和目的RNA 的富集、RNA 建库及测序、Reads mapping 和后继分析等(图1)。
首先进行总RNA 的提取和目的RNA 的富集。细菌mRNA 的半衰期很短, 又极易降解, 因此防止其降解是十分重要的。仪器用具都要经过严格的处理, 最好是RNA 实验专用; 材料一定要新鲜, 切忌使用反复冻融的材料; 整个RNA 提取过程中动作应迅速, 并需要加入RNA 酶抑制剂。细菌mRNA 一般只占总RNA 的1%?5%, 因此, 总RNA 提取完成后一般要进行mRNA 的富集, 主要有以下4种方法[54]: (1) rRNA 捕获法。根据16S rRNA 和23S rRNA 保守区域序列设计探针并将探针固定在磁珠上, 利用探针和rRNA 杂交去除rRNA 。在一般情况下该方法可以去掉大部分rRNA, 但效果因不同的基因组而异。(2) 降解rRNA 和tRNA 法。在原核生物中rRNA 和tRNA 等加工过的RNA 含有5′单磷酸(5′P)而mRNA 含有5′三磷酸(5′PPP), 利用特异性降解5′单磷酸RNA 分子的核酸外切酶(5′→3′)降解rRNA 和tRNA 。该方法一般仅可以去掉细菌10%?20%的rRNA 。(3) 多腺苷酸mRNA 选择法。利用大肠杆菌polyA 聚合酶能够在mRNA 末尾添加polyA 而不能在rRNA 末尾添加polyA 的特性, 在mRNA 末尾加上polyA, 然后利用Oligo (dT)探针捕获处理后的mRNA 。该方法简单快速, 但mRNA 中会有初级转录本的加工产物和未翻译的mRNA 降解产物。(4) 抗体法。通过抗体捕获与特定蛋白质相互作用的RNA, 比如Small RNA 可以和Hfq 蛋白质相互作用, 可以利用这一特性采取免疫共沉淀的方法获得Small RNA 。该方法特异性好, 效率高, 可用于Small RNA 的分析。4种mRNA 富集方法各有特点, rRNA 捕获法需要预先知道rRNA 序列; 多腺苷酸mRNA 选择法依赖polyA 聚合酶; 抗体法比较适合Small RNA 等的分析; 而降解rRNA 和tRNA 法也依赖特异性核酸外切酶。
第二步, 选择合适的建库方法和测序平台进行
测序。测序文库包括5′末端测序文库、3′末端测序文库、完整转录本测序文库等, 5′末端测序文库和3′末端测序文库适合UTR 区和操纵子的研究, 而完整转录本测序文库适合进行不同样本或者不同时期转录本之间表达量的比较分析以及新基因鉴定等。测序平台主要有SOLiD 、Solexa 和454三种。SOLiD 具有链特异性, 适合研究Small RNA 、发现新基因等, Solexa 测序成本较低, 而454适合进行基因组序列未知物种的转录组分析。大家要根据研究对象和研究目的选择合适的建库方法和测序平台, 比如鉴定转录起始位点应该构建5′末端测序文库和采用链特异性测序方法(SOLiD), 而研究操纵子要构建完整转录本测序文库。需要说明的是链特异性测序方法越来越受欢迎。此方法可以明确转录本的方向、解决正负链基因重叠问题和提高基因(包括操纵子)注释的准确性, 特别适合鉴定转录起点、反义RNA 和发现新基因。目前链特异性测序方法主要分为两类, 一类是在mRNA 的两端分别连接不同的接头, 使5'和3'易于区分便于寻找转录模板; 另一类是对cDNA 的一条链进行化学修饰产生标记, 如用重亚硫酸盐处理RNA 或者利用dUTP 来合成cDNA 的第二条链[55]。
第三步, Reads mapping 和后继分析。首先将Reads 进行Mapping, 从而获得Reads 在基因组上的位置, 常用的Mapping 软件有BWA [56]、Bowtie [57]、SOAP2[58]等。其次, 根据Reads mapping 结果, 进行后继分析。TSS 鉴定: 根据基因组上的Reads cov-erage 来鉴定转录起始位点; 5′UTR 和3′UTR 鉴定: 鉴定编码基因中转录但不翻译的区域, 即5′UTR 和3′UTR; Operon 鉴定: 根据TSS 、Reads coverage 等鉴定Operon (也就是细菌中的转录单元); 新基因鉴定: 找出之前没有注释但是表达的区域, 并根据是否存在ORF 区分为蛋白质编码基因(Coding gene)和非蛋白质编码基因(Small RNA gene); Antisense RNA 鉴定: 根据Small RNA 与其他基因的位置关系确定是否是Antisense RNA; Pseudogenes 分析: 比如研究假基因的表达情况; 保守结构域的鉴定: 利用MEME [59]等软件鉴定保守结构域; ncRNA 预测和鉴定: 可以利用sRNAFinder [60]、nocoRNAc [61] 等软件
刘万飞等: 基于第二代测序技术的细菌基因组与转录组研究策略简介
1711
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
进行预测, 也可以通过与Rfam database 比较来预测Small RNA [25]。另外, 对那些比较重要的或者感兴趣的基因, 可以通过Real-time PCR 来验证RNA-seq 的结果。
此外, 为了方便科学家直观地了解细菌基因组及转录组图谱, 生物信息学家开发了许多基因组可视化软件, 比如Artemis [62]和Integrated Genome Browser (IGB)[63]等。
3 细菌基因组学与转录组学研究的机遇与挑战
3.1 细菌基因组学研究机遇与挑战
伴随着国际人类基因组计划的进行, 细菌基因组学也获得了快速的发展。随着第二代测序技术的出现, 细菌基因组学研究迎来了第二次高峰。目前, 细菌基因组测序多采用454或者454加Solexa 的方式, 不但加快了数据产出, 而且有利于基因组拼接。获得细菌完整基因组后, 就要进行细菌基因组的分析和注释。常用的细菌基因组分析和注释工具如上文所述, 我们也可以参考Pau Stothard 等的综述[64]。细菌的研究现在多集中于模式细菌(如大肠杆菌)和致病菌, 主要研究细菌的毒素、运动、粘附和生物膜形成、分泌系统、细胞表面蛋白、代谢及应激反应等[65]。此外, 通过相似物种基因组比较分析来揭示病原菌相关遗传线索[66], 也是细菌基因组学研究的一个重要方向。
虽然第二代测序技术给细菌基因组学研究带来了新的机遇, 但是也带来了一些新的问题, 比如基于焦磷酸测序的454测序方法常在单碱基重复序列区域出现插入/缺失, 会导致注释基因的移码突变; Solexa 测序法获得的Reads 长度较短而影响拼接结果; 细菌基因组组装及分析流程较繁琐, 亟待新的方法或高度整合型的处理流程来加快分析过程等。
3.2 细菌转录组学研究机遇与挑战
转录组学是研究生物体基因表达和RNA 调控的有力工具。伴随着第二代测序技术的出现, 在真核生物中进行了大量基于高通量测序技术的转录组学研究[67]。然而, 由于科学家普遍认为细菌转录组
比较简单、细菌mRNA 不易富集等原因, 细菌转录组学被大大忽视。随着测序能力的大幅度提高和一些特定细菌mRNA 富集方法的出现[68?71], 细菌转录组越来越受到关注。
虽然细菌转录组学领域的研究目前还处于发展阶段, 但是它提供了在基因组水平上研究细菌RNA 调控机制的重要手段。通过深度测序的转录组研究, 人们发现许多调控元件(如Small RNA 、Riboswitches 和Cis-antisense 调控因子等)[72?76]参与了原核生物的生理和病理过程。此外, 有效的转录组分析可以改善基因组的注释, 转录组分析将来可能会成为基因组注释的一个标准构件。
目前原核转录组研究表明原核生物的调控复杂性和冗余性远远超过了最初的预料[54]。进一步的原核转录组研究可能揭示Small RNA 调控网络、顺式作用元件(Cis-acting element)、环境依赖性功能开关(Riboswitch)和长反义转录本等的重要作用。转录组学方法的局限性在于它们需要成千上万的细胞作为材料, 这就无法确定正义转录本和反义转录本是互相排斥的还是可以同时转录的。因此, 单细胞转录组研究是转录组研究的新方向, 同时单细胞转录组将促进非培养细菌的研究和更精确地研究不同时间、不同环境下转录组的变化。此外, 随着转录组学研究的深入, 便捷的转录组学分析流程及软件会大大促进细菌转录组学的发展。
4 总结
细菌基因组学和转录组学的发展, 不但可以用来研究生命形成、生物进化、基础代谢、疾病发生、药物靶点等, 同时也可以相互促进各自学科的发展。比如基因组序列为转录组数据注释提供了参考, 同时转录组数据可以用来校正基因组注释信息、发现新基因和促进功能基因组学的发展。细菌基因组学和转录组学研究策略为大多数细菌研究提供了一个相对完整的研究路线, 同时也会促进单个实验室或者研究组进行细菌基因组及转录组的研究。此外, 细菌转录组学还需要科学家们进一步的深入研究, 并把细菌转录组分析变成为细菌的常规分析。
1712
微生物学通报
2011, Vol.38, No.11
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
参 考 文 献
[1] Fleischmann RD, Adams MD, White O, et al.
Whole-genome random sequencing and assembly of Haemophilus influenzae Rd[J]. Science, 1995, 269(5223): 496?512.
[2] Bult CJ, White O, Olsen GJ, et al. Complete genome se-quence of the methanogenic archaeon, Methanococcus jannaschii[J]. Science, 1996, 273(5278): 1058?1073. [3] Blattner FR, Plunkett G 3rd, Bloch CA, et al. The com-plete genome sequence of Escherichia coli K-12[J]. Sci-ence, 1997, 277(5331): 1453?1462.
[4] NCBI: http: //www. ncbi. nlm. nih. gov/genomes/lproks.
Cgi.
[5] Sayers EW, Barrett T, Benson DA, et al. Database re-sources of the National Center for Biotechnology Infor-mation[J]. Nucleic Acids Res, 2009, 37(Database issue): D5?D15.
[6] Margulies M, Egholm M, Altman WE, et al. Genome se-quencing in microfabricated high-density picolitre reac-tors[J]. Nature, 2005, 437(7057): 376?380.
[7] Turcatti G, Romieu A, Fedurco M, et al. A new class of
cleavable fluorescent nucleotides: synthesis and optimiza-tion as reversible terminators for DNA sequencing by synthesis[J]. Nucleic Acids Res, 2008, 36(4): e25.
[8] Shendure J, Porreca GJ, Reppas NB, et al. Accurate mul-tiplex polony sequencing of an evolved bacterial ge-nome[J]. Science, 2005, 309(5741): 1728?1732.
[9] Rothberg JM, Hinz W, Rearick TM, et al. An integrated
semiconductor device enabling non-optical genome se-quencing[J]. Nature, 2011, 475(7356): 348?352. [10] 454 sequencing: http://454. com.
[11] Pop M, Phillippy A, Delcher AL, et al. Comparative ge-nome assembly[J]. Briefings in Bioinformatics, 2004, 5(3): 237?248.
[12] Ewing B, Green P. Base-calling of automated sequencer
traces using phred. II. Error probabilities[J]. Genome Re-search, 1998, 8(3): 186?194.
[13] Gordon D. Viewing and editing assembled sequences us-ing Consed[J]. Curr Protoc Bioinformatics, 2003, Chapter 11.
[14] Zerbino DR, Birney E. Velvet: Algorithms for de novo
short read assembly using de Bruijn graphs[J]. Genome Research, 2008, 18(5): 821?829.
[15] Darling ACE, Mau B, Blattner FR, et al. Mauve: multiple
alignment of conserved genomic sequence with rear-rangements[J]. Genome Research, 2004, 14(7): 1394? 1403.
[16] Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open
software for comparing large genomes[J]. Genome Biol-
ogy, 2004, 5(2): R12.
[17]
Delcher AL, Bratke KA, Powers EC, et al. Identifying bacterial genes and endosymbiont DNA with Glimmer[J]. Bioinformatics, 2007, 23(6): 673?679.
[18]
Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in mi-crobial genomes. Implications for finding sequence motifs in regulatory regions[J]. Nucleic Acids Research, 2001, 29(12): 2607?2618.
[19]
Hyatt D, Chen GL, Locascio PF, et al. Prodigal: prokary-otic gene recognition and translation initiation site identi-fication[J]. BMC Bioinformatics, 2010, 11(1): 119.
[20]
Guo FB, Ou HY, Zhang CT. ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes[J]. Nucleic Acids Res, 2003, 31(6): 1780?1789. [21]
Ou HY, Guo FB, Zhang CT. GS-Finder: a program to find bacterial gene start sites with a self-training method[J]. Int J Biochem Cell Biol, 2004, 36(3): 535?544.
[22]
Ermolaeva MD, White O, Salzberg SL. Prediction of op-erons in microbial genomes[J]. Nucleic Acids Res, 2001, 29(5): 1216?1221.
[23]
Lagesen K, Hallin P, R?dland EA, et al. RNAmmer: con-sistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Research, 2007, 35(9): 3100?3108.
[24]
Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs[J]. Nucleic Acids Research, 2005, 33(Issue suppl 2):W686?W689.
[25]
Gardner PP, Daub J, Tate JG, et al. Rfam: updates to the RNA families database[J]. Nucleic Acids Research, 2009, 37(Database issue): D136?D140.
[26] splitter: http: //emboss. bioinformatics. nl/cgi ?bin/emboss/ splitter.
[27]
Hunter S, Apweiler R, Attwood TK, et al. InterPro: the integrative protein signature database[J]. Nucleic Acids Research, 2009, 37(Database issue): D211?D215.
[28]
Tatusov RL, Koonin EV, Lipman DJ. A genomic perspec-tive on protein families[J]. Science, 1997, 278(5338): 631?637.
[29] KEGG: http: //www. genome. jp/kegg/.
[30]
Bendtsen JD, Nielsen H, von Heijne G, et al. Improved prediction of signal peptides: SignalP 3. 0[J]. Journal of
Molecular Biology, 2004, 340(4): 783?795.
[31] Krogh A, Larsson B, von Heijne G, et al. Predicting
transmembrane protein topology with a hidden Markov model: application to complete genomes[J]. Journal of Molecular Biology, 2001, 305(3): 567?580.
[32] Siguier P, Perochon J, Lestrade L, et al. ISfinder: the ref-erence centre for bacterial insertion sequences[J]. Nucleic Acids Research, 2006, 34(Database issue): D32?D36.
刘万飞等: 基于第二代测序技术的细菌基因组与转录组研究策略简介
1713
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
[33] Yang J, Chen LH, Sun LL, et al. VFDB 2008 release: an
enhanced web-based resource for comparative pathoge-nomics[J]. Nucleic Acids Research, 2008, 36(Database issue): D539?D542.
[34] Mantri Y, Williams KP. Islander: a database of integrative
islands in prokaryotic genomes, the associated integrases and their DNA site specificities[J]. Nucleic Acids Res, 2004, 32(Database issue): D55?D58.
[35] Ou HY, He XY, Harrison EM, et al. MobilomeFINDER:
web-based tools for in silico and experimental discovery of bacterial genomic islands[J]. Nucleic Acids Res, 2007, 35(Web Server issue): W97?W104.
[36] Langille MGI, Brinkman FSL. IslandViewer: an integrated
interface for computational identification and visualiza-tion of genomic islands[J]. Bioinformatics, 2009, 25(5): 664?665.
[37] Yoon SH, Park YK, Lee S, et al. Towards pathogenomics:
a web-based resource for pathogenicity islands[J]. Nucleic Acids Res, 2007, 35(Database issue): D395?D400.
[38] Benson G. Tandem repeats finder: a program to analyze
DNA sequences[J]. Nucleic Acids Research, 1999, 27(2): 573?580.
[39] Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web
tool to identify clustered regularly interspaced short pal-indromic repeats[J]. Nucleic Acids Research, 2007, 35(Web Server issue): W52?W57.
[40] Fouts DE. Phage_Finder : Automated identification and
classification of prophage regions in complete bacterial genome sequences[J]. Nucleic Acids Research, 2006, 34(20): 5839?5851. [41] Saier MH Jr, Yen MR, Noto K, et al. The Transporter
Classification Database: recent advances[J]. Nucleic Ac-ids Research, 2009, 37(Database issue): D274?D278. [42] Gao F, Zhang CT. Ori-Finder: a web-based system for
finding oriCs in unannotated bacterial genomes[J]. BMC Bioinformatics, 2008, 9: 79.
[43] Liu B, Pop M. ARDB-Antibiotic Resistance Genes Data-base[J]. Nucleic Acids Res, 2009, 37(Database issue): D443?447.
[44] Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a
CLAssification of Mobile genetic Elements, update 2010[J]. Nucleic Acids Res, 2010, 38(Database issue): D57?D61.
[45] Shao YC, Harrison EM, Bi DX, et al. TADB: a web-based
resource for Type 2 toxin-antitoxin loci in bacteria and archaea[J]. Nucleic Acids Res, 2011, 39(Database issue): D606?D611.
[46] Zhang R, Lin Y. DEG 5. 0, a database of essential genes in
both prokaryotes and eukaryotes[J]. Nucleic Acids Res, 2009, 37(Database issue): D455?D458.
[47] Microbial Genome Submission Check: http://www. ncbi.
https://www.doczj.com/doc/b315427538.html,/genomes/frameshifts/frameshifts.cgi.
[48] Carver TJ, Rutherford KM, Berriman M, et al. ACT: the
Artemis Comparison Tool[J]. Bioinformatics, 2005, 21(16): 3422?3423.
[49] Dehal PS, Joachimiak MP, Price MN, et al. Microbe-sOnline: an integrated portal for comparative and func-tional genomics[J]. Nucleic Acids Res, 2010, 38(Database issue): D396?D400.
[50] Shao YC, He XY, Harrison EM, et al. mGenomeSubtrac-tor: a web-based tool for parallel in silico subtractive hy-bridization analysis of multiple bacterial genomes[J]. Nu-cleic Acids Res, 2010, 38(Web Server issue): W194?W200.
[51] Chaudhuri RR, Loman NJ, Snyder LAS, et al. xBASE2: a
comprehensive resource for comparative bacterial ge-nomics[J]. Nucleic Acids Res, 2008, 36(Database issue): D543?D546.
[52] Abbott JC, Aanensen DM, Rutherford K, et al. We-bACT-an online companion for the Artemis Comparison Tool[J]. Bioinformatics, 2005, 21(18): 3665?3666.
[53] Xu Z, Hao BL. CVTree update: a newly designed phy-logenetic study platform using composition vectors and whole genomes[J]. Nucleic Acids Res, 2009, 37(Web Server issue): W174?W178.
[54] Sorek R, Cossart P. Prokaryotic transcriptomics: a new
view on regulation, physiology and pathogenicity[J]. Na-ture Reviews Genetics, 2010, 11(1): 9?16.
[55] Levin JZ, Yassour M, Adiconis X, et al. Comprehensive
comparative analysis of strand-specific RNA sequencing methods[J]. Nat Methods, 2010, 7(9): 709?715.
[56] Li H, Durbin R. Fast and accurate short read alignment
with Burrows-Wheeler transform[J]. Bioinformatics, 2009, 25(14): 1754?1760.
[57] Langmead B, Trapnell C, Pop M, et al. Ultrafast and
memory-efficient alignment of short DNA sequences to the human genome[J]. Genome Biology, 2009, 10(3): R25. [58] Li RQ, Yu C, Li YR, et al. SOAP2: an improved ultrafast
tool for short read alignment[J]. Bioinformatics, 2009, 25(15): 1966?1967.
[59] Bailey TL, Elkan C. Fitting a mixture model by expecta-tion maximization to discover motifs in biopolymers // Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. 1994, 2: 28?36.
[60] Tjaden B. Prediction of small, noncoding RNAs in bacte-ria using heterogeneous data[J]. J Math Biol, 2008, 56(1/2): 183?200.
[61] Herbig A, Nieselt K. nocoRNAc: characterization of
non-coding RNAs in prokaryotes[J]. BMC Bioinformatics,
1714
微生物学通报
2011, Vol.38, No.11
https://www.doczj.com/doc/b315427538.html,/wswxtbcn
2011, 12: 40.
[62]
Rutherford K, Parkhill J, Crook J, et al. Artemis: sequence visualization and annotation[J]. Bioinformatics, 2000, 16(10): 944?945.
[63]
Nicol JW, Helt GA, Blanchard SG Jr, et al. The Integrated Genome Browser: free software for distribution and ex-ploration of genome-scale datasets[J]. Bioinformatics, 2009, 25(20): 2730?2731.
[64]
Stothard P, Wishart DS. Automated bacterial genome analysis and annotation[J]. Current Opinion in Microbiology, 2006, 9(5): 505?510.
[65]
Duchaud E, Boussaha M, Loux V, et al. Complete genome sequence of the fish pathogen Flavobacterium psychro-philum [J]. Nature Biotechnology, 2007, 25(7): 763?769. [66]
Guzmán E, Romeu A, Garcia-Vallve S. Completely se-quenced genomes of pathogenic bacteria: a review[J]. Enferm Infecc Microbiol Clin, 2008, 26(2): 88?98.
[67]
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolution-ary tool for transcriptomics[J]. Nature Reviews Genetics, 2009, 10(1): 57?63.
[68]
Yoder-Himes DR, Chain PSG, Zhu Y, et al. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing[J]. Proceedings of the Na-tional Academy of Sciences of the United States of Amer-ica, 2009, 106(10): 3976?3981.
[69]
Sharma CM, Hoffmann S, Darfeuille F, et al. The primary
transcriptome of the major human pathogen Helicobacter pylori [J]. Nature, 2010, 464(7286): 250?255.
[70]
Frias-Lopez J, Shi YM, Tyson GW, et al. Microbial com-munity gene expression in ocean surface waters[J]. Pro-ceedings of the National Academy of Sciences of the United States of America, 2008, 105(10): 3805?3810. [71]
Sittka A, Lucchini S, Papenfort K, et al. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq[J]. PLoS Genetics, 2008, 4(8): e1000163.
[72]
Perkins TT, Kingsley RA, Fookes MC, et al. A strand-specific RNA-Seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi [J]. PLoS Genet, 2009, 5(7): e1000569.
[73]
Wurtzel O, Sapra R, Chen F, et al. A single-base resolution map of an archaeal transcriptome[J]. Genome Res, 20(1): 133?141.
[74]
Güell M, van Noort V, Yus E, et al. Transcriptome com-plexity in a genome-reduced bacterium[J]. Science, 2009, 326(5957): 1268?1271.
[75]
Toledo-Arana A, Dussurget O, Nikitas G, et al. The Lis-teria transcriptional landscape from saprophytism to viru-lence[J]. Nature, 2009, 459(7249): 950?956.
[76]
Passalacqua KD, Varadarajan A, Ondov BD, et al. Struc-ture and complexity of a bacterial transcriptome[J]. J Bacteriol, 2009, 191(10): 3203?3211.
JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ
稿件书写规范
论文中有关正、斜体的约定
物种的学名: 菌株的属名、种名(包括亚种、变种)用拉丁文斜体。属的首字母大写, 其余小写, 属以上用拉丁文正体。病毒一律用正体, 首字母大写。
限制性内切酶: 前3个字母用斜体, 后面的字母和编码正体平排, 例如: Bam H I 、Msp I 、Sau 3A I 等。 氨基酸和碱基的缩写: 氨基酸缩写用3个字母表示时, 仅第一个字母大写, 其余小写, 正体。碱基缩写为大写正体。
基因符号用小写斜体, 蛋白质符号首字母大写, 用正体。
DNA测序技术的发展及其最新进展 摘要:自从诺贝尔奖得主桑格于1977年发明了第一代DN测序技术以来,DNA测序技术已经作为重要的实验技术广泛的应用于现代生物学研究当中。经过了几十年的发展,DNA测序技术日臻成熟,并且以单分子测序为特点的第三代测序技术也已经诞生。本文主要就每一代测序技术原理和特点及其最新进展做简要介绍。 关键词:DNA测序技术;第三代DNA测序技术;最新进展 The Development and New Progress of DNA Sequencing Technology Abstract: Since Nobel Prize Winner Sanger have founded the first generation of DNA Sequence technology in 1977, DNA sequencing technology has been widely used in modern biological researches as an important experimental. Over decades of year’s development, DNA sequence technology mature gradually and the third generation sequencing technologies characterized by single-molecule sequencing have also emerged. The mechanisms and features of each generation of sequencing technology and their latest progress will be discussed here. Key Words: DNA Sequence technology ; third generation DNA sequencing ;latest development 1.引言 DNA测序技术是分子生物学研究中最常用的技术,它的出现极大地推动了生物学的发展。自从1953年Watson和Crick发现DNA双螺旋结构后[1],人类就开始了对DNA序列的探索,在世界各地掀起了DNA测序技术的热潮。1977年Maxam和Gilbert报道了通过化学降解测定DNA序列的方法[2]。同一时期,Sanger发明了双脱氧链终止法[3]。20世纪90年代初出现的荧光自动测序技术将DNA测序带入自动化测序的时代。这些技术统称为第一代DNA测序技术。最近几年发展起来的第二代DNA测序技术则使得DNA测序进入了高通量、低成本的时代。目前,基于单分子读取技术的第三代测序技术已经出现,该技术测定DNA序列更快,并有望进一步降低测序成本,推进相关领域生物学研究。本文主要介绍DNA测序技术的发展历史及不同发展阶段各种主要测序技术的特点,并针对目前新一代DNA测序技术及目前国际DNA测序最新进展做简要综述。
高通量测序基础知识简介 陆桂 什么是高通量测序? 高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变,一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。 什么是Sanger法测序(一代测序) Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。 什么是基因组重测序(Genome Re-sequencing) 全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。 什么是de novo测序 de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。 什么是外显子测序(whole exon sequencing) 外显子组测序是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等。
全基因组重测序数据分析 1. 简介(Introduction) 通过高通量测序识别发现de novo的somatic和germ line 突变,结构变异-SNV,包括重排 突变(deletioin, duplication 以及copy number variation)以及SNP的座位;针对重排突变和SNP的功能性进行综合分析;我们将分析基因功能(包括miRNA),重组率(Recombination)情况,杂合性缺失(LOH)以及进化选择与mutation之间的关系;以及这些关系将怎样使 得在disease(cancer)genome中的mutation产生对应的易感机制和功能。我们将在基因组 学以及比较基因组学,群体遗传学综合层面上深入探索疾病基因组和癌症基因组。 实验设计与样本 (1)Case-Control 对照组设计; (2)家庭成员组设计:父母-子女组(4人、3人组或多人); 初级数据分析 1.数据量产出:总碱基数量、Total Mapping Reads、Uniquely Mapping Reads统计,测序深度分析。 2.一致性序列组装:与参考基因组序列(Reference genome sequence)的比对分析,利用贝叶斯统计模型检测出每个碱基位点的最大可能性基因型,并组装出该个体基因组的一致序列。3.SNP检测及在基因组中的分布:提取全基因组中所有多态性位点,结合质量值、测序深度、重复性等因素作进一步的过滤筛选,最终得到可信度高的SNP数据集。并根据参考基 因组信息对检测到的变异进行注释。 4.InDel检测及在基因组的分布: 在进行mapping的过程中,进行容gap的比对并检测可信的short InDel。在检测过程中,gap的长度为1~5个碱基。对于每个InDel的检测,至少需 要3个Paired-End序列的支持。 5.Structure Variation检测及在基因组中的分布: 能够检测到的结构变异类型主要有:插入、缺失、复制、倒位、易位等。根据测序个体序列与参考基因组序列比对分析结果,检测全基因组水平的结构变异并对检测到的变异进行注释。
转录组学主要技术及其应用研究 姓名:梁迪 专业:微生物学 年级:2013 学号:3130179 二零一四年六月十五日
转录学主要技术及其应用研究 摘要:转录组(transcriptome)是特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的集合。转录组学研究能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程以及疾病发生过程中的分子机理。目前,转录组学研究技术主要包括两种:基于杂交技术的微阵列技术(microarray)和基于测序技术的转录组测序技术,包括表达序列标签技术(Expression Sequence Tags Technology,EST)、基因表达系列分析技术(Serial analysis of gene expression,SAGE)、大规模平行测序技术(Massively parallel signature sequencing,MPSS)、以及RNA 测序技术(RNA sequencing,RNA-seq)。文章主要介绍了以上转录组学主要研究技术的原理、技术特点及其应用,并就这些技术面临的挑战和未来发展前景进行了讨论,为其今后的研究与应用提供参考。 关键词:转录组学;微阵列技术;转录组测序技术;应用 Study on the main technologies of transcriptomics and their application Abstract: The transcriptome is the complete set of transcripts for certain type of cells or tissues in a specific developmental stage or physiological condition. Transcriptome analysis can provide a comprehensive understanding of molecularmechanisms involved in specific biological processes and diseases from the information on gene structure and function. Currently, transcriptomics technology mainly includes microarry -based on hybridization technology and transcriptome sequencing-based on sequencing technology, involving Expression sequence tags technology, Serial analysis of gene expression, Massively parallel signature sequencing and RNA sequencing. The detailed principles, technical characteristics and applications of the main transcriptomics technologies are reviewed here, and the challenges and application potentials of these technologies in the future are also discussed. This will present the useful information for other researchers. Keywords: transcriptomics ; microarray ; transcriptome sequencing; application 随着后基因组时代的到来,转录组学、蛋白质组学、代谢组学等各种组学技术相继出现,其中转 录组学是率先发展起来以及应用最广泛的技术[1]。
双脱氧链终止法又称为Sanger法 原理是:核酸模板在DNA聚合酶、引物、4 种单脱氧核苷三磷酸 ( d NTP,其中的一种用放射性P32标记 )存在条件下复制时,在四管反应系统中分别按比例引入4种双脱氧核苷三磷酸 ( dd NTP ),因为双脱氧核苷没有3’-O H,所以只要双脱氧核苷掺入链的末端,该链就停止延长,若链端掺入单脱氧核苷,链就可以继续延长。如此每管反应体系中便合成以各自 的双脱氧碱基为3’端的一系列长度不等的核酸片段。反应终止后,分4个泳道进行凝胶电泳,分离长短不一的核酸片段,长度相邻的片段相差一个碱基。经过放射自显影后,根据片段3’端的双脱氧核苷,便可依次阅读合成片段的碱基排列顺序。Sanger法因操作简便,得到广泛的应用。后来在此基础上发展出多种DNA 测序技术,其中最重要的是荧光自动测序技术。 荧光自动测序技术荧光自动测序技术基于Sanger 原理,用荧光标记代替同位素标记,并用成像系统自动检测,从而大大提高了D NA测序的速度和准确性。20世纪80 年代初Jorgenson 和 Lukacs提出了毛细管电泳技术( c a p il l ar y el ect r ophor es i s )。1992 年美国的Mathies实验室首先提出阵列毛细管电泳 ( c a p il l ar y ar r a y el ectr ophor es i s ) 新方法,并采用激光聚焦荧光扫描检测装置,25只毛细管并列电泳,每只毛细管在1.5h内可读出350 bp,DNA 序列,分析效率可达6 000 bp/h。1995年Woolley研究组用该技术进行测序研究,使用四色荧光标记法,每个毛细管长3.5cM,在9min内可读取150个碱基,准确率约 97 % 。目前, 应用最广泛的应用生物系统公司 ( ABI ) 37 30 系列自动测序仪即是基于毛细管电泳和荧光标记技术的D NA测序仪。如ABI3730XL 测序仪拥有 96 道毛细管, 4 种双脱氧核苷酸的碱基分别用不同的荧光标记, 在通过毛细管时 不同长度的 DNA 片段上的 4 种荧光基团被激光激发, 发出不同颜色的荧光, 被 CCD 检测系统识别, 并直接翻译成 DNA 序列。 杂交测序技术杂交法测序是20世纪80年代末出现的一种测序方法, 该方法不同于化学降解法和Sanger 法, 而是利用 DNA杂交原理, 将一系列已知序列的单链寡核苷酸片段固定在基片上, 把待测的 DN A 样品片段变性后与其杂交, 根据杂交情况排列出样品的序列
一代、二代、三代测序技术 (2014-01-22 10:42:13) 转载 第一代测序技术-Sanger链终止法 一代测序技术是20世纪70年代中期由Fred Sanger及其同事首先发明。其基本原理是,聚丙烯酰胺凝胶电泳能够把长度只差一个核苷酸的单链DNA分子区分开来。一代测序实验的起始材料是均一的单链DNA分子。第一步是短寡聚核苷酸在每个分子的相同位置上退火,然后该寡聚核苷酸就充当引物来合成与模板互补的新的DNA链。用双脱氧核苷酸作为链终止试剂(双脱氧核苷酸在脱氧核糖上没有聚合酶延伸链所需要的3-OH基团,所以可被用作链终止试剂)通过聚合酶的引物延伸产生一系列大小不同的分子后再进行分离的方法。测序引物与单链DNA模板分子结合后,DNA聚合酶用dNTP延伸引物。延伸反应分四组进行,每一组分别用四种ddNTP(双脱氧核苷酸)中的一种来进行终止,再用PAGE分析四组样品。从得到的PAGE胶上可以读出我们需要的序列。 第二代测序技术-大规模平行测序 大规模平行测序平台(massively parallel DNA sequencing platform)的出现不仅令DNA测序费用降到了以前的百分之一,还让基因组测序这项以前专属于大型测序中心的“特权”能够被众多研究人员分享。新一代DNA测序技术有助于人们以更低廉的价格,更全面、更深入地分析基因组、转录组及蛋白质之间交互作用组的各项数据。市面上出现了很多新一代测序仪产品,例如美国Roche Applied Science公司的454基因组测序仪、美国Illumina公司和英国Solexa technology公司合作开发的Illumina测序仪、美国Applied Biosystems公司的SOLiD测序仪。Illumina/Solexa Genome Analyzer测序的基本原理是边合成边测序。在Sanger等测序方法的基础上,通过技术创新,用不同颜色的荧光标记四种不同的dNTP,当DNA聚合酶合成互补链时,每添加一种dNTP就会释放出不同的荧光,根据捕捉的荧光信号并经过特定的计算机软件处理,从而获得待测DNA的序列信息。以Illumina测序仪说明二代测序的一般流程,(1)文库制备,将DNA用雾化或超声波随机片段化成几百碱基或更短的小片段。用聚合酶和外切核酸酶把DNA片段切成平末端,紧接着磷酸化并增加一个核苷酸黏性末端。然后将Illumina测序接头与片段连接。(2)簇的创建,将模板分子加入芯片用于产生克隆簇和测序循环。芯片有8个纵向泳道的硅基片。每个泳道内芯片表面有无数的被固定的单链接头。上述步骤得到的带接头的DNA 片段变性成单链后与测序通道上的接头引物结合形成桥状结构,以供后续的预扩增使用。通过不断循环获得上百万条成簇分布的双链待测片段。(3)测序,分三步:DNA聚合酶结合荧光可逆终止子,荧光标记簇成像,在下一个循环开
全基因组从头测序(de novo测序) https://www.doczj.com/doc/b315427538.html,/view/351686f19e3143323968936a.html 从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动物、植物、细菌、真菌的全基因组序列,从而推进该物种的研究。一个物种基因组序列图谱的完成,意味着这个物种学科和产业的新开端!这也将带动这个物种下游一系列研究的开展。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。华大科技利用新一代高通量测序技术,可以高效、低成本地完成所有物种的基因组序列图谱。包括研究内容、案例、技术流程、技术参数等,摘自深圳华大科技网站 https://www.doczj.com/doc/b315427538.html,/service-solutions/ngs/genomics/de-novo-sequencing/ 技术优势: 高通量测序:效率高,成本低;高深度测序:准确率高;全球领先的基因组组装软件:采用华大基因研究院自主研发的SOAPdenovo软件;经验丰富:华大科技已经成功完成上百个物种的全基因组从头测序。 研究内容: 基因组组装■K-mer分析以及基因组大小估计;■基因组杂合模拟(出现杂合时使用); ■初步组装;■GC-Depth分布分析;■测序深 度分析。基因组注释■Repeat注释; ■基因预测;■基因功能注释;■ ncRNA 注释。动植物进化分析■基因家族鉴定(动物TreeFam;植物OrthoMCL);■物种系统发育树构建; ■物种分歧时间估算(需要标定时间信息);■基因组共线性分析; ■全基因组复制分析(动物WGAC;植物WGD)。微生物高级分析 ■基因组圈图;■共线性分析;■基因家族分析; ■CRISPR预测;■基因岛预测(毒力岛); ■前噬菌体预测;■分泌蛋白预测。 熊猫基因组图谱Nature. 2010.463:311-317. 案例描述 大熊猫有21对染色体,基因组大小2.4 Gb,重复序列含量36%,基因2万多个。熊猫基因组图谱是世界上第一个完全采用新一代测序技术完成的基因组图谱,样品取自北京奥运会吉祥物大熊猫“晶晶”。部分研究成果测序分析结果表明,大熊猫不喜欢吃肉主要是因为T1R1基因失活,无法感觉到肉的鲜味。大熊猫基因组仍然具备很高的杂合率,从而推断具有较高的遗传多态性,不会濒于灭绝。研究人员全面掌握了大熊猫的基因资源,对其在分子水平上的保护具有重要意义。 黄瓜基因组图谱黄三文, 李瑞强, 王俊等. Nature Genetics. 2009. 案例描述国际黄瓜基因组计划是由中国农业科学院蔬菜花卉研究所于2007年初发起并组织,并由深圳华大基因研究院承担基因组测序和组装等技术工作。部分研究成果黄瓜基因组是世界上第一个蔬菜作物的基因组图谱。该项目首次将传
转录组测序技术的应用及发展综述 摘要:转录组测序(RNA-Seq)作为一种新的高效、快捷的转录组研究手段正在改变着人们对转录组的认识。RNA-Seq利用高通量测序技术对组织或细胞中所有RNA 反转录而成cDNA文库进行测序,通过统计相关读段(reads)数计算出不同RNA的表达量,发现新的转录本;如果有基因组参考序列,可以把转录本映射回基因组,确定转录本位置、剪切情况等更为全面的遗传信息,已广泛应用于生物学研究、医学研究、临床研究和药物研发等。文章主要比较近年来转录组研究的几种方法和几种RNA-Seq的研究平台,着重介绍RNA-Seq的原理、用途、步骤和生物信息学分析,并就RNA-Seq技术面临的挑战和未来发展前景进行了讨论及在相关领域的应用等内容,为今后该技术的研究与应用提供参考。 关键词: RNA-Seq;原理应用;方法;挑战;发展前景 Abstract:Transcriptome sequencing (RNA-Seq) is a kind of high efficiency, quick transcriptome research methods are changing our understanding of transcriptome. RNA-Seq to use high-throughput sequencing of tissues or cells of all RNA reverse transcription into cDNA library were sequenced, through statistical correlation read paragraph (reads) numbers were calculated from the expression of different RNA transcripts, find new; if the genome reference sequence, the transcripts mapped to genomic, determine the position of the transcription shear condition, more genetic information, has been widely used in biological research, medical research, clinical research and drug development. This paper compared several methods of platform transcriptome studies and several kinds of RNA-Seq in recent years, RNA-Seq focuses on the principle, purpose, steps and bioinformatics analysis, and discusses the RNA-Seq technology challenges and future development prospect and the application in related field and other content, provide the reference for the research and application of the technology future. Key word:RNA-Seq ;application; principle; method; challenge; development prospects
(内部资料,请勿外传) 动植物转录组 (Transcriptome ) 产品说明书 科技服务体系 动植物研究方向
版本信息: 2011年07月08日
目录 1产品概述 (1) 1.1 什么是转录组测序 (1) 1.2 转录组测序的产品功能 (1) 1.3 转录组测序产品优势 (1) 1.4 转录组测序产品发展史 (1) 1.5 项目执行时间 (3) 1.6 产品交付结果 (3) 2转录组测序研究方法 (4) 2.1 产品策略 (4) 2.2 样品准备 (5) 2.2.1 RNA样品要求 (5) 2.2.2 RNA样品送样标准 (6) 2.2.3 RNA提取的组织用量建议 (6) 2.3 样品运输要求 (7) 2.3.1 样品包装 (7) 2.3.2 样品标识 (8) 2.3.3 样品运输条件 (8) 2.4 文库的构建及测序 (9) 2.4.1 实验流程 (9) 2.4.2 测序及数据处理 (10) 2.5 转录组生物信息学分析 (10) 2.5.1 没有参考序列的转录组De novo (10) 2.5.2 有参考序列的转录组Re-sequencing (18) 2.5.3 参考文献 (24) 3成功案例 (25)
3.1 华大成功案例 (25) 3.2 相关文献解读 (26)
1产品概述 1.1什么是转录组测序? 转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。转录组测序是指用新一代高通量测序技术对物种或者组织的转录本进行测序并得到相关的转录本信息。 1.2转录组测序的产品功能 1.获得物种或者组织的转录本信息; 2.得到转录本上基因的相关信息,如:基因结构,功能等; 3.发现新的基因; 4.基因结构优化; 5.发现可变剪切; 6.发现基因融合; 7.基因表达差异分析。 1.3转录组测序产品优势 覆盖度高:检测信号是数字信号,几乎覆盖所有转录本; 检测精度高:几十到数十万个拷贝精确计数; 分辨率高:可以检测到单碱基差异,基因家族中相似基因及可变剪切造成的不同转录本的表达; 完成速度快:整个项目周期只需要50个工作日时间; 成本低:基本上每个实验室可以承担相关研究经费。 1.4转录组测序产品发展史 转录组的研究手段大体包括:EST序列构建及研究,芯片研究,运用第二代测序技术研究等。EST是从一个随机选择的cDNA 克隆进行5’端和3’端单一次sanger测序获得的短的cDNA 部分序列,代表一个完整基因的一小部分,在
高通量测序:第二代测序技 术详细介绍 -标准化文件发布号:(9456-EUATWK-MWUB-WUNN-INNUL-DDQTY-KII
在过去几年里,新一代DNA 测序技术平台在那些大型测序实验室中迅猛发展,各种新技术犹如雨后春笋般涌现。之所以将它们称之为新一代测序技术(next-generation sequencing),是相对于传统Sanger 测序而言的。Sanger 测序法一直以来因可靠、准确,可以产生长的读长而被广泛应用,但是它的致命缺陷是相当慢。十三年,一个人类基因组,这显然不是理想的速度,我们需要更高通量的测序平台。此时,新一代测序技术应运而生,它们利用大量并行处理的能力读取多个短DNA 片段,然后拼接成一幅完整的图画。 Sanger 测序大家都比较了解,是先将基因组DNA 片断化,然后克隆到质粒载体上,再转化大肠杆菌。对于每个测序反应,挑出单克隆,并纯化质粒DNA。每个循环测序反应产生以ddNTP 终止的,荧光标记的产物梯度,在测序仪的96 或384 毛细管中进行高分辨率的电泳分离。当不同分子量的荧光标记片断通过检测器时,四通道发射光谱就构成了测序轨迹。 在新一代测序技术中,片断化的基因组DNA 两侧连上接头,随后运用不同的步骤来产生几百万个空间固定的PCR 克隆阵列(polony)。每个克隆由单个文库片段的多个拷贝组成。之后进行引物杂交和酶延伸反应。由于所有的克隆都是系在同一平面上,这些反应就能够大规模平行进行。同样地,每个延伸所掺入的荧光标记的成像检测也能同时进行,来获取测序数据。酶拷问和成像的持续反复构成了相邻的测序阅读片段。
Solexa 高通量测序原理 --采用大规模并行合成测序法(SBS, Sequencing-By-Synthesis)和可逆性末端终结技术(Reversible Terminator Chemistry) --可减少因二级结构造成的一段区域的缺失。 --具有高精确度、高通量、高灵敏度和低成本等突出优势 --可以同时完成传统基因组学研究(测序和注释)以及功能基因组学(基因表达及调控,基因功能,蛋白/核酸相互作用)研究 ----将接头连接到片段上,经 PCR 扩增后制成 Library 。 ----随后在含有接头(单链引物)的芯片( flow cell )上将已加入接头的 DNA 片段变成单链后通过与单链引物互补配对绑定在芯片上,另一端和附近的另外一个引物互补也被固定,形成“桥” ----经30伦扩增反应,形成单克隆DNA簇 ----边合成边测序(Sequencing By Synthesis)的原理,加入改造过的DNA 聚合酶和带有4 种荧光标记的dNTP。这些dNTP是“可逆终止子”,其3’羟基末端带有可化学切割的基团,使得每个循环只能掺入单个碱基。此时,用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类。之后,将这些基团化学切割,恢复3'端粘性,继续聚合第二个核苷酸。如此继续下去,直到每条模板序列都完全被聚合为双链。这样,统计每轮收集到的荧光信号结果,就可以得知每个模板DNA 片段的序列。目前的配对末端读长可达到2×50 bp,更长的读长也能实现,但错误率会增高。读长会受到多个引起信号衰减的因素所影响,如荧光标记的不完全切割。 Roche 454 测序技术 “一个片段 = 一个磁珠 = 一条读长(One fragment =One bead = One read)”
原理是:核酸模板在DNA聚合酶、引物、4 种单脱氧核苷三磷酸 ( d NTP,其中的一种用放射性P32标记 )存在条件下复制时,在四管反应系统中分别按比例引入4种双脱氧核苷三磷酸 ( dd NTP ),因为双脱氧核苷没有3’-O H,所以只要双脱氧核苷掺入链的末端,该链就停止延长,若链端掺入单脱氧核苷,链就可以继续延长。如此每管反应体系中便合成以各自 的双脱氧碱基为3’端的一系列长度不等的核酸片段。反应终止后,分4个泳道进行凝胶电泳,分离长短不一的核酸片段,长度相邻的片段相差一个碱基。经过放射自显影后,根据片段3’端的双脱氧核苷,便可依次阅读合成片段的碱基排列顺序。Sanger法因操作简便,得到广泛的应用。后来在此基础上发展出多种DNA 测序技术,其中最重要的是荧光自动测序技术。 荧光自动测序技术荧光自动测序技术基于Sanger 原理,用荧光标记代替同位素标记,并用成像系统自动检测,从而大大提高了D NA测序的速度和准确性。20世纪80 年代初Jorgenson 和 Lukacs提出了毛细管电泳技术( c a p il l ar y el ect r ophor es i s )。1992 年美国的Mathies实验室首先提出阵列毛细管电泳 ( c a p il l ar y ar r a y el ectr ophor es i s ) 新方法,并采用激光聚焦荧光扫描检测装置,25只毛细管并列电泳,每只毛细管在内可读出350 bp,DNA 序列,分析效率可达6 000 bp/h。1995年Woolley研究组用该技术进行测序研究,使用四色荧光标记法,每个毛细管长,在9min内可读取150个碱基,准确率约 97 % 。目前, 应用最广泛的应用生物系统公司 ( ABI ) 37 30 系列自动测序仪即是基于毛细管电泳和荧光标记技术的D NA测序仪。如ABI3730XL 测序仪拥有 96 道毛细管, 4 种双脱氧核苷酸的碱基分别用不同的荧光标记, 在通过毛细管时 不同长度的 DNA 片段上的 4 种荧光基团被激光激发, 发出不同颜色的荧光, 被 CCD 检测系统识别, 并直接翻译成 DNA 序列。 杂交测序技术杂交法测序是20世纪80年代末出现的一种测序方法, 该方法不同于化学降解法和Sanger 法, 而是利用 DNA杂交原理, 将一系列已知序列的单链寡核苷酸片段固定在基片上, 把待测的 DN A 样品片段变性后与其杂交, 根据杂交情况排列出样品的序列
1 技术优势 全基因组测序(Whole Genome Sequencing,WGS)是利用高通量测序平台对人类不同个体或群体进行全基因组测序,并在个体或群体水平上进行生物信息分析。可全面挖掘DNA 水平的遗传变异,为筛选疾病的致病及易感基因,研究发病及遗传机制提供重要信息。 全基因组测序 平台优势 HiSeq X 测序平台 读长:PE150 通量:1.8T/run 测序周期:3 天 专为人全基因组测序准备、测序周期短、通量高
生物信息分析 技术路线 技术参数 样品要求 样本类型:DNA 样品 样本总量:≥1.0 μg DNA (提取自新鲜及冻存样本) ≥1.5 μg DNA (提取自FFPE 样本)样品浓度:≥ 20 ng/μl 测序平台及策略HiSeq X PE150 测序深度 肿瘤:癌组织(50X),癌旁组织/血液样本(30X)遗传病:30~50 X 项目周期37天
3 案例解析 该研究选取3个家系中6个患者和1个正常个体,首先使用基因芯片寻找纯合突变位点,然后对其中无亲缘关系的2例患者采用全基因组测序研究,在2例患者非编码区域均发现相同的变异,10号染色体PTF1A 末端发生一个点突变(chr10:23508437 A>G),且变异在患病人群和细胞试验中均得到了验证。研究解释了生长发育启动子隐性变异是罕见孟德尔遗传病的常见致病原因,同时说明许多疾病的致病突变也可能位于非编码区。 图1 检出的变异信息 智力障碍是影响新生儿心智发育的一类疾病。这项研究选取50个经过基因芯片和全外显子测序未确诊致病因子的trio 家系,全基因组测序检出84个de novo SNVs 和8个de novo CNVs,及一些结构变异(如VPS13B、STAG1、IQSEC2-TENM3),检出率为42%。揭示编码区的de novo SNVs 和de novo CNVs 是导致智力障碍的主要因素,全基因组测序可以作为可靠的遗传性检测应用工具。 案例一 单基因病研究——全基因组测序鉴定PTF1A末端增强子常染色体隐性突变导致胰腺 发育不全[1] 案例二 复杂疾病研究——全基因组测序解析智力障碍的主要致病因素[2] 图2 PTF1A 的家系图谱
基因组组装 摘要 基因组测序是生物信息学的核心,有着极其重要的应用价值。新的测序技术大量涌现,产生的reads长度更短,数量更多,覆盖率更大,能直接读取的碱基对序列长度远小于基因组长度。所以测序之前DNA分子要经过复制若干份、随机打断成短片段。要获取整个DNA片段,需要把这些片段利用重合部分信息组织连接。如何在保证组装序列的连续性、完整性和准确性的同时设计耗时短、内存小的组装算法是本题的关键。 本文建立改进后OLC算法模型。该模型首先使用了特定的编码规定,通过C++程序对庞大的数据先后进行十进制和二进制的处理,不改变数据准确性的前提下尽可能减小内存和缩短计算机操作时间,并引入解决碱基识别错误问题的一般思路消除初始reads中的碱基错误。然后通过深度优先算法,设定适当的阈值,找出具有重叠关系的碱基片段并形成一有向赋权图,其中点是碱基片段,边代表具有重叠关系,权值代表片段重叠的多少,将问题转化为图论中寻找最大赋权通路的问题,从而对OLC算法进行改进,采用图论的方法更直观和更具操作性的解决DNA的拼接问题,从而对OLC算法进行改进。最后再根据OLC算法对Hamilton 路径进行拼接,生成共有序列,通过多序列比对等方法,获得最终的基因组序列。 关键词:基因组测序 OLC算法深度优先算法Hamilton路径
一问题的重述 1.1 问题背景 快速和准确地获取生物体的遗传信息对于生命科学研究具有重要的意义。对每个生物体来说,基因组包含了整个生物体的遗传信息,这些信息通常由组成基因组的DNA或RNA分子中碱基对的排列顺序所决定。获得目标生物基因组的序列信息,进而比较全面地揭示基因组的复杂性和多样性,成为生命科学领域的重要研究内容。 1.2 问题提出 确定基因组碱基对序列的过程称为测序。目前能直接读取的碱基对序列长度远小于基因组序列长度,因此需要利用一定的方法将测序得到的短片段序列组装成更长的序列。通常的做法是,将基因组复制若干份,无规律地分断成短片段后进行测序,然后寻找测得的不同短片段序列之间的重合部分,并利用这些信息进行组装。例如,若有两个短片段序列分别为 ATACCTT GCTAGCGT GCTAGCGT AGGTCTGA 则有可能基因组序列中包含有ATACCTT GCTAGCGT AGGTCTGA这一段。 由于技术的限制和实际情况的复杂性,最终组装得到的序列与真实基因组序列之间仍可能存在差异,甚至只能得到若干条无法进一步连接起来的序列。对组装效果的评价主要依据组装序列的连续性、完整性和准确性。连续性要求组装得到的(多条)序列长度尽可能长;完整性要求组装序列的总长度占基因组序列长度的比例尽可能大;准确性要求组装序列与真实序列尽可能符合。 利用现有的测序技术,可按一定的测序策略获得长度约为50–100个碱基对的序列,称为读长(reads)。基因组复制份数约为50–100。基因组组装软件可根据得到的所有读长组装成基因组,这些软件的核心是某个组装算法。一个好的算法应具备组装效果好、时间短、内存小等特点。新一代测序技术在高通量、低成本的同时也带来了错误率略有增加、读长较短等缺点,现有算法的性能还有较大的改善空间。具体解决问题如下: (1)建立数学模型,设计算法并编制程序,将读长序列组装成基因组。你的算法和程序应能较好地解决测序中可能出现的个别碱基对识别错误、基因组中存在重复片段等复杂情况。 (2)现有一个全长约为120,000个碱基对的细菌人工染色体,采用Hiseq2000测序仪进行测序,测序策略以及数据格式的简要说明见附录一和附录二,测得的读长数据见附录三,测序深度约为70×,即基因组每个位置平均被测到约70次。试利
在过去几年里,新一代DNA 测序技术平台在那些大型测序实验室中迅猛发展,各种新技术犹如雨后春笋般涌现。之所以将它们称之为新一代测序技术(next-generation sequencing),是相对于传统Sanger 测序而言的。Sanger 测序法一直以来因可靠、准确,可以产生长的读长而被广泛应用,但是它的致命缺陷是相当慢。十三年,一个人类基因组,这显然不是理想的速度,我们需要更高通量的测序平台。此时,新一代测序技术应运而生,它们利用大量并行处理的能力读取多个短DNA 片段,然后拼接成一幅完整的图画。 Sanger 测序大家都比较了解,是先将基因组DNA 片断化,然后克隆到质粒载体上,再转化大肠杆菌。对于每个测序反应,挑出单克隆,并纯化质粒DNA。每个循环测序反应产生以ddNTP 终止的,荧光标记的产物梯度,在测序仪的96或384 毛细管中进行高分辨率的电泳分离。当不同分子量的荧光标记片断通过检测器时,四通道发射光谱就构成了测序轨迹。 在新一代测序技术中,片断化的基因组DNA 两侧连上接头,随后运用不同的步骤来产生几百万个空间固定的PCR 克隆阵列(polony)。每个克隆由单个文库片段的多个拷贝组成。之后进行引物杂交和酶延伸反应。由于所有的克隆都是系在同一平面上,这些反应就能够大规模平行进行。同样地,每个延伸所掺入的荧光标记的成像检测也能同时进行,来获取测序数据。酶拷问和成像的持续反复构成了相邻的测序阅读片段。
Solexa高通量测序原理 --采用大规模并行合成测序法(SBS,Sequencing-By-Synthesis)和可逆性末端终结技术(ReversibleTerminatorChemistry) --可减少因二级结构造成的一段区域的缺失。 --具有高精确度、高通量、高灵敏度和低成本等突出优势 --可以同时完成传统基因组学研究(测序和注释)以及功能基因组学(基因表达及调控,基因功能,蛋白/核酸相互作用)研究 ----将接头连接到片段上,经PCR扩增后制成Library。 ----随后在含有接头(单链引物)的芯片(flowcell)上将已加入接头的DNA片段变成单链后通过与单链引物互补配对绑定在芯片上,另一端和附近的另外一个引物互补也被固定,形成“桥” ----经30伦扩增反应,形成单克隆DNA簇 ----边合成边测序(Sequencing By Synthesis)的原理,加入改造过的DNA 聚合酶和带有4 种荧光标记的dNTP。这些dNTP是“可逆终止子”,其3’羟基末端带有可化学切割的基团,使得每个循环只能掺入单个碱基。此时,用激光扫描反应板表面,读取每条模板序列第一轮反应所聚合上去的核苷酸种类。之后,将这些基团化学切割,恢复3'端粘性,继续聚合第二个核苷酸。如此继续下去,直到每条模板序列都完全被聚合为双链。这样,统计每轮收集到的荧光信号结果,就可以得知每个模板DNA 片段的序列。目前的配对末端读长可达到2×50 bp,更长的读长也能实现,但错误率会增高。读长会受到多个引起信号衰减的因素所影响,如荧光标记的不完全切割。 Roche 454 测序技术 “一个片段= 一个磁珠= 一条读长(One fragment =One bead = One read)”
转录组测序(RNA-seq)技术 转录组是某个物种或者特定细胞类型产生的所有转录本的集合。转录组研究能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程以及疾病发生过程中的分子机理,已广泛应用于基础研究、临床诊断和药物研发等领域。基于Illumina高通量测序平台的转录组测序技术使能够在单核苷酸水平对任意物种的整体转录活动进行检测,在分析转录本的结构和表达水平的同时,还能发现未知转录本和稀有转录本,精确地识别可变剪切位点以及cSNP(编码序列单核苷酸多态性),提供最全面的转录组信息。相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。 技术优势: 数字化信号:直接测定每个转录本片段序列,单核苷酸分辨率的精确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题。 高灵敏度:能够检测到细胞中少至几个拷贝的稀有转录本。 任意物种的全基因组分析:无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析。同时能够检测未知基因,发现新的转录本,并精确地识别可变剪切位点及cSNP,UTR区域。 更广的检测范围:高于6个数量级的动态检测范围,能够同时鉴定和定量稀有转录本和正常转录本。 应用领域:转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP研究),非编码区域功能研究(Non-coding RNA研究、microRNA前体研究等),基因表达水平研究以及全新转录本发现。 图1 RNA-seq获得的数据能够进行全面的数据挖掘,既能够进行基因结构分析,鉴定UTR、可变剪切位点,也能够发现新的转录本及非编码RNA,比较样本间的表达水平差异