
植物RNA 原位杂交方法
一、制片
1. 材料的固定和包埋
固定液:FAA: 10%甲醛,5%冰醋酸,50%乙醇(均为RNase-free, DEPC 水配制)
Day 1:
取植物材料,茎尖或花原基(如花原基,不要剥除最外层苞片),立即放入FAA,抽真
空15min,缓慢放气,换一次固定液,4℃过夜。
Day 2:
脱水:
50%乙醇30min
50%乙醇60min
60%乙醇60min
70%乙醇60min 组织块可在70%乙醇中4℃保存达几个月
Day 3:
85%乙醇60min
95%乙醇60min
100%乙醇30min
100%乙醇30min
100%乙醇60min
25%二甲苯+75%乙醇60min 以下有二甲苯,在通风橱中操作
50%二甲苯+50%乙醇60min
75%二甲苯+25%乙醇60min
100%二甲苯60min
100%二甲苯60min
100%二甲苯60min 组织透明,也可两次100%二甲苯,第二次至组织透明为止
50%二甲苯+50%石蜡58℃过夜
Day 4:
换两次100%石蜡(58℃),倾倒时避免产生起泡
Day 5:
换两次100%石蜡(58℃),倾倒时避免产生起泡
Day 6:
换两次100%石蜡(58℃),倾倒时避免产生起泡
用干净较硬的纸(如打印纸)折好纸盒,薄的可双层折。
包埋。
2. 杂交探针的制备
1)将要转录的DNA(通常50-300bp,无polyA 尾)克隆在PGEM T-easy vector 上,摇菌,提质粒,送样测序,确定目的片段的连入方向(以T7 或者SP6 引物测序出的连入方向,即是用T7 或是SP6 转录酶转录出的sense 探针的方向。例如,某片段T7 引物的测序结果是反向连入,则用T7 转录出的探针是antisense,才是正确探针)
2)用T7,SP6 引物从质粒上将目的片段扩增出来(大体积扩增,通常200-400ul)。若产物
干净则可直接回收,不干净切胶回收。回收用RNase-free 的管子,NFW 水溶解(30-40ul, 浓度>200ng/ul)
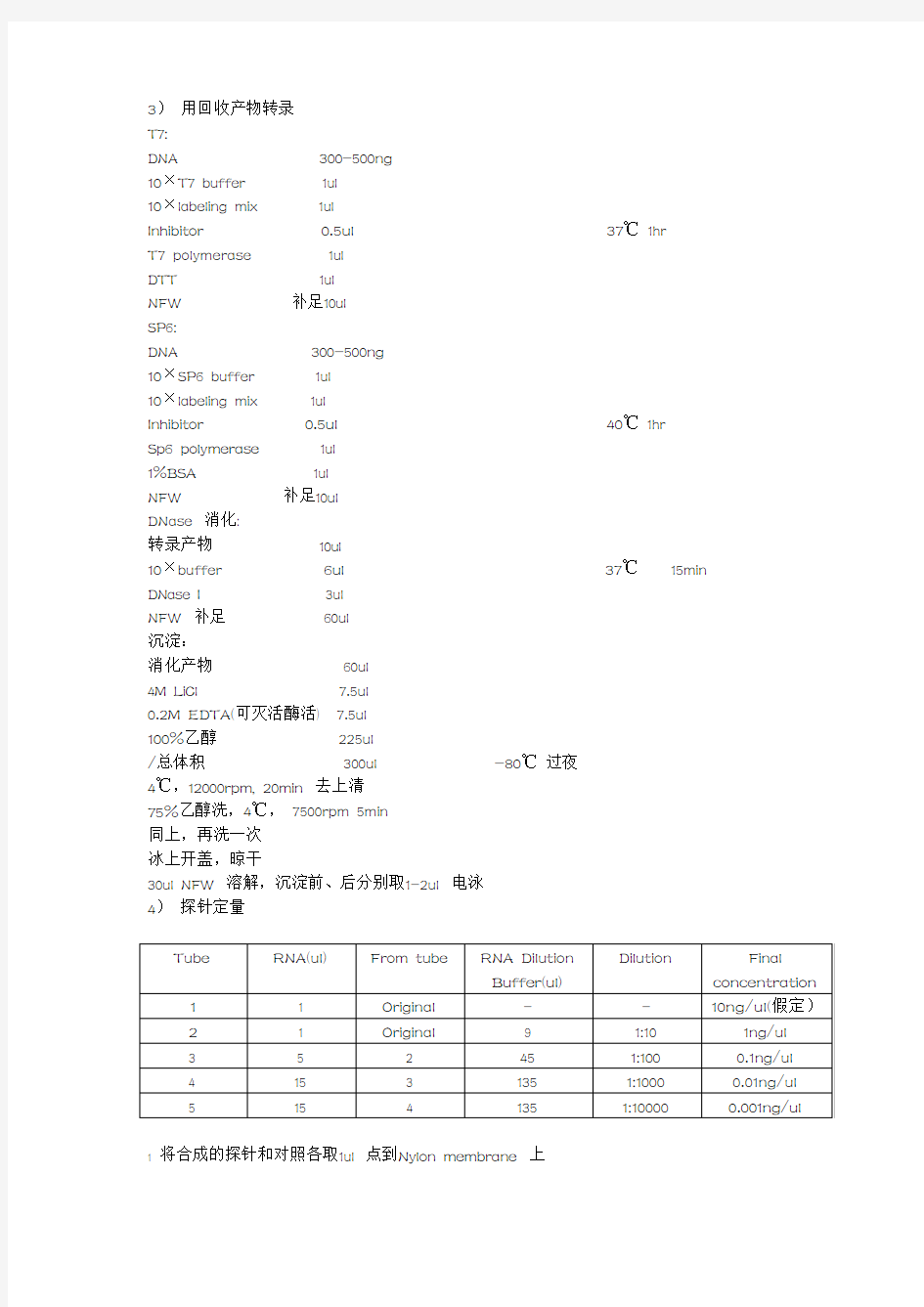
3)用回收产物转录
T7:
DNA 300-500ng
10×T7 buffer 1ul
10×labeling mix 1ul
Inhibitor 0.5ul37℃1hr
T7 polymerase 1ul
DTT 1ul
NFW 补足10ul
SP6:
DNA 300-500ng
10×SP6 buffer 1ul
10×labeling mix 1ul
Inhibitor 0.5ul40℃1hr
Sp6 polymerase 1ul
1%BSA 1ul
NFW 补足10ul
DNase 消化:
转录产物10ul
10×buffer 6ul37℃15min DNase I 3ul
NFW 补足60ul
沉淀:
消化产物60ul
4M LiCl 7.5ul
0.2M EDTA(可灭活酶活) 7.5ul
100%乙醇225ul
/总体积300ul -80℃过夜
4℃,12000rpm, 20min 去上清
75%乙醇洗,4℃,7500rpm 5min
同上,再洗一次
冰上开盖,晾干
30ul NFW 溶解,沉淀前、后分别取1-2ul 电泳
4)探针定量
1 将合成的探针和对照各取1ul 点到Nylon membrane 上
2 UV-cross linker(2400)
3 将膜放到2ml 冻存管中
4 2ml Washing buffer 2min
5 2ml Blocking solution 30min
6 2ml Antibody solution 30min
7 2ml Washing buffer 15min
8 2ml Washing buffer 15min
9 2ml Detection buffer 2-5min
10 2ml Detection buffer+20ulNBT/BCIP(2:1)锡箔纸包裹冻存管,5min 看一次
探针定量所需试剂:
3. 切片
将已用石蜡包埋好的植物材料修成梯形,切成8um 厚的蜡带材料(尽量少带石蜡,
但不能过少,因为材料周围的石蜡太少,则容易切断,不易形成蜡条,而且梯形要规整,否则蜡带会绕圈,不方便展片)。按顺序摆放,正面朝上。
在解剖镜下用暗视野观察蜡带,以决定取舍。
在聚赖氨酸化的载玻片(Fisher)上面铺满DEPC 水,然后挑选合适的蜡带小心地
放在水面上(可在一张片上摆上从大到小不同时期)(注意正反面!!!),蜡带完全展开后(42℃烫板上数分钟至几十分钟不等,注意观察以免展过,以组织不开裂为度),用
一张镜头纸轻轻压在蜡带上,然后用吸水纸(枪头)吸掉载玻片上多余的水,使材料与
载玻片之间无水存在,置于42℃烫板上烘烤干燥36-48hr 后备用,制好的片子最好在一星期内进行杂交)
二、片子预处理
1. Deparaffine and rehydration
100% xylene 二甲苯20min——100% xylene 20min——75% xylene+25% ethanol 2min ——25% xylene+75% ethanol——100% ethanol——95% ethanol——80% ethanol——60%
ethanol——30% ethanol——DEPC H2O( each step 2 min after the 1st and 2nd steps)
2. 0.25M HCl 20min (862ul HCl→40ml H2O)
3. DEPC-H2O 5min
4. 2×SSC 20min
5. DEPC-H2O 5min
6. Proteinase K: Proteinase K buffer (37℃pre-warmed,40ml) + Proteinase K (8ul,
20mg/ml) 37℃,30min
K buffer: 32ml DEPC-H2O + 4ml 1M Tris-HCl pH7.5 + 4ml 0.5M EDTA pH7.5
7. RT, 1×PBS, 2min
8. RT, 0.2% glycine in 1×PBS, 2min (0.08g glycine 现称in 40ml 1×PBS)
9. RT, 1×PBS, 2×2min
10. RT, 4% formaldehyde, 10min( (4.3ml 甲醛溶液(浓度37%-40%)+35.7ml 1×PBS)
11. RT, 1×PBS, 2×5min
12. To reduce background: (现用现加)
0.1M triethanolamine pH8.0(三乙醇胺,不调pH)with 0.5% acetic anhydride(乙酸
酐),with constant stirring.
Add 536ul triethanolamine +160ul HCl pH8.0(不调pH),and 200ul acetic anhydide
in 40ml PBS and immediately add slides. Incubate at RT for 5min.
13. RT, 1×PBS, 2×5min
三、杂交
1.杂交体系
每张片子大约用100-150ul 杂交液,杂交液由A 和B 组成,比例为150ul 杂交液由
115.8ul 杂交液A 加34.2ul 杂交液B 组成
杂交液A 15.44ml: 10ml 去离子甲酰胺
2ml 50% dextran sulfate (1g 溶于2ml formamide)
2ml 10×blocking solution
1.2ml 5M NaCl
0.2ml 1M Tris-HCl pH7.5
0.04ml 0.5M EDTA pH7.5
杂交液B 34.2ul: 2.2ul tRNA
32ul probe+DEPC-H2O
杂交液B 80℃5min 变性后立即放置冰上5min
2.杂交
用parafilm 膜或者盖玻片轻轻覆盖预处理后的片子,42℃(42℃-50℃)在湿室中杂交
过夜(湿室中垫有用0.3M NaCl-2×SSC-50%甲酰胺饱和的吸水纸,放在一个大平皿中,每皿需要50ml)
四、冲洗
1. 准备冲洗缓冲液2×SSC,50%甲酰胺(国产),准备2 次用量
20×SSC 50ml 5ml 4ml 8ml
甲酰胺(国产)250ml 25ml 20ml 40ml
DEPC-H2O 200ml 20ml 16ml 32ml
总体积500ml(大)50ml(小)40ml 80ml
20×SSC (pH7.0) 1L: NaCl 173.5g
Na3citrate 柠檬酸钠88.2g
2. 染缸中加入冲洗缓冲液,放入载玻片,50℃水浴中温育30min(此时盖玻片会落下)
3. 在冲洗过程中准备NTE 溶液,准备5 次用量。
5×NTE (pH7.5) 1L: NaCl 146.1g
Tris 6.05g
EDTA 1.86g
5 次
6 次
5×NTE 500ml 60ml
DEPC-H2O 2000ml 240ml
总体积2500ml(大)300ml(小染缸)
4. 用NTE 溶液,在37℃水浴中冲洗两次,每次5min
5. 准备RNA 酶A 溶液(20ug/ml),将载玻片放入溶液中37℃水浴温育30min
RNA 酶A 储备液10mg/ml 1ml 0.1ml
NTE 溶液500ml(大染缸)50ml(小染缸)
注:(1)RNA 酶A 溶液应提前放入37℃水浴中预热5min
(2) 加入RNA 酶以后各步使用蒸馏水即可
(3)加入RNA 酶A 之前和之后使用的玻璃缸一定要分开放置,切勿混用!
(4)加入RNA 酶A 之前使用的玻璃缸,每次用完后冲洗干净,加入部分100%乙醇
溶液,下次使用在通风橱中吹干即可。
6. 准备溶液:
1)1×SSC:
20×SSC 25ml 2.5ml 2ml
水475ml 47.5ml 38ml
总体积500ml(大染缸)50ml(小染缸)40ml
2) 10×PBS( 1×PBS: 130mM NaCl, 7mM Na2HPO4.7H2O, 3mM NaH2PO4.H2O)
NaCl 15.194g 22.791g 37.985g Na2HPO4.7H2O(Na2HPO4.12H2O) 3.752g(5.012g) 5.628g(7.518g) (12.53g) NaH2PO4.H2O(NaH2PO4.2H2O) 0.828g(0.936g) 1.242g(1.404g) (2.34g) 体积200ml 300ml 500ml 3) 1×PBS:
10×PBS(pH6.5-7.0)50ml5ml
水450ml45ml
总体积500ml(大染缸)50ml(小染缸)
7. NTE 溶液,室温冲洗两次,每次5min,37℃
8. 冲洗缓冲液,50℃水浴,温育1hr
9. 1×SSC,RT,2min
10. PBS,RT,5min
五、抗体染色
1.准备溶液:
10×DIG 1 (pH8.0) 1L: Tris 121.1g
NaCl 87.7g
1)DIG1:(配10 个,DIG2,DIG3 均在DIG1 基础上配制)
10×DIG1 400ml 50ml 40ml
水3600ml 450ml360ml
总体积4000ml(大染缸)500ml(小染缸)400ml
2)DIG2:
10×Blocking reagent 50ml 5ml 4ml
DIG1 450ml 45ml 36ml
总体积500ml(大染缸)50ml(小染缸)40ml
注:新鲜配制,在60-70℃水浴中溶解1hr,溶液仍显浑浊。Blocking reagent 本身有很
多类似核酸的片段,可用于封闭核酸。这一步的目的是防止非特异性的吸附
3)DIG3:(准备5-6 次用量)
牛血清白蛋白(BSA) 25g 3g 0.5g
DIG1 2500ml 300ml50ml
Triton X-1007.5ml 0.9ml0.15ml
总体积2500ml(大)300ml(小,6 次)50ml(1 次)注:当天新鲜配制,这一步的目的是封闭蛋白
4)DIG4:(不用染缸,直接点在片上,节约)
每张片150ul, 10 张1.5ml
加入抗体anti-digoxingenin-AP(1:3000) 0.5ul
注:使用前2-3min 前再配制
5)DIG5:(准备2-3 次用量,两小盒染色)
0.5M Tris pH9.5 20ml 30ml
5M NaCl 2ml 3ml
1M MgCl2(MgCl2.6H2O)1.016g 1.524g
SDW 78ml 117ml
体积100ml 150ml
6)DIG6:25ml/枪头盒
DIG5 75ml(3 盒或大盒)150ml(6 盒)
NBT(50mg/ml) 225ul 450ul 总浓度0.15mg/ml BCIP(50mg/ml) 112.5ul 225ul0.075mg/ml
注:使用前再配制,配制时应避光
2.染色
DIG1 5min
DIG2 60min(封闭,可超过1hr) RT
DIG3 30min
DIG4 90min-2hr RT
3.冲洗
DIG3 4×20min (2-4 次,摇床上摇)
4.平衡
DIG1 5min
DIG5 5min
5.显色
在每一个枪头盒中加入25ml DIG6,将片子放入其中,显色液没过片子即可。
6.将盒子盖好,密封(避免蒸发),用锡箔纸包好(避光),置黑暗处显色36hr 或更长,每
隔12hr 检查显色情况,以免背景过深(可以放在28℃烘箱中)
(可以在此时拍照,滴加无水乙醇,盖上盖玻片,注意要快一些照,以防信号消失)
7.终止酶反应,洗去背景,将载玻片放回染缸中进行冲洗(此时应根据切片的染色情况决定
冲洗的时间,如果背景染色不深的话,越短越好)
停显液:PBS (pH7.4)+20mM EDTA
PBS (pH7.4): 131mM NaCl, 2.7mM KCl, 10mM Na2HPO4.12H2O, 2mM KH2PO4
10×PBS(pH7.4) 1L:NaCl 80g
KCl 2g
Na2HPO435.8g
KH2PO4 2.7g
HCl 调pH 至7.4,定容至1L
蒸馏水5s
70%乙醇5s
95%乙醇5s
100%乙醇5s
95%乙醇5s
70%乙醇5s
蒸馏水5s
注:(1)冲洗的时间需镜检后再决定,若背景较弱则处理时间相应减少。
(2)终止前信号呈粉红色,经乙醇脱水复水后变为紫色。
六、荧光染色
1.用水配置0.1%的荧光染料(Fluorescent brightener, Sigma)
注:(1)用一次配一次
(2)溶解一段时间后,低俗离心,取上清液去染色(也可用细菌过滤免菌期过滤以除去颗粒)
(3)每8 片载玻片染色后,不必换新的荧光染料
2.载玻片在荧光染料中染色5 分钟
3.很快用水冲洗一下,放在超净台上吹干
七、封片
八、封片
滴加50ul(视材料而定)国产中性树胶,轻轻盖上盖玻片,避免产生气泡。
注:封片后一定要在通风橱中过夜干燥,才能在荧光显微镜下观察
△多聚赖氨酸,工作浓度1mg/ml(配的母液浓度5mg/ml)
每张片子涂10ul__
原位杂交组织(或细胞)化学(In situ Hybridization Histochemistry,ISHH)简称原位杂交(In Situ Hybridization),属于固相分子杂交的范畴,它是用标记的DNA或RNA为探针,在原位检测组织细胞内特定核酸序列的方法。根据所用探针和靶核酸的不同,原位杂交可分为DNA-DNA杂交,DNA-RNA杂交和RNA-RNA 杂交三类。 根据探针的标记物是否直接被检测,原位杂交又可分为直接法和间接法两类。直接法主要用放射性同位素、荧光及某些酶标记的探针与靶核酸进行杂交,杂交后分别通过放射自显影、荧光显微镜术或成色酶促反应直接显示。间接法一般用半抗原标记探针,最后通过免疫组织化学法对半抗原定位,间接地显示探针与靶核酸形成的杂交体。 原位杂交最初是以同位素标记探针进行的。尽管同位素标记(如35S,3H,32P等)仍然广泛使用,但非同位素标记探针的迅速发展(尤其是生物素标记探针和地高辛标记探针),更引起科技工作者的极大兴趣。 一、基本要求 1. 组织取材:组织取材应尽可能新鲜。由于组织RNA降解较快,所以新鲜组织和培养细胞最好在30 min 内固定。 2. 固定目的是: (1)保持细胞结构; (2)最大限度地保持细胞内DNA或RNA的水平; (3)使探针易于进入细胞或组织。 最常用的固定剂是多聚甲醛,与其它醛类固定剂(如戊二醛)不同,多聚甲醛不会与蛋白质产生广泛的交叉连接,因而不会影响探针穿透入细胞或组织。 3. 增强组织的通透性和核酸探针的穿透性: (1)稀酸处理和酸酐处理:为防止探针与组织中碱性蛋白之间的静电结合,以降低背景,杂交前标本可用0.25%乙酸酐处理10 min,经乙酸酐处理后,组织蛋白中的碱性基团通过乙酰化而被阻断。组织和细胞标本亦可用0.2 M HCl处理10 min,稀酸能使碱性蛋白变性,结合蛋白酶消化,容易将碱性蛋白移除。 (2)去污剂处理:去污剂处理的目的是增加组织的通透性,利于杂交探针进入组织细胞,最常应用的去污剂是Triton X-100。注意:过度的去污剂处理不仅影响组织的形态结构,而且还会引起靶核酸的丢失。
荧光原位杂交(FISH)综述 摘要 本文简单介绍了荧光原位杂交(FISH)技术的一些基础理论知识以及常用操作方法和步骤。 关键词:荧光原位杂交; 1.发展 荧光原位杂交(fluorescent in situ hybridization,FISH)是一种细胞遗传学技术,可以用来对核酸进行检测和定位。荧光标记的核酸探针只和具有高度相似性的核酸杂交,可用于染色体上基因的定位,或在分子生态学中用来标记不同分类细菌或古菌中的核糖体RNA[1]。1969年,Pardue等和John两个研究小组发明了原位杂交技术,放射性标记的DNA 或28s RNA 被杂交到细胞制备物上,通过放射自显影技术(m icroautoradiography, MAR)检测杂交位点,这一技术可以在保持细胞形态完整性的条件下,使核酸序列在细胞内被检测[2]。 2.原理 通过特定分子的荧光标记探针在细胞内与染色体上特意的互补核酸序列原位杂交,通过激发杂交探针的荧光来检测信号。由于荧光燃料收到一定波长的(即激发波长)的光激发后会发射荧光(即发射波长),所以就滤光镜选择合适的激发波长的光,即可显示某一特定的荧光染料,于是就可以直接显示特定细胞核中或染色体上的DNA序列间相互位置关系[2]。 原位杂交的处理:染色体上杂交的位点提供了DNA探针序列的定位信息。所以应用该方法时,需打开维持染色体DNA双螺旋结构的碱基配对以使其形成单链分子(这称为DNA变性)。只有这样染色体DNA才能与探针杂交。变性染色体DNA而不破坏其形态的标准方法是将染色体干燥在玻璃载玻片上,再用甲酰胺处理[1]。
3.关于探针的发展 早期原位杂交技术中探针是放射性标记的,但这个方法并不令人满意,因为放射性标记很验证同时满足灵敏度和分辨率这两个原位杂交成功的必要条件。灵敏度要求放射性标记具有高中辐射能(例如用32P标记),当标记物能量过高时,会因为信号散射导致分辨率过低。如果使用低辐射能的放射性标记物,如 3H可以得到较高的分辨率,但由于灵敏度低而需要长时间曝光,并由此导致背景过高,难以分辨出真正的信号。20世纪80年代后期,非放射性DNA荧光标记技术的发展解决了上述问题,这些标记将高灵敏度与高分辨率结合起来,适用于原位杂交。 现已设计出具有不同发光特性的荧光标记物,因此有可能将一组不同的探针与单个染色体杂交,并分辨出每种杂交信号,从而测定出各探针序列的相对位置。为了得到最高的灵敏度,探针的标记需要尽可能大一些。在过去这就意味着探针必须是相当长的DNA分子,通常是至少40kb的克隆片段。现在已发展出将较短的DNA分子进行标记的技术,对长度的要求已不那么重要。 构建物理学图谱时,克隆的DNA片段可被简单地看作另一种类型的标记物,但在实际应用,由于克隆的DNA片段确定了DNA序列,将其作为标记应用则具有另一层含义。因此,克隆间位置关系的确定提供了基因组图谱与其DNA序列间的直接联系。如果探针是长的DNA片段,至少对于高等真核生物,就可能产生这样一个问题:探针中可能含有一些重复的DNA序列,因此探针可能与染色体上多个位点杂交,探针在使用前要与来自被研究组织的未标记DNA混合。这种DNA可以是总的核DNA(即代表了全基因组),但如果使用富集重复序列的片段更好。加入未标记DNA的目的在于与探针中的重复序列结合并将其封闭,使随后的原位杂交完全由单一序列驱动(Lichter et al.,1990)。这样非特异性杂交即可被减少或完全消除[1]。 4.荧光染料 常用的荧光染料的DAPI(在UV 光激发下发出蓝色荧光)、FITC和荧光素(蓝光 下发出绿色荧光)以及罗丹明和德克萨斯红(绿光激发下产生红色荧光)。由于FISH 的信号空间分辨率高。不同的DNA 探针可以用不同的半抗原标。再用不
原位杂交原理及具体操作
原位杂交实验原理与方法 一、目的 本实验的目的是学会原位杂交的使用方法。了解各种原位杂交的基本原理和优缺点。 二、原理 原位杂交组化(简称原位杂交,in situ hybridization histochemistry;ISHH)属于分子杂交的一种,是一种应用标记探针与组织细胞中的待测核酸杂交,再应用标记物相关的检测系统,在核酸原有的位置将其显示出来的一种检测技术。原位杂交的本质就是在一定的温度和离子浓度下,使具有特异序列的单链探针通过碱基互补规则与组织细胞内待测的核酸复性结合而使得组织细胞中的特异性核酸得到定位,并通过探针上所标记的检测系统将其在核酸的原有位置上显示出来。 当然杂交分子的形成并不要求两条单链的碱基顺序完全互补,所以不同来源的核酸单链只要彼此之间有一定程度的互补顺序(即某种程度的同源性)就可以形成杂交双链。 探针的种类按所带标记物可分为同位素标记探针和非同位素标记探针两大类。目前,大多数放
射性标记法是通过酶促反应将标记的基因掺入DNA中,常用的同位素标记物有3H、35S、125I 和32P。同位素标记物虽然有灵敏性高,背底较为清晰等优点,但是由于放射性同位素对人和环境均会造成伤害,近来有被非同位素取代的趋势。非同位素标记物中目前最常用的有生物素、地高辛和荧光素三种。 探针的种类按核酸性质不同又可分为DNA探针、cDNA探针、cRNA探针和合成寡核苷酸探针。cDNA 探针又可分为双链cDNA探针和单链cDNA探针。原位杂交又可分为菌落原位杂交和组织原位杂交。 菌落原位杂交(Colony in situ hybridization)菌落原位杂交是将细菌从培养平板转移到硝酸 纤维素滤膜上,然后将滤膜上的菌落裂菌以释出DNA。将NDA烘干固定于膜上与32P标记的探针杂交,放射自显影检测菌落杂交信号,并与平板上的菌落对位。 组织原位杂交(Tissue in situ hybridization)组织原位杂交简称原位杂交,指组织或细胞的原位杂交,它与菌落的原位杂交不同。菌落原位杂交需裂解细菌释出DNA,然后进行杂交。而原位
原位杂交实验操作步骤 一质粒制备 1质粒的转化和扩增 1.1制备XL1-Blue感受态细菌 1.取400uLXL1-Blue菌种加入到含200mlLB培养基的锥形瓶中,37℃、100rpm 培养4h,离心,倒置,以冰冷的0.1mol/LCaCl_2重悬细菌,冰浴30min,离心,弃上清,倒置,再加4ml(含15%甘油)冰冷的CaCl2重悬细菌,分装(200μ/tube),-80℃保存。 2.转化:在冰浴中将1管XL1-Blue感受态菌解冻,将浓度为2ng/μ1的质粒DNA4μ1加入到8Oμ1感受态菌中。 3.轻轻摇匀,冰浴30min。 4.42℃热激9O秒,然后迅速冰浴2min。 5.加入LB培养液(无氨苄青霉素)0.8ml,在37℃,100转/min水浴孵育60min。 6.取200μl菌液铺于琼脂板上(涂有X-Gal(20mg/ml)-IPTG(200mg/ml)的LB-氨苄青霉素50mg/ml,1μl/ml培养基),待菌液全部被吸收后,倒置平板于37℃培养12-16h。 1.2鉴定和挑选含重组质粒的菌落 1.用无菌牙签挑取单菌落,接种到10ml含氨苄青霉素的LB培养液的离心管中,于37℃,200转/分培养2h,取1ml之一Eppendorf离心管,加 50μl10mmol/L EDTA(pH8.0)。 2.加入50μl新配置的0.2mol/LNaOH、0.5%SDS、20%蔗糖溶液后,振荡30秒。 3.在70℃温育5min,然后冷却到室温。 4.加1.5μ14mol/LKCl和0.5μ1含0.4%溴酚兰染液,振荡3O秒后,冰浴5min。 5.12000g,4℃离以3min,以除去细菌碎片。 6.制备1%的琼脂糖凝胶(含EB0.5μg/ml),取50μl上清液加入到样品孔中,其中一孔加入中等分子量DNAMarker。恒压50V,进行电泳。 7.当溴酚兰迁移到凝胶全长的2/3-3/4时,停止电泳,在紫外灯下检查质粒DNA 分于量的大小是否与转入质粒相符。
原位杂交组织化学技术的基本方法 一、核酸分子杂交技术 1961年Hall开拓了液相核酸杂交技术的研究,其基本原理是利用核酸分子单链之间有互补的碱基顺序,通过碱基对之间非共价键的形成,出现稳定的双链区,形成杂交的双链。自此以后,由于分子生物学技术的迅猛发展,特别是70年代末到80年代初,分子克隆">克隆、质粒和噬菌体DNA的构建成功,核酸自动合成仪的诞生,大大丰富了核酸探针的来源,新的核酸分子杂交类型和方法不断涌现。按其作用方式可大致分为固相杂交和液相杂交两种:液相杂交是指参加反应的两条核酸链都游离在溶液中,而固相杂交是将参加反应的一条核酸链固定在固体的支持物上常用的有硝酸纤维素滤膜,其它如尼龙膜、乳胶颗粒和微孔板等),另一条参加反应的核酸链游离在溶液中。固相杂交有菌落原位杂交(colony in situ hybri dization)、斑点杂交法(Dot blot)、Southern印迹杂交(Southern blot)、Northern印迹杂交( N orthern blot)和组织原位杂交(Tissue in situ hybridization),即原位杂交组织化学技术和原位杂交免疫细胞化学技术。液相分子杂交技术包括吸附杂交、发光液相杂交、液相夹心杂交和复性速率液相分子杂交等。 二、原位杂交组织化学技术的由来及发展 原位杂交组织(或细胞)化学技术简称原位杂交(In situ hybridization),如上所述,属于固相核酸分子杂交的范畴。但它区别于固相核酸分子杂交中的任何一种核酸分子杂交技术。菌落杂交系细菌裂解释放出DNA,然后进行杂交。Southern印迹杂交法是以鉴定DNA中某一特定的基因片段,而Norhtern印迹杂交法是用以检测某一特定的RNA片段的。它们都只能证明该病原体、细胞或组织中是否存在待测的核酸而不能证明该核酸分子在细胞或组织中存在的部位。1969年美国耶鲁大学Gall和Pardue首先用爪蟾核糖体基因探针与其卵母细胞杂交,确定该基因定位于卵母细胞的核仁中。与此同时,Buongiorno– Nardell i和Amaldi, John及其同事等相继利用同位素标记核酸探针进行了细胞或组织的基因定位,从而创造了原位杂交细胞或组织化学技术。Orth(1970)应用3H标记的兔乳头状瘤病毒cRNA探针与兔乳头状瘤组织的冰冻切片进行杂交,首次用原位杂交检测了病毒DNA在细胞中的定位,但当时的工作多采用冰冻组织切片或培养细胞,探针均采用同位素标记。 由于同位素标记探针具有放射性既污染环境,又对人体有害,且受半衰期限制等缺点,科学工作者们开始探索用非放射性的标记物标记核酸探针进行原位杂交。Bauman(1981)等首先应用荧光素标记cRNA探针做原位杂交,然后用荧光显微镜观察获得成功。Shroyer(1982)报道用2,4二硝基苯甲醛(DNP)标记DNA探针,使该DNA探针具有抗原性,然后用兔抗DNP的抗体来识别杂交后的探针,最后经免疫过氧化物酶的方法来定位杂交探针。这两种方法至今仍有采用,但因敏感度不够高,应用不够普遍。 Pezzella(1 987)创建了用磺基化DNA探针来做细胞或组织原位杂交的方法,其基本原理是使DNA探针的胞嘧啶碱基磺基化,利用单克隆">克隆抗体识别磺基化探针,再通过免疫组化方法显示结合的单克隆抗体,从而对杂交结合的探针进行定位。本法的优点是磺基化DAN探针标记简便,不需作缺口平移标记,敏感度也较高。但自生物素和高辛标记探针技术建立后,已有取而代之的趋势。生物素标记探针技术是Brigat(1983)首先建立的,它利用生物素标记的探针在组织切片上检测了病毒DNA,通过生物素与抗生物素结合,过氧化物酶-抗过氧化物酶显示系统显示病毒DNA在细胞中的定位。生物素标记探针技术目前已被广泛应用,特别是在病毒学和病理学的临床诊断中。这种生物素标记技术又叫酶促生物素标记技术。另一种叫光促生物素标记核酸技术,该技术是用光敏生物素(Photobiotin)标记核酸。目前应用的光敏生物素有乙酸盐和补骨脂素生物素,它们都是由三个部分组成:光敏基团、连结臂和生物素(图20-1)。在强光下,不需酶反应,光敏生物素的光敏基团即可与核酸中的碱基相结合。光敏生物素标记核酸,方法简单,灵敏度也不低,但标记效率不高,每100~150个碱基才能标记一个生物素,对于短的基因探针特别是寡核苷酸探针不宜使用,以免因标记数过少而影响灵敏度(Forster et al 1985)。 近年来,地高辛(Digoxigonin)标记技术引起科技工作者的极大兴趣。Boeringer Mannhem Bio-ch emisca于1987年将地高辛标记的有关试剂及药盒投放市场。和其它非放射性标记物一样,地高辛较放射性标记系统安全,方便、省时间。同时在敏感性和质量控制方面比生物素标记技术要优越,可以检测出人基因组DNA中的单拷贝基因。地高辛标记法显示的颜色为紫蓝色(标记碱性磷酸酶-抗碱性磷酸酶显色系统),有较好的反差背景。 核酸探针根据标记方法的不同可粗略分为放射性探针和非放射性探针两类。根据探针的核酸性质不同可分为DNA探针、RNA探针、cDNA探针、cRNA探针和寡核苷酸探针等。DNA探针还有单链DNA(Single st randed, ssDNA)和双链DNA(Double stranded, dsDNA)之分(详见十九章)。早期应用的主要是DNA探
荧光原位杂交技术及其应用 摘要:荧光原位杂交技术是一种非常有用的分子细胞遗传学工具,特别是对一些染色体数目异常和复杂染色体异常的诊断,架起了染色体显带技术和分子遗传学之间的桥梁。本文主要就荧光原位杂交技术的发展历程、探针制备和临床应用做一简单的综述。 关键词:FISH;荧光原位杂交;临床应用;产前诊断;肿瘤 Fluorescence in situ hybridization and applications SUN Jingjing,YAN Shouqing (College of Animal Science and Veterinary Medicine,Jilin University,Changchun 130062,China) Abstract:FISH is a powerful molecular cytogenetic technique which allows rapid detection of numerical and complex chromosome aberrations on interphase cells and metaphase spreads, bridging the gap between conventional chromosome banding analysis and molecular genetic DNA studies. This review gives a brief overview of the historical developments of FISH techniques and applications in clinic genetic diagnostics. Key words:FISH; Fluorescence in situ hybridization; Clinical applications; Prenatal diagnosis; Tumor DNA荧光原位杂交(fluorescence in situ hybridization,FISH)技术是一种应用非放射性荧光物质依靠核酸探针杂交原理在核中或染色体上显示DNA序列位置的方法[1]。该技术具有快速、安全、灵敏度高以及探针可长期保存等特点,目前已广泛应用于细胞遗传学、肿瘤生物学、基因定位、基因作图、基因扩增,产前诊断及哺乳动物染色体进化研究等领域。 1 FISH技术的产生 1969年Gall和Pardue利用放射性同位素标记的DNA探针检测细胞制片上非洲爪蟾细胞核内的rDNA获得成功之后,Pardue等同年又以小鼠卫星DNA为模板,利用体外合成的含3H 的RNA为探针成功地与中期染色体标本进行了原位杂交,从而开创了RNA-DNA的同位素原位杂交技术[2],但是没有得到广泛应用。1974年Evans第一次将染色体显带技术和原位杂交技术结合起来,提高了基因定位的准确性。1981年,Langer等首次采用生物素标记的核苷酸探针(bio-dUTP)成功地进行了染色体原位杂交,建立了非放射性原位杂交技术(Nonisotopic in situ hybridization),至此,以荧光标记的探针在细胞制片上进行基因原位杂交的技术建立起来。1985年这项技术被引进到植物。1986年,Cremer与Licher等分别证实了荧光原位杂交技
原位杂交操作流程 1、使用地高辛标记的核酸探针进行石蜡切片的RNA原位杂交第一天 1)二甲苯于37℃脱蜡2次,每次15分钟; 2)无水乙醇浸泡2次,每次3分钟; 3) 95%乙醇浸泡2次,每次3分钟; 4) PBS清洗3分钟; 5) 2%焦碳酸二乙酯室温下浸泡10分钟; 6) PBS清洗10分钟; 7)加入胃蛋白酶25ul/ml,37℃孵育15分钟; 8) PBS清洗2次,每次3分钟; 9) 0.2N的HCl孵育30分钟; 10)PBS清洗2次,每次3分钟; 11)0.25%无水乙酸和0.1M三乙醇胺孵育10分钟; 12)PBS清洗2次,每次5分钟; 13)预杂交缓冲液孵育30分钟; 14)准备核酸探针混合物:使用预杂交缓冲液稀释探针,85℃加热5分钟,置于冰块中10分钟; 15)杂交;第二天 16)将玻片置于SSC中2次,每次5分钟以去除封片; 17)PBS清洗3分钟; 18)RNA酶A溶液中(或0.1-1ng/mlPBS中),37℃孵育30分钟; 19)PBS清洗5分钟; 20)室温,2×SSC清洗10分钟; 21)37℃,1×SSC清洗10分钟; 22)37℃,0.5×SSC清洗10分钟; 23)缓冲液A孵育10分钟; 24)缓冲液A(1%正常绵羊血清和0.03%三重氢核X-100)孵育30分钟; 25)加入抗地高辛抗体(1/200的上述缓冲液,来自Boehringer Mannheim),37℃孵育3 小时; 26)缓冲液A清洗2次,每次10分钟; 27)缓冲液B清洗2次,每次5分钟; 28)制成NBT/BCIP暗处保存30-60分钟,显微镜下进行观察,如果背景尚佳,显色时间可延长到16小时;29)停止缓冲液B的反应,用水进行简单的清洗; 30)固红,脱水以及封片进行核的复染。 2、使用地高辛标记的寡核苷酸探针进行石蜡切片的原位DNA杂交第一天 1)二甲苯于37℃脱蜡2次,每次15分钟; 2)无水乙醇浸泡2次,每次5分钟; 3) 95%乙醇浸泡2次,每次5分钟; 4) PBS清洗5分钟; 5) 2%焦碳酸二乙酯室温下浸泡10分钟; 6) PBS清洗5分钟; 7)加入胃蛋白酶25ul/ml,37℃孵育10分钟; 8) PBS清洗2次,每次5分钟; 9) 0.2N的HCl孵育30分钟; 10)PBS清洗2次,每次5分钟; 11)0.25%无水乙酸和0.1M三乙醇胺孵育10分钟;
原位杂交技术(ISH)步骤与注意事项 材料 (一)仪器:光学显微镜、烤箱、温箱、水浴箱、电吹风机 (二)器皿:塑料反应盒、玻片架、洗涤杯、微量移液器、Eppendorf 管 (三)试剂: 1.DEPC (所有的试剂以此配制,注:DEPC致癌,应在通风下进行,并避免接触皮肤;含有Tris 中不能用DEPC) 3.0.2mol/L HCl 1.72ml 浓HCl,加ddH2O至1000ml 4.20%冰醋酸20ml 冰醋酸,加ddH2O至100ml 5.蛋白酶(50μg/ml) 6.溶液I: 含0.1mol/L Tris、0.1mol/L NaCl、2mmol/L MgCl2、0.05% Triton X-100 取12.1g Tris; 5.85g NaCl; 0.41g MgCl2·6H2O,溶于800ml ddH2O中,再加入0.5ml Triton X-100,用HCl调pH至7.5,加ddH2O至1000ml。8×105Pa高压蒸汽灭菌15min 7.溶液II: 含0.1 mol/L Tris、0.1mol/L NaCl、50mol/L MgCl2。方法:取12.1gTris;5.85g NaCl ; 10.15g MgCl2·6H2O,溶于800ml ddH2O中,用HCl调至9.5 ddH2O至1000ml。8×105Pa高压蒸汽灭菌15min。 8.4%多聚甲醛:A液:称4g多聚甲醛,加40ml ddH2O,加温至60℃后,边摇边滴加1mol/L NaOH至全部溶解。B:液:1.69g NaH2PO4·2H2O,加30ml ddH2O至溶。C液:0.39gNaOH 溶于20ml ddH2O中。先将B液与C液混合后再加入A液,以1mol/L NaOH或HCl调pH 值至7.2~7.4,加ddH2O至100ml。4℃保存。 9.去离子甲酰胺:新的。 10.20×SSC:含有3mol/L NaCl, 0.3mol/L SSC。取175.32g NaCl,加ddH2O,充分溶解后,加88.2gSSC,溶解后加ddH2O至1000ml。8×105Pa高压蒸汽灭菌15min。 11.硫酸葡聚糖(1mg/ml),取硫酸葡聚糖1g溶于1ml灭菌水,-20℃下保存。 12.50×Denhart液:含1% BSA,1% Ficoll-400(水溶性聚蔗糖),1% PVP-40(聚乙烯吡咯烷酮),分别称取1g BSA,1g PVP-40,1g Ficoll-400,用少量的ddH2O溶解混合,加ddH2O 至100ml。过滤灭菌,贮存于-20℃下备用。 13.亲和素---碱性磷酸酶(Av-AP)200U/200μl 14.底物显色液 15.50%甲酰胺50ml甲酰胺,加50ml 2×SSC,混匀后备用。 16.0.1mol/L pH 7.4 PBS.。称取43.2g Na2PO4·2H2O,溶于少量ddH2O,分别溶解后将二者合并,加入127.5g NaCl至完全溶解,加ddH2O至1500ml。8×105Pa高压蒸汽灭菌15min。使用时作10倍稀释。 17.2%BSA。取0.1g BSA,溶于5ml溶液I中。 杂交前标本处理 试剂配制与准备 A 0.1mol/L PBS, pH 7.0 B 0.2% DEPC C 4%多聚甲醛 D 0.1mol/L HCl E 200μg/ml RNase A F 蛋白酶K,稀释成含0.5-1μg/ml, 必要时用1-3或5μg/ml(用10 mg/ml贮存液稀释) G 0.1%甘氨酸, 用0.1mol/L PBS, pH 7.5配制 H 2×SSC, 1×SSC, 用20×SSC贮存液稀释配制
原位杂交 1 探针的设计与合成 1)根据实验室已有的p8基因cDNA全长序列,用premier primer5.0设计引物p81和p82, 以卤虫cDNA为模板,PCR扩增得到346bp的产物,用Takara胶回收试剂盒回收纯化。引物编号引物序列长度 p81 TGCGGACGAAACAGGAAG 18 bp p82 GCTCAAACAGTGA TGCCAGT 20 bp 2)目的片段克隆 a. 在无菌离心管中加入连接载体的各种成分,载体与片段的摩尔比控制在1:3-1:8,根据凝胶电泳检测后的浓度及载体与片段分子大小来计算摩尔比。加入成分及比例如下: 目的PCR片段 5 μl pGM-T载体(约50ng/uL) 1 μl 10×T4 DNA Ligation Buffer 1 μl T4 DNA Ligase(3U/uL) 1 μl 无菌去离子水 3 μl 总体积10 μl b. 轻轻弹动离心管以混合内容物,短暂离心。置于PCR仪中16℃过夜连接,反应结束后将离心管置于冰上。 c. 向铺好的含有氨苄青霉素的固体平板表面加入16 μl的IPTG(50mg/ml)、40 μl的X-gal (20mg/ml),使用无菌的弯头玻璃棒将其均匀的涂开,避光置于37℃培养箱1-3小时,使溶解X-gal的二甲基甲酰挥发干净。 d. 将10 μl的连接产物加到100 μl DH5 感受态细胞中,轻弹混匀,冰浴半小时,将离心管置于42℃水浴90秒,取出管后立即置于冰浴上放置2-3分钟,其间不要摇动离心管。向离心管加入500 μl 37℃预热的LB(不含抗生素)培养基,150rpm摇床37℃振荡培养45分钟。目的是使质粒上相关的抗性标记基因表达,使菌体复苏。将菌液于4000g下离心10分钟,去掉上清,加入100 μl培养液重溶并加入到配制好的LB固体培养基上,用无菌的弯头玻璃棒轻轻将细胞均匀涂开。待平板表面干燥后,倒置平板,37℃培养12-16小时。 e. 挑取白色菌落直接进行PCR检测,筛选转化子。 f. 将转化子接种于LB液体培养基中培养24小时,吸取1mL菌液送至大连宝生物公司进行序列测序。 3)重组质粒的线性化 取6 μl以测序的重组质粒,选取NcoI内切酶37℃酶切4h。酶切反应体系为20 μl: 质粒 6 μl 10xK Buffer 2 μl NcoI酶 1 μl 0.1% BSA 2 μl 灭菌水9 μl 终体积20 μl 取酶切前后的质粒各4 μl,经1%琼脂糖电泳检测,确认酶切完全,将酶切产物用Takara胶回收试剂盒回收纯化,作为探针合成的模板。 4)探针合成 按罗氏DIG RNA Labeling Kit (SP6/T7)试剂盒使用指南,标记反义RNA探针。 使用的所有试剂和器皿均经去RNase处理,合成方法如下:先准备反应体系。冰上向RNase-Free的微离心管中顺序加入下列试剂:
1.For each probe (control and experimental), set up a separate 100-ml PCR in a 0.5-ml sterile tube, as tabulated below. Either.cDNA inserted in plasmids or genomic DNA can be used as templates for the PCR (see REAGENT SETUP for details on primer design). 每个探针(实验组和对照组),在0.5 -ml无菌管设立一个独立的100毫升PCR,正如下面的表。另外。互补脱氧核糖核酸插入到基因组DNA质体或可用作模板PCR(见试剂设置有关底漆设计)。 **注意关键: 1.很多版本的实验反义核酸探针可以作为一种控制背景染色(见试剂设置)。然而,我们相信最好的方法来演示特异性是获取相同的空间限制表达模式使用不同的非重叠探测器相同的基因。 2.小心不要污染pcr.使用无菌试管和过滤器的技巧和戴手套 3.另外,PCR扩增,cDNAs质粒中可以使用约束线性化酶,独特的站点位于5¢(反义核酸探针)或3¢(对感官探测)来插入。净化的线性DNA可以通过苯酚/氯仿萃取乙醇沉淀紧随其后。 2| Run the PCR using the conditions tabulated below. 使用下面列出的条件运行PCR **暂停点:把扩增好的pcr产品放4℃降温和在-20℃贮藏几个星期。 3| Add the 100-ml PCR to a Microcon YM-50 column and add 400 ml of sterile water. Centrifuge for 15–20 min at 1,000 g at room temperature. 加入100毫升PCR到Microcon YM-50列并加入400毫升的无菌水。在室温下1000g离心15 - 20分钟。 **注意关键:膜应该是干的。如果没有再离心 4|Place the Microcon column into a new microfuge tube (provided in the kit), add 20ml of sterile water, vortex briefly and then turn the Microcon column upside down. Spin for 1 min at 1,000 g at room temperature to recover the DNA. 把小层析柱放在一个新的离心管(在这个工具包中提供),增加20毫升的无菌水、短暂离心,然后颠倒层析柱。自旋1分钟1000 g在室温下恢复了DNA **注意关键:离心的步骤应该快速。离心机1分钟只是为了避免样本太干。 5|Check the quality, quantity and size of the PCR amplification product by loading 1/20 of the preparation on a 1% (wt/vol) agarose gel in 1 TBE buffer. DNA should appear as a band and not as a smear. The 1/20 of the preparation should contain at least 40 ng of DNA. 通过装载1/20的稀释液在1*的TBE buffer缓冲液中的1% (wt/vol)的琼脂糖凝胶检查PCR的扩增产物的质量
原位杂交组织(或细胞)化学(In situ Hybridization Histochemistry,ISHH) 简称原位杂交(In Situ Hybridization),属于固相分子杂交的范畴,它是用标记的DNA或RNA为探针,在原位检测组织细胞内特定核酸序列的方法。根据所用探针和靶核酸的不同,原位杂交可分为DNA-DNA杂交,DNA-RNA杂交和RNA-RNA杂交三类。根据探针的标记物是否直接被检测,原位杂交又可分为直接法和间接法两类。直接法主要用放射性同位素、荧光及某些酶标记的探针与靶核酸进行杂交,杂交后分别通过放射自显影、荧光显微镜术或成色酶促反应直接显示。间接法一般用半抗原标记探针,最后通过免疫组织化学法对半抗原定位,间接地显示探针与靶核酸形成的杂交体。原位杂交最初是以同位素标记探针进行的。尽管同位素标记(如35S,3H,32P等)仍然广泛使用,但非同位素标记探针的迅速发展(尤其是生物素标记探针和地高辛标记探针),更引起科技工作者的极大兴趣。 一、基本要求 组织取材:组织取材应尽可能新鲜。由于组织RNA降解较快,所以新鲜组织和培养细胞最好在30min内固定。 固定目的是: (1)保持细胞结构; (2)最大限度地保持细胞内DNA或RNA的水平; (3)使探针易于进入细胞或组织。 最常用的固定剂是多聚甲醛,与其它醛类固定剂(如戊二醛)不同,多聚甲醛不会与蛋白质产生广泛的交叉连接,因而不会影响探针穿透入细胞或组织。 增强组织的通透性和核酸探针的穿透性: (1)稀酸处理和酸酐处理:为防止探针与组织中碱性蛋白之间的静电结合,以降低背景,杂交前标本可用0.25%乙酸酐处理10min,经乙酸酐处理后,组织蛋白中的碱性基团通过乙酰化而被阻断。组织和细胞标本亦可用0.2M HCl处理10min,稀酸能使碱性蛋白变性,结合蛋白酶消化,容易将碱性蛋白移除。 (2)去污剂处理:去污剂处理的目的是增加组织的通透性,利于杂交探针进入组织细胞,最常应用的去污剂是Triton X-100。注意:过度的去污剂处理不仅影响组织的形态结构,而且还会引起靶核酸的丢失。 (3) 蛋白酶处理:蛋白酶消化能使经固定后被遮蔽的靶核酸暴露,以增加探针对靶核酸的可及性。常用的蛋白酶有蛋白酶K(proteinase K),还有链霉蛋白酶(pronase)和胃蛋白酶(pepsin)等。 杂交缓冲液孵育: 杂交前用不含探针的杂交缓冲液在杂交温度下孵育2hr,以阻断玻片和标本中可能与探针产生非特异性结合的位点,达到减低背景的目的。 防止污染: 由于在手指皮肤及实验室用玻璃器皿上均可能含有RNA酶,为防止其污染影响实验结果,在整个杂交前处理过程中都需要戴消毒手套,实验所用玻璃器皿及镊子都应于实验前一日置高温烘烤(180℃)以达到消除RNA酶的目的。杂交前及杂交时所用的溶液均需经高压消毒处理。 双链DNA探针和靶DNA的变性: 杂交反应进行时,探针和靶核酸都必须是单链的。如果用双链DNA探针进行杂交(包括检测RNA时),双链DNA探针在杂交前必须进行变性。探针变性后要立即进行杂交反应,不然解链的探针又会重新复性。 杂交液: 杂交液内除含一定浓度的标记探针外,还含有较高浓度的盐类、甲酰胺、硫酸葡聚糖、牛血
实验四荧光原位杂交实验 一、实验目的 1.掌握核酸杂交的基本原理、过程和操作; 2.熟练掌握荧光显微镜的使用方法; 3.掌握Image J软件进行图像融合的方法; 4.理解荧光原位杂交技术的应用。 二、实验原理 核酸杂交是以碱基互补为基础、以双链核酸分子在特定条件下的变性和复性为手段,对核酸分子进行定性和定量检测。两条互补的核苷酸单链在特定条件下退火形成异质双链;应用荧光标记的探针与待检测材料中的单链核酸进行特异性结合,形成可悲检测的杂交双链核酸。在显微镜下观察并记录结果。 本实验所用探针是位点特异性标记探针GSP myc,是由SpectrumOrange (552/576)直接标记的荧光DNA探针,长度为200bp,能识别8号染色体的着丝粒部位的α卫星序列。 三、实验器材和试剂 防脱载玻片(premiere公司),医用砂轮,20×20mm盖玻片,橡皮胶,平板加热器,杂交盒,恒温水浴锅,玻璃染色缸,香柏油,荧光显微镜拍照系统,MYC基因扩增检测试剂盒。 四、实验步骤 1.样品处理 1)取细胞培养悬液5ml(107 cell),2000rpm离心5min,去上清; 2)加入10ml的0.075M KCl,吹打混匀,静置3min; 3)37℃水浴箱低渗30min; 4)加固定液2ml,吹打混匀,室温预固定10min; 5)2000rpm离心5min,去上清; 6)沉淀加固定液5~10ml,吹打混匀,-20℃冰箱静置30min; 7)2000rpm离心5min,去上清。 2.制片 1)取一张干净的载玻片,用砂轮在背面划“一”标记,尽量在中间或靠下
端标记; 2)加50ul固定液重悬细胞,取4ul悬液滴加到载玻片上的标记位置,室温晾干; 3)平板加热器上烤片,60℃30min,显微镜下观察细胞密度; 4)PBS洗涤3min; 5)1%多聚甲醛室温固定10min; 6)PBS洗涤3min; 7)70%、90%、100%梯度乙醇脱水2min; 8)取出玻片,室温晾干。 3.样品和探针同时变性,杂交 1)取出杂交液,混匀,瞬离; 2)加10ul杂交液到杂交区域,迅速盖上盖玻片,轻压使杂交液均匀分布,赶走气泡; 3)用橡皮胶沿盖玻片边缘封片; 4)平板加热器上78℃变性3min,放入湿盒; 5)37℃温箱中杂交10~18h。 4.杂交后洗涤 1)将载玻片取出,轻轻撕去橡皮胶; 2)载玻片放入2×SSC中,1min左右微微晃动,以移去盖玻片,37℃水浴10min,不时上下晃动载玻片; 3)取出载玻片,37℃,0.1%NP-40/2×SSC中6min; 4)取出玻片,70%,90%,100%乙醇各2min脱水; 5)取出玻片,暗处自然晾干。 5.DAPI复染细胞核 室温滴加10ul DAPI复染剂到盖玻片,载玻片目标区域朝下,请放于盖玻片上,轻压,避免产生气泡,在暗处存放,待观察。 6.荧光显微镜观察拍照 出现2G2O的细胞计为FISH阴性细胞,出现≥3O的细胞计为FISH阳性细胞。 7.Image J进行图片融合处理
原位杂交实验操作步骤 撰写人:范为民 一、实验原理 原位杂交是指借助于核酸分子杂交的方法,在显微镜水平检测和定位特异的核苷酸片段。现在原位杂交已经成为在分子水平研究肿瘤和遗传性疾病的发生,发展和调控等根本性问题的有力工具。 二、试剂盒 本实验室常用的原位杂交试剂盒是博士德公司生产的敏感加强型原位杂交检测试剂盒,此试剂盒分为两种,一种为过氧化物酶(POD)检测(MK1030型),一种为碱性磷酸酶(AP)检测系统(MK1032型),用于mRNA的杂交。过氧化物酶(POD)检测的最终信号为棕黄色,而碱性磷酸酶(AP)检测的信号为紫色,因后者信号比较突出,所以一般采用后一种检测方法。两种检测方法的实验步骤相差不多,所用洗脱缓冲液也大同小异。用于杂交的探针也可以分为两种,一种是DNA探针,即是用DNA与组织中的mRNA杂交,另一种是RNA 探针,即用RNA与组织中的mRNA杂交。DNA探针处理操作简单,但杂交信号一般不如RNA探针强烈,所以条件允许的话一般采用RNA探针。下面先介绍碱性磷酸酶(AP)检测试剂盒,采用RNA作为探针的操作步骤。 三、实验步骤 原位杂交实验主要包括三大部分,即组织冰冻切片、RNA探针标记、原位杂交三部分。 (一)组织冰冻切片 1. 实验准备 (1)原位杂交专用载玻片:用多聚赖氨酸处理后的载玻片,使切片紧密粘附在玻片上,可以用于后面的洗脱。一般一张载玻片上可以贴至多十张切片(可以是不同组织的切片),所以需要玻片的数目需要根据实验的要求而定。这种专用载玻片可以从中杉金桥公司购买,目前价格是每片2.2元,玻片有一面的一端是毛玻璃,用于标记组织名称等,切片应该贴在此面,切勿贴到反面。 (2)缓冲液配备 1.1器具准备 剪刀、镊子各三把,开壳钳一把,100ml量筒一个,磁力搅拌子一个;100ml试剂瓶一个,250ml试剂瓶三个。以上器具均洗净后置于180摄氏度以上烘烤6小时以上。铅笔、显微镜、冰、吸水纸、一次性塑料手套等。 1.2 溶液配制 0.1M PB缓冲液:Na2HPO4?12H2O 5.8021g,NaH2PO4?2 H2O 0.5928 g,加入200ml ddH2O溶解于之前准备的250ml试剂瓶中,再加入200ūl DEPC,充分摇匀后过夜,高压灭菌。以上溶液配制两份,其中一份瓶中放入磁力搅拌子。
原位杂交实验原理与方法 一、目的 本实验的目的是学会原位杂交的使用方法。了解各种原位杂交的基本原理和优缺点。 二、原理 原位杂交组化(简称原位杂交,in situ hybridization histochemistry;ISHH)属于分子杂交的一种,是一种应用标记探针与组织细胞中的待测核酸杂交,再应用标记物相关的检测系统,在核酸原有的位置将其显示出来的一种检测技术。原位杂交的本质就是在一定的温度和离子浓度下,使具有特异序列的单链探针通过碱基互补规则与组织细胞内待测的核酸复性结合而使得组织细胞中的特异性核酸得到定位,并通过探针上所标记的检测系统将其在核酸的原有位置上显示出来。 当然杂交分子的形成并不要求两条单链的碱基顺序完全互补,所以不同来源的核酸单链只要彼此之间有一定程度的互补顺序(即某种程度的同源性)就可以形成杂交双链。 探针的种类按所带标记物可分为同位素标记探针和非同位素标记探针两大类。目前,大多数放射性标记法是通过酶促反应将标记的基因掺入DNA中,常用的同位素标记物有3H、35S、125I和32P。同位素标记物虽然有灵敏性高,背底较为清晰等优点,但是由于放射性同位素对人和环境均会造成伤害,近来有被非同位素取代的趋势。非同位素标记物中目前最常用的有生物素、地高辛和荧光素三种。 探针的种类按核酸性质不同又可分为DNA探针、cDNA探针、cRNA探针和合成寡核苷酸探针。cDNA探针又可分为双链cDNA探针和单链cDNA探针。 原位杂交又可分为菌落原位杂交和组织原位杂交。 菌落原位杂交(Colony in situ hybridization)菌落原位杂交是将细菌从培养平板转移到硝酸纤维素滤膜上,然后将滤膜上的菌落裂菌以释出DNA。将NDA烘干固定于膜上与32P 标记的探针杂交,放射自显影检测菌落杂交信号,并与平板上的菌落对位。 组织原位杂交(Tissue in situ hybridization)组织原位杂交简称原位杂交,指组织或细胞的原位杂交,它与菌落的原位杂交不同。菌落原位杂交需裂解细菌释出DNA,然后进行杂交。而原位杂交是经适当处理后,使细胞通透性增加,让探针进入细胞内与DNA或RNA 杂交。 (一)探针的选择 根据不同的杂交实验要求,应选择不同的核酸探针。在大多数情况下,可以选择克隆的DNA 或cDNA双链探针。但是在有些情况下,必须选用其它类型的探针如寡核苷酸探针和RNA探针。例如,在检测靶序列上的单个碱基改变时应选用寡核苷酸探针,在检测单链靶序列时应选用与其互补的DNA单链探针(通过克隆人M13噬菌体DNA获得)或RNA探针,寡核苷酸探针也可。长的双链DNA探针特异性较强,适宜检测复杂的靶核苷酸序列和病原体,但不适宜于组织原位杂交,因为它不易透过细胞膜进入胞内或核内。在这种情况下,寡核苷酸探针和短的PCR标记探针(80~150bp)具有较大的优越性。 在选用探针时经常会受到可利用探针种类的限制。如在建立DNA文库时,手头没有筛选特定基因的克隆探针,这时就可用寡核苷酸探针来代替。但必须首先纯化该基因的编码蛋白,并测定6个以上的末端氨基酸序列,通过反推的核苷酸序列合成一套寡核苷酸探针。如果已有其它动物的同种基因克隆,因为人类和动物间在同一基因的核苷酸顺序上存在较高的同源性,因此可利用已鉴定的动物基因作探针来筛选人类基因克隆。对于基因核苷酸序列背景清楚而无法获得克隆探针时,可采用PCR方法扩增某段基因序列,并克隆人合适的质粒载体中,