热力学、动力学、计算化学软件安装总结
i.预软件安装
一、Gaussian View5.0的安装
1.找到压缩文件解压后出现;
2.点击打开看到;
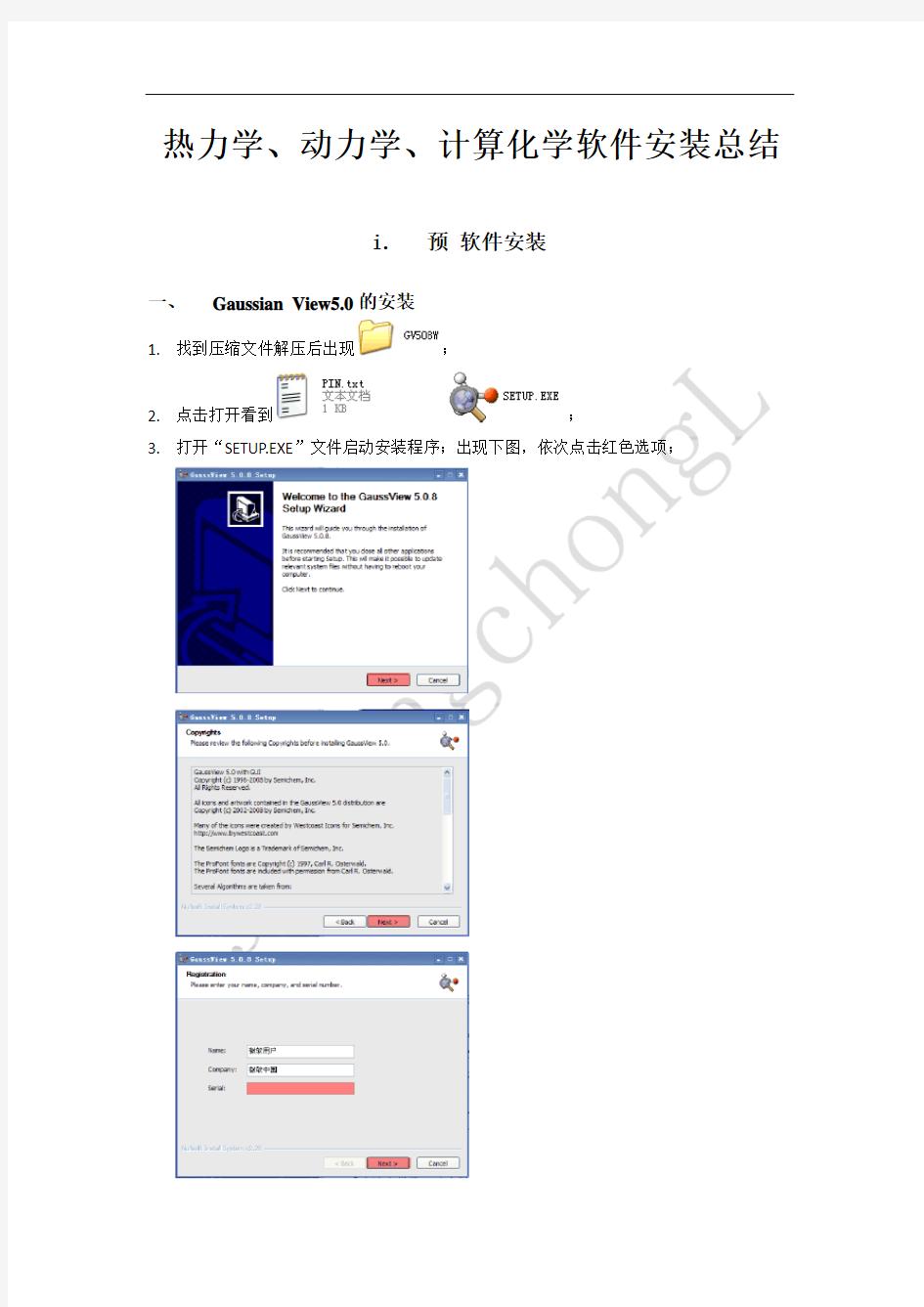
3.打开“SETUP.EXE”文件启动安装程序;出现下图,依次点击红色选项;
4.填写“serial”(打开左侧文件复制粘贴即可),继
续“next”;
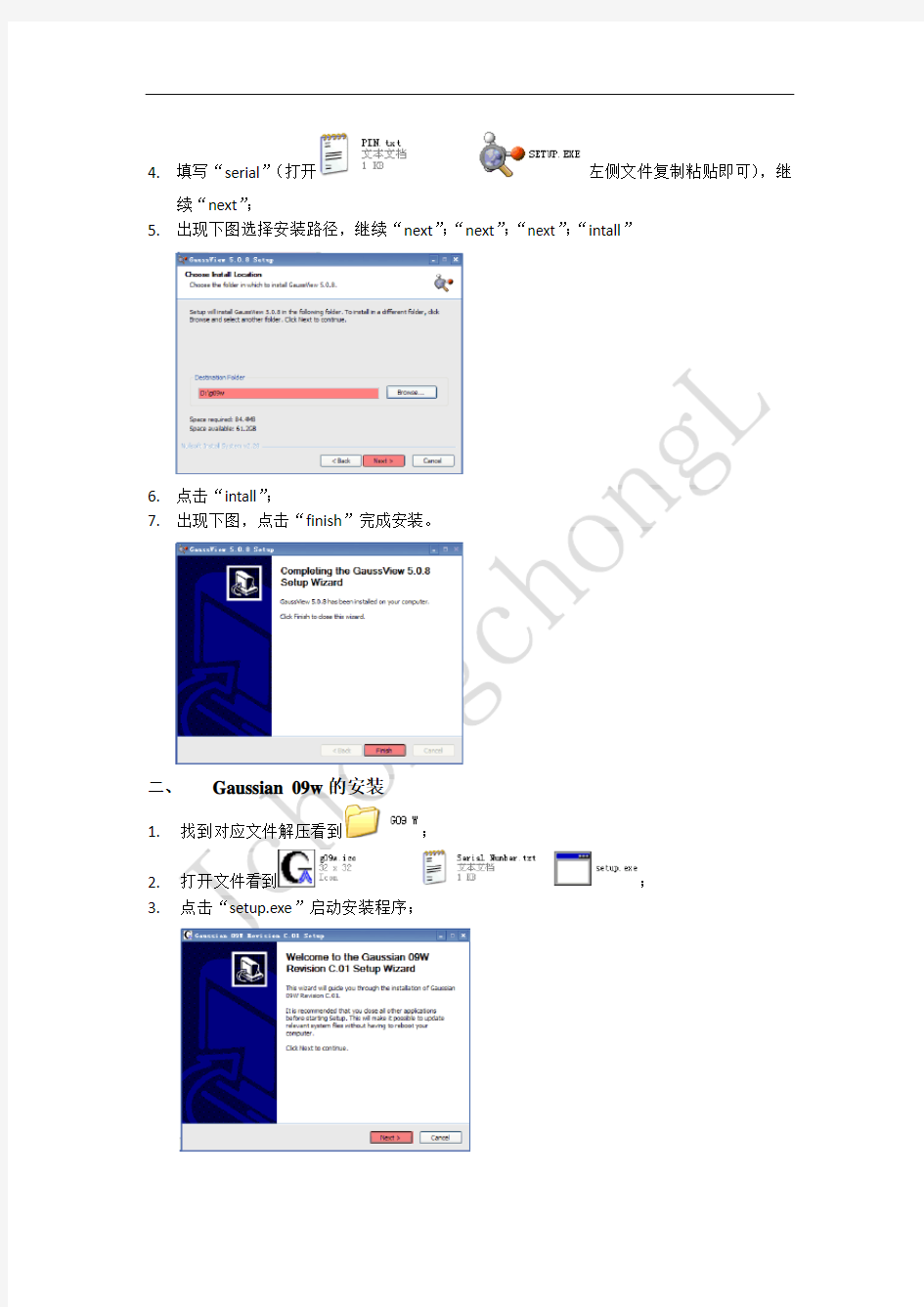
5.出现下图选择安装路径,继续“next”;“next”;“next”;“intall”
6.点击“intall”;
7.出现下图,点击“finish”完成安装。
二、Gaussian 09w的安装
1.找到对应文件解压看到;
2.打开文件看到;
3.点击“setup.exe”启动安装程序;
4.点击上图“Next”下一步看到下图;打开复制粘贴到“Serial”继续“Next”;
5.选择安装路径点击“Next”如下图;
6.点击“Intall”安装程序,出现下图;
7.点击“Next”完成安装并选择“Scratch”文件的存放位置如下图(直接点击确定即可);
三、UltraEdit的安装
1.找到对应文件解压后如图;
2.打开后找到“.exe”文件点击启动安装程序如下图;
3.点击“下一步”;
4.选择“我同意许可协议中的条款”继续“下一步”;
5.选择安装路径后点击“下一步”如下图;
6.点击“安装”完成安装。
7.软件注册,点击“注册程序”;
8.找到文件打开任选一个填写后点击“确定”;
9.弹出提示框点击“确定”;
10.再次打开就会看到注册成功提示框!
四、ChemOffice-2010的安装
1.找到ChemOffice压缩包,解压后如图;
2.首先点击dotnetfx2.0.exe完成安装;
3.完成安装后点击CambridgeSoft_ChemBioOffice_Ultra_2010.msi启动安装程序;
4.到此程序已安装成功,现在打开找到
注册表应用扩展程序,复制;
5.打开ChemOffice-2010安装的文件夹打开“Common”文件夹,打开“DLLs”文件夹,
将刚才复制的文件粘贴,即安装完成。
ii.计算化学及软件使用
一.反应动力学计算流程
1.判断一个反应能否发生(热力学范畴)
1)计算一个反应中每种物质的几何构形(键长、键角、频率),并与标准数值进行比较(误差不超过5%时则认为比较准确)。
2)计算每种物质的能量,看反应的吉布斯自由能(ΔG)的正负。
2.
3.
k
4.
二
1.
1).“New”→“Creat Molecule Group”建立一个新定的文件。
2).“Open”打开已有的输入或输出文件。
3).“recent file”记录最近的文件输出和输入路径。
4).“save”保存文件,格式为“.gjf”或“.com”。
5).“Preference”单击出现对话框。“Colors”中第一行设置背景颜色;“Element colrs”查看或改变元素周期表中每个元素的代表颜色或改变颜色。
2.“Edit”菜单栏中的使用
1).“updo”撤销操作。
2).“cut”剪切。
3).“copy”复制。
4).“paste”粘贴。
5).“atom list…”出现如图一,图二的示例。
“Tag”为原子的序号,“Symbol”为元素符号。
如:第四行Tag=4,NA=1,NB=2,NC=3;“第几行即表示tag下的竖着第几个元素”
(1).Tag=4,NA=1,即第四行的元素与第一行的元素形成的键长=1.1171462(Bond键长)
(2).Tag=4.NA=1,NB=2。即第四行元素与第一行和第二行元素所形成的键角=110.7248351(Angle键角)。(3).Tag=4.NA=1,NB=2,NC=3。即表示第四行元素与第一行,第二行和第三行元素所形成的二面角=119.9945406(Dihedral)。
3.“view”菜单栏的使用
1).“Add view”增加一个文件。
2).“Buider”构造一个分子,单击“”后分别点击分子中的两个原子即可得到两个原子
间的键长;单击“”给原子加氢原子;单击“”给原子减去氢原子。
单击“”会出现元素周期表,而后构造分子;单击“”出现C链等;单击“”
出现官能团;单击“”出现氨基酸。单击“”标后点击分子上的三个原子即可出
现其形成的键角大小。
三、在Gaussian 09w中计算优化加频率
下面的表列出了Gaussian 09中可以使用的任务类型:
结果如上图所示:
需对其做相关内容的补充,补充后如下图:
注意事项:结果中标题前后需空两行,默认已空出两行,只需检查即可。
保存好上述结果文件,进入Gaussian 09w软件进行计算,计算流程如下:
在“File”中“open…”打开上述文件,如图示,
点击图示右侧“√”符,如出现下图所示界面,则说明前面构建模型准确无误。
下一步点击“OK”符既可进入计算页面。
3.如图点击”Begin Processing”即开始计算,结果保存至D盘G09w文件夹(也可根据
需求存入其他文件夹)。
结果如图示:
再次进入Gaussview软件,打开上述文件,即可看到优化好的H2O分子构型。
点击“Result”→“Summary”,即可看到相关总结。
4.采集数据:
应用窗口快捷方式,点击后再点击相关原子,既可看到相关键长,键角,二面角等数据。
将采集数据与数据库进行比较,数据库网址:https://www.doczj.com/doc/b71041089.html,
四、Gaussian 运算中的错误及解决方法
1.2070错误
最基本的解释就是:你的系统在安装了g09之后,运行时对内存调用出错。
1.1解决方法
(1).更换稳定版本的系统,采用完整的正版系统。
(2)对于一个新手,你不必管什么是2070错误。是什么错误应该看最后终止在哪一步,例如L1就是命令行里有错误,L301通常是自选多重度错误等。
2.不收敛错误(520错误)
不收敛错误可分为scf不收敛和几何构型不收敛两种情况。
2.1 scf不收敛
是自洽场叠代不收敛,,可以认为是对指定结构的波函数不断优化的过程,是为了找到这个某个指定结构下能量最低的波函数。
2.1.1 scf不收敛解决方法
(1)可以加大scf的循环次数,默认的循环次数是128次,通过scf=(maxcycle=n)来设置最大循环次数n。建议不要超过512,更多的循换没有必要。
(2)如果加大循环次数不管用,在分子有对称性的情况下,使用scf=dsymm关键词来强制密度对称,有时可以收敛,或者使用scf=symm关键词,有时也可以收敛。
(3)如果(2)步都不行,可以将对称的分子中的某几个原子的位置微调,使分子丧失对称性。
(4)如果还不行,只能拿出杀手锏了,就是使用qc,但不建议直接使用,而是使用xqc关键词,比如
scf=(maxcycle=80,xqc),意思是如果scf正常计算(dc)在80个循环之内不收敛才进行昂贵的qc计算。
(5)中级用户可以在输入文件的井号“#”开头那一行井号后面加上字母"p"来输出更多的信息,其中就有自洽场叠代的信息,分析原因可能会对采用什么方法提供指导。
(6)如果用用小基组计算,scf可以收敛,那么保存好检查点文件,换成大基组的时候从检查点文件中读取初始猜测(使用guess=read关键词),有时可以算过去。
(7)上述方法有时可以组合使用,如果经过各种组合处理都不收敛,那么放弃吧,你的分子的电子结构太差了。
2.2几何构型不收敛
是对结构的优化的过程,是为了找到某个指定的组分下能量极小结构。
2.2.1几何构型不收敛解决方法
(1)如果是很小的分子(10原子以内),初始结构可能离平衡结构比较远,又在输出文件优化的最后一步判断是否收敛的位置(可以通过查找Threshold字段找到,它下面有四个判断项,都是YES才代表优化收敛)看到了“-- Number of steps exceeded, NStep=xxx”,可以通过加大优化的循环次数来解决问题。
(2)优化到如果四个收敛标准中前两项早就收敛了,而后两项尤其是第三项不收敛,这需要判断原因。如果每一步优化过程中的能量始终在下降,那么可以继续让他算,超过了最大步数停掉了的话把结构拿出来,重新提交就行了。如果能量忽大忽小出现跳跃,说明遇到了比较平坦的势能面,或者优化的算法不好。此时建议按如下顺序处理:
a. 使用opt=maxstep=n来缩小最大步长,即原子移动的距离,n的默认值是30,只能设置整数。
b. 使用关键词opt=gdiis,很多时候可以很快收敛。
c. 使用opt=calcfc关键词,在几何优化的第一步计算力常数,为优化定一个大方向,类似在第一个三岔路口选一条路。
d. #P产生额外输出。包括每一执行模块在开始和结束时与计算机系统有关的各种信息(包括执行时间数据),以及SCF计算的收敛信息。
e.使用opt=calcall关键词,在几何优化计算的每一步都计算力常数,类似于在每一个三岔路口都来选一条路。这个关键词不到万不得已不要使用,非常耗时。
(3) 四个判断标准的后两项早就收敛了,但前两项似乎固定在某个数值上了。遇到这种情况,首先
去检查你优化第一步的时候给出的每个轨道得本征值部分,也就是轨道的能量,如果HOMO和LUMO的能量相同或者非常接近,那么你的设置有问题,即自旋多重度不对。如果HOMO-LUMO能量没问题,那么将目前的计算停掉,提出结构,使用opt=gdiis关键词,很多时候管用。
(4) 如果几何优化过程中发现键变得异常短,而且此时几何优化总是不收敛。遇到这种情
况,问题出在你的基组使用,肯定是使用了赝势基组却没有读入赝势。由于赝势基组将内层电子
和原子核用一个有效核心势来表示,你没有读入赝势就说明没有读入这个有效核心势,那么相当
于把原子的内层电子都给除掉了,巨大的核引力把原子之间的距离变得异常短也不足为奇。
五、收敛判据
六、过渡态的寻找
:故无法分离出来,也是无法观测到的。
过渡态这一概念,对于理解有机反应机理具有很重要的作用。过渡态理论认为,化学反应不是通过反应物分子的简单碰撞就可以完成的,而是在反应物到生成物的过程中,经过了一个高能量的过渡态。这与爬山类似,山的最高点便是过渡态。过渡态是一种不稳定的反应物原子组合体,不可逆反应中,它可以很快地分解为产物。通常反应中间体的能量与过渡态相差不大,两者很难区分。借助于飞秒红外光谱,目前已经可以观测到接近过渡态时的分子构型结构。
确定方法的可行性:通过键长,键角,二面角,频率与数据库中数据对比,误差小于5%,则成立 寻找过渡态影响程度大小:键长>键角>二面角 强制弥散:scf=qc
#p opt=(ts,calcall,maxcycle=300,noeigen) hf/3-21g
iop(2/16=1,3/15=1,9/6=500)
3.过渡态寻找的步骤
(1).构建并优化反应物,产物(分别优化),获取键长,键角,二面角等有用信息;
(2).根据所获取得信息在GaussView重新建立模型;
(3). 用Gaussian进行计算,获得过渡态;
(4). 若寻找过渡态失败,则重新调整构型,计算,直至结束!
4.过渡态的计算
Gaussian 计算过渡态是一个相对复杂的过程,其计算性的工作也是一个相对难解决的问题,针对不同的体系和研究对象,有不同的方法和途径。Gaussian计算过渡态的大体原则如下:
(1).计算之前,一定要多查阅文献,看看是否有类似的计算和过渡态的构型构象等等,这对猜测过渡态结构很是有用的。文献之外,应对反应的类型、途径尽可能考虑周全,对各种过渡态的空间结构有较深的了解。
(2).对体系不熟悉时,尽量用QST3方法去做,即要输入反应物、构想的过渡态、产物进行计算,而非直接用TS或QST2方法。
(3).鉴于计算的复杂性,首次计算建议采用HF方法,基组为3-21g即可,意思是先采用小基组小方法,这对计算来说很有必要,否则多半会因大方法和基组一棍子打死。
(4).计算出现错误是很正常的,处理的方法是:
A、修改结构(一般修改的幅度为反应部位±0.1-0.2A)
B、增加循环次数(此针对Link9999错误),方法是OPT(Maxcycle=500)SCF(maxcycle=3000)
C、减小步长(IOP的1/8适当降低,这有利于震荡收敛的情形)
D、将计算结果(XYZ坐标重新输入作为计算初始值)......
总之,用HF方法必须找到过渡态,即有且只一个虚频,否则,下面的叙述将失去其价值。
(5). 用HF计算出来的结果是欠佳的,因为HF方法未考虑到电子的相关效应,也是得不到承认的(冯浩语),因而我们应当用更为高级一些的方法去计算,比如DFT、CCSD(T)等等。然而经过HF计算的磨难之后,接着的问题会更加麻烦,原因是计算出错用上述方法做处理有时很难奏效,于是乎我想到了一下解决途径:A.将HF方法计算的坐标重新做输入卡,并再次用QST3方法,使用的方法和基组为:b3lyp/6-311++g(d,p) 直接进行计算,也可以用先用b3lyp/6-31g 计算,之后用b3lyp/6-311++g(d,p)接着计算,我可以肯定的说,后者先优化的时间并不是很长,虽然步骤是麻烦些,但有时确实能够解决前者无法计算的过渡态。
B、除此之外,我想说几句,计算对机器的要求很高,平均而言,用QST3计算过渡态很费时间,我是计算17-21个原子,计算时间在1Day and 10 hours to 3 Days and 11hours .对一般的电脑而言,配置不是很差。
C、如果方法A中仍不奏效,不妨直接将HF计算结果放在TS中做计算,这样或许有可能,原因是你的反应物和产物编写的结构不是很好,其对反应的指导意义也大大降低甚至有负作用。当然,如果QST3计算时老是犯同样的错误,用原则4中的方法均已失灵,那么就直接用TS计算。
D、要经常查看计算的结果和预期设想的是否差别很大,不要闷着头搞计算,这也是所谓的,既要埋头拉车,又要抬头看路!
E、如果上述方法再次连连无效,收效情况目前看来还算可以:HF方法计算的过渡态通过IRC(反应路径分析),计算两个方向的反应情况,找出与反应位置极为相似之处的反应物,找出其对应的坐标,之后找出与产物极其相似的结构,如此将找到反应物、过渡态、产物的HF方法下的结构,纳入QST3计算,因为这些结构要比起初我们自己绘制的好得多!
(6).最后牵涉到用IRC计算来验证过渡态,寻找其他的过渡态,来分析反应的路径,来处理反应的Gibbs 自由能和反应的焓变,进一步分析反应的热力学和动力学。
5.优化过渡态的关键词
a) # B3LYP/6-31G* opt=(ts,calcfc) iop(1/11=1)
b) # B3LYP/6-31G* opt=(ts,,calcfc,noeingentest) * a,b 实际上相同,计算力常数
c) # B3LYP/6-31G* opt=(ts,calchffc)iop(1/11=1) * 计算HF力常数,用MP2或CI方法寻找过渡态时,用这个比较好,节省很多时间。
d) # B3LYP/6-31G* opt=(ts,readfc)iop(1/11=1) * 当之前有做freq的check提供力常数时。实际上也可以用在ts优化中断后重接,此时的力常数是上一步优化后估算的。
e) # UB3LYP/6-31G* opt=(ts,modredundant) iop(1/11=1) * 无需力常数的计算,不过需要分子描述后指定modredundant
f) # UB3LYP/6-31G* opt=qst2 *无需iop(1/11)选项,需要给出反应物和产物构型,并要求构型间原子标号一致。
g) # UB3LYP/6-31G* opt=qst3 *无需iop(1/11)选项,需要给出反应物,产物,过渡态构型,并要求构型间原子标号一致。
opt=qst2和opt=qst3也可以和modredundant 结合使用
注意的问题:
(1).一般过渡态都涉及到键的断裂与生成,因此即使是自旋多重度为1的体系,也应该用非限制性的理论方法来做。经常看到有人用rb3lyp之类的找过渡态,实际上是不对的,虽然有时得到的过渡态结构也算合理。设想一个键的均裂反应,原来成对的两个电子在反应过程中是要分别属于两个自由基的,不会占据同一个空间轨道了。
(2).在做SQT2时,一般都不会直接输入反应物和产物的坐标,而是会相应作些调整,比如将active bond 伸长15%~20%左右,这样做有利于加快计算速度,还有,transition state对基组和理论的选取非常敏感,要谨慎。
6.过渡态命令及其解释
%mem=100MW
%nprocshared=2
#p opt=(ts,maxcycle=200,noeigen,internal) freq=numerical
b3lyp/6-311+g(2df,2p) guess=nosymm iop(2/16=1,3/15=1,9/6=200
“%mem”——所需运存大小
“%nproc”——进存数
“#”——Gaussian 输入文件的执行路径部分需要以#符号开头,作为这一行的第一个非空格字符。这一行的其它部分使用自由格式。对于大部分的计算工作,所有的信息都可位于这一行,但也可以超过一行(超过的行前的#符号可用也可不用)。执行路径部分必须以空行作为结果。
“#p”——产生额外输出。包括每一执行模块在开始和结束时与计算机系统有关的各种信息,(包括执行时间数据),以及SCF 计算的收敛信息。
“opt”——优化
“ts”——过渡态
“maxcycle”——最大循环次数
“freq”——频率
“b3lyp/6-311”——算法
“+”——弥散
“*”——极化
“g(2df,2p)”——轨道
“guss” 控制Hartree-Fock 波函的初始轨道猜测。Guess 不加选项是没有意义的。
“iop(2/16=1,3/15=1,9/6=200”——初始结构(在分子说明部分给出)是过渡态的结构,反应路径可以是从该点的一个或全部两个方向。当相位的最大分量为正时,默认为向前的方向,是过渡矢量指定的方向;它可以直接用Phase 选项定。定义过渡矢量的相位,沿着过渡矢量正向,也就是指定的内坐标增加的方向运动,
需要四个原子序号指定。如果只给出两个原子序号,坐标为两个原子间的键伸展;三个原子序号指定弯曲的角度;四个原子指定二面角。
七、集群计算
1.连接到集群
a.打开SSH软件如图
b.点击“Quick Connect”即可看到登录对话框,输入完成后点击“Connect”
c.在下图输入密码点击“OK”即可登录成功!
2.利用集群进行计算
首先要找到一个输入文件,且要改“.gjf”格式为“.com”在此处要注意编译,编译有两种方式:一是直接
a.双击打开编译,二在vi xxx(文件)下进行编译。
b.输入cd xxx(文件夹名)回车。
c.在输入ls(不是数字1)回车即可出现此文件夹下的文件。
d.此时可输入vi xxx(文件)进行编译。
vi命令下,键入“i“进入修改状态;“ESC”退出修改状态;键入“:wq”保存并退出修改状态
e.输入g09 -qa-t 72000:00:00 –i xxx(文件名)回车,此时即可进入计算。(注意加空格)
f.输入“qstat”即可进行任务查看。
g.计算完成时,输出文件为“log”型(同文件名共有4个不同的文件类型,分别为.chk;.com;.log;.o )。
h.把输出文件(.log)拉入GaussView即可进行查看得到我们所需要的数据。
(3).常用集群命令【注意加空格!!!!】
1.
2.
a.
b.构建输入文件:
工作类型(Job Type):IRC,分别再选Forward和Reverse进行计算。具体计算方法(不同精度的方法)可据硬件条件进行选择。
其余各选项与以前要求相同。也可根据如下命令行进行编辑:IRCR命令:
%mem=100MW
%nproc=2
#p ub3lyp/6-311+g(2df,2p)(此处计算方法根据条件选择)irc=(reverse,calcfc,stepsize=2(可选择1.2.3.4.5.10,
热一定律总结 一、 通用公式 ΔU = Q + W 绝热: Q = 0,ΔU = W 恒容(W ’=0):W = 0,ΔU = Q V 恒压(W ’=0):W =-p ΔV =-Δ(pV ),ΔU = Q -Δ(pV ) → ΔH = Q p 恒容+绝热(W ’=0) :ΔU = 0 恒压+绝热(W ’=0) :ΔH = 0 焓的定义式:H = U + pV → ΔH = ΔU + Δ(pV ) 典型例题:3.11思考题第3题,第4题。 二、 理想气体的单纯pVT 变化 恒温:ΔU = ΔH = 0 变温: 或 或 如恒容,ΔU = Q ,否则不一定相等。如恒压,ΔH = Q ,否则不一定相等。 C p , m – C V , m = R 双原子理想气体:C p , m = 7R /2, C V , m = 5R /2 单原子理想气体:C p , m = 5R /2, C V , m = 3R /2 典型例题:3.18思考题第2,3,4题 书2.18、2.19 三、 凝聚态物质的ΔU 和ΔH 只和温度有关 或 典型例题:书2.15 ΔU = n C V , m d T T 2 T 1 ∫ ΔH = n C p, m d T T 2 T 1 ∫ ΔU = nC V , m (T 2-T 1) ΔH = nC p, m (T 2-T 1) ΔU ≈ ΔH = n C p, m d T T 2 T 1 ∫ ΔU ≈ ΔH = nC p, m (T 2-T 1)
四、可逆相变(一定温度T 和对应的p 下的相变,是恒压过程) ΔU ≈ ΔH –ΔnRT (Δn :气体摩尔数的变化量。如凝聚态物质之间相变,如熔化、凝固、转晶等,则Δn = 0,ΔU ≈ ΔH 。 101.325 kPa 及其对应温度下的相变可以查表。 其它温度下的相变要设计状态函数 不管是理想气体或凝聚态物质,ΔH 1和ΔH 3均仅为温度的函数,可以直接用C p,m 计算。 或 典型例题:3.18作业题第3题 五、化学反应焓的计算 其他温度:状态函数法 Δ H m (T ) = ΔH 1 +Δ H m (T 0) + ΔH 3 α β β α Δ H m (T ) α β ΔH 1 ΔH 3 Δ H m (T 0) α β 可逆相变 298.15 K: ΔH = Q p = n Δ H m α β Δr H m ? =Δf H ?(生) – Δf H ?(反) = y Δf H m ?(Y) + z Δf H m ?(Z) – a Δf H m ?(A) – b Δf H m ?(B) Δr H m ? =Δc H ?(反) – Δc H ?(生) = a Δc H m ?(A) + b Δc H m ?(B) –y Δc H m ?(Y) – z Δc H m ?(Z) ΔH = nC p, m (T 2-T 1) ΔH = n C p, m d T T 2 T 1 ∫
化学热力学与动力学在有机化学中的应用 一.化学热力学: 一个反应能否自发发生及反应平衡时反应物和产物之间的相对比例是一个化学热力学问题。可以解决,一个反应的能否自发进行及反应的限度问题,是用自由能ΔG 来判断的,ΔG<0反应可以自发进行,直到平衡即ΔG=0,相反如果大于0时,反应是不能自发进行的,由平衡常数与ΔG 的关系可以知道,此时K 很小所以往往是可逆的。S T H G ?-?=?,Δ H 反应的是反应的热效应,在反应中焓的变化反映了反应物键的断裂与生成物键的生成能量 之差(其中包括张力能,离域能等),即断裂键的键能之和减去生成键的键能之和(键能为正值)。当ΔS 可以忽略不计的时候,ΔG ≈ΔH ,当反应是放热的时候,即ΔH<0,则ΔG<0,即反应可以自发进行。ΔS 的判断:1.分子在体系中的自由度越大,她的熵值也就越大。即气>液>固。在一个反应中如果反应物都是液相的,而产物至少有一个是气相的,那么在热力 学上由于熵增大所以是有利的。2.产物的分子数目等于反应物的分子数目的反应,熵变通常是不大的,但是如果生成物的分子数增加,通常会有较大的熵值增加。所以分解,分裂的反应在热力学上是有利的。但是注意,有些时候分解反应的热焓变比较大抵消了ΔS 的增大,最终ΔG 仍>0,反应仍不自发。3.链状分子比对应的环状分子有更大的熵值,因而分子的打开是有利的,闭环意味着熵值的减少。如果弱键断裂,强健生成,则反映放热,ΔH<0,在放热反应中,焓变对G 有一个负的的贡献,所以反应易于由弱键生成强键,反之,由强键生成 弱键,会消耗能量,H>0对G 有正的贡献,不易发生。综上焓减少是反应的推动力,熵增加是反应的推动力。 化学动力学: 对反应速度的处理研究涉及到化学动力学问题。有机化学中主要应用过渡态理论。过渡态是反应途径中能量最高点时所存在的结构。它和反应物、产物或中间体不同,并不是一个化学实体,无法分离和实验观察,仅是一个有一定几何形状的和电荷分布的高度不稳定状态。 微观可逆原理::(1)一个基元反应的逆反应也必然是基元反应,即任何基元反应都是可逆的;(2)正反应与逆反应经过相同的过渡态即正逆反应途径一样,机理一样。 当我们研究一个有机反应时,最希望了解的是这一反应将向产物方向进行到什么样程度?一般来说,任何体系都有转变成它们最稳定状态的趋势(即自发的趋势都是体系自由能减小的方向,ΔG<0),因此,可以预料当产物的稳定性愈大于反应物的稳定性时,则平衡愈移向产物一侧。这句话的意思可由下式看出来。△G=-RTlnK ,当两个物质的稳定性差很大的时候,即自由能差很大,如果生成物的自由能比反应物的小,即ΔG 很负,所以K 很大,平衡常数大,说明向稳定的方向进行的趋势很大,即平衡移向产物一侧,反应进行的很完全。 要使反应发生,产物的自由能必须低于反应物的自由能,即△G 必须是负值。说白了对于一个自发反应,反应物与产物之间自由能差别越大,或者说稳定性差别越大,反应进行的趋势完全程度也越大。 过渡态理论:1.起反应的物质结合时需要通过比原始和终了的状态较高的势能,具有较高势能的状态较过渡态。即假设一个反应先达到一个过渡态,然后从过渡态以极快的速度变成产物。2.反应有几种产物时,每一种产物都从不通过过渡态过来的,主要产物是过渡态能量最低的转化而来的。3反应物与过渡态之间存在一个平衡,反应的速度(生成过渡态的速度)依赖于平衡常数,而K 又与活化自由能有关。过渡态的自由能的高低成为衡量反应速率的重要标志。 在一步反应的图中能量最高点是活化络合物,在它的左边,所有络合物都被认为同反应物处于平衡中;而在它右边,所有络合物则被认为是同产物处于平衡中。 A+B 反应物 D 产物△G 双步 反 应G ΔG 1≠ΔG 2≠ C 中间体 A+B 反应物 C 产物过渡态△G 单 步 反 应G ΔG 1≠a b
化工热力学复习题 一、选择题 1. T 温度下的纯物质,当压力低于该温度下的饱和蒸汽压时,则气体的状态为( C ) A. 饱和蒸汽 超临界流体 过热蒸汽 2. 纯物质的第二virial 系数B ( A ) A 仅是T 的函数 B 是T 和P 的函数 C 是T 和V 的函数 D 是任何两强度性质的函数 3. 设Z 为x ,y 的连续函数,,根据欧拉连锁式,有( B ) A. 1x y z Z Z x x y y ?????????=- ? ? ?????????? B. 1y x Z Z x y x y Z ?????????=- ? ? ?????????? C. 1y x Z Z x y x y Z ?????????= ? ? ?????????? D. 1y Z x Z y y x x Z ?????????=- ? ? ?????????? 4. 关于偏离函数M R ,理想性质M *,下列公式正确的是( C ) " A. *R M M M =+ B. *2R M M M =- C. *R M M M =- D. *R M M M =+ 5. 下面的说法中不正确的是 ( B ) (A )纯物质无偏摩尔量 。 (B )任何偏摩尔性质都是T ,P 的函数。 (C )偏摩尔性质是强度性质。 (D )强度性质无偏摩尔量 。 6. 关于逸度的下列说法中不正确的是 ( D ) (A )逸度可称为“校正压力” 。 (B )逸度可称为“有效压力” 。 (C )逸度表达了真实气体对理想气体的偏差 。 (D )逸度可代替压力,使真实气体的状态方程变为fv=nRT 。 (E )逸度就是物质从系统中逃逸趋势的量度。 7. 二元溶液,T, P 一定时,Gibbs —Duhem 方程的正确形式是 ( C ). a. X 1dlnγ1/dX 1+ X 2dlnγ2/dX 2 = 0 b. X 1dlnγ1/dX 2+ X 2 dlnγ2/dX 1 = 0 ` c. X 1dlnγ1/dX 1+ X 2dlnγ2/dX 1 = 0 d. X 1dlnγ1/dX 1– X 2 dlnγ2/dX 1 = 0 8. 关于化学势的下列说法中不正确的是( A ) A. 系统的偏摩尔量就是化学势 B. 化学势是系统的强度性质 C. 系统中的任一物质都有化学势 D. 化学势大小决定物质迁移的方向 9.关于活度和活度系数的下列说法中不正确的是 ( E ) (A )活度是相对逸度,校正浓度,有效浓度;(B) 理想溶液活度等于其浓度。 (C )活度系数表示实际溶液与理想溶液的偏差。(D )任何纯物质的活度均为1。 (E )r i 是G E /RT 的偏摩尔量。 10.等温等压下,在A 和B 组成的均相体系中,若A 的偏摩尔体积随浓度的改变而增加,则B 的偏摩尔体积将(B ) A. 增加 B. 减小 C. 不变 D. 不一定 " 11.下列各式中,化学位的定义式是 ( A ) 12.混合物中组分i 的逸度的完整定义式是( A )。 A. d G ___i =RTdln f ^i , 0lim →p [f ^i /(Y i P)]=1 B. d G ___i =RTdln f ^i , 0lim →p [f ^ i /P]=1 C. dG i =RTdln f ^i , 0lim →p f i =1 ; D. d G ___i =RTdln f ^i , 0lim →p f ^ i =1 j j j j n nS T i i n T P i i n nS nV i i n nS P i i n nU d n nA c n nG b n nH a ,,,,,,,,])([.])([.])([.])([.??≡??≡??≡??≡μμμμ
工程热力学与传热学课 程总结与体会 Company number:【WTUT-WT88Y-W8BBGB-BWYTT-19998】
工程热力学与传热学 题目:工程热力学与传热学课程总结与体 会 院系:水利建筑工程学院给排水科学与工 程 班级:给排水科学与工程一班 姓名:张琦文 指导老师:姚雪东 日期:2016年5月1日 认识看法地位作用存在问题解决措施未来 发展展望 传热学在高新技术领域中的应用 摘要: 热传递现象无时无处不在【2】它的影响几乎遍及现代所有的工业部门【1】也渗透到农业、林业等许多技术部门中。本文介绍了航空航天、核能、微电子、材料、生物
医学工程、环境工程、新能源以及农业工程等诸多高新技术领域在不同程度上应用传热研究的最新成果。可以说除了极个别的情况以外,很难发现一个行业、部门或者工业过程和传热完全没有任何关系。不仅传统工业领域,像能源动力、冶金、化工、交通、建筑建材、机械以及食品、轻工、纺织、医药等要用到许多传热学的有关知识【1】而且诸如航空航天、核能、微电子、材料、生物医学工程、环境工程、新能源以及农业工程等很多高新技术领域也都在不同程度上有赖于应用传热研究的最新成果,并涌现出像相变与多相流传热、(超)低温传热、微尺度传热、生物传热等许多交叉分支学科。在某些环节上,传热技术及相关材料设备的研制开发甚至成为整个系统成败的关键因素。 前言:通过对传热学这门课程的学习,了解了传热的基本知识和理论。发现传热学是一门基础学科应用非常广泛,它会解决许许多多的实际问题更是与机械制造这门学科息息相关。传热学是研究由温度差异引起的热量传递过程的科学。传热现象在我们的日常生活中司空见惯。早在人类文明之初人们就学会了烧火取暖。随着工业革命的到来,蒸汽机、内燃机等热动力机械相继出现,传热研究更是得到了飞速的发展,被广泛地应用于工农业生产与人们的日常生活之中。当今世界国与国之间的竞争是经济竞争,而伴随着经济的高速发展也带来了资源、人口与环境等重大国
第一章 基本概念及定义 一、填空题 1、热量与膨胀功都是 量,热量通过 差而传递热能,膨胀功通过 差传递机械能。 2、使系统实现可逆过程的条件是:(1) ,(2) 。 3、工质的基本状态参数有 、 、 。 4、热力过程中工质比热力学能的变化量只取决于过程的___________而与过程的路经无关。 5、热力过程中热力系与外界交换的热量,不但与过程的初终状态有关,而且与_______有关。 6、温度计测温的基本原理是 。 二、判断题 1、容器中气体的压力不变则压力表的读数也绝对不会改变。( ) 2、无论过程是否可逆,闭口绝热系统的膨胀功总是等于初、终态的内能差。( ) 3、膨胀功的计算式?= 2 1 pdv w ,只能适用于可逆过程。 ( ) 4、系统的平衡状态是指系统在无外界影响的条件下(不考虑外力场作用),宏观热力性质不随时间而变化的状态。( ) 5、循环功越大,热效率越高。( ) 6、可逆过程必是准静态过程,准静态过程不一定是可逆过程。( ) 7、系统内质量保持不变,则一定是闭口系统。( ) 8、系统的状态参数保持不变,则系统一定处于平衡状态。( ) 9、孤立系统的热力状态不能发生变化。( ) 10、经历一个不可逆过程后,系统和外界的整个系统都能恢复原来状态。( ) 三、选择题 1、闭口系统功的计算式21u u w -=( )。 (A )适用于可逆与不可逆的绝热过程 (B )只适用于绝热自由膨胀过程 (C )只适用于理想气体绝热过程 (D )只适用于可逆的绝热过程 2、孤立系统是指系统与外界( )。 (A )没有物质交换 (B )没有热量交换 (C )没有任何能量交换 (D )没有任何能量传递与质交换 3、绝热系统与外界没有( )。 (A )没有物质交换 (B )没有热量交换 (C )没有任何能量交换 (D )没有功量交换
第一章 化学热力学基础 公式总结 1.体积功 We = -Pe △V 2.热力学第一定律的数学表达式 △U = Q + W 3.n mol 理想气体的定温膨胀过程 .定温可逆时: Wmax=-Wmin= 4.焓定义式 H = U + PV 在封闭体系中,W ′= 0,体系发生一定容过程 Qv = △U 在封闭体系中,W ′= 0,体系发生一定压过程 Qp = H2 – H1 = △H 5.摩尔热容 Cm ( J·K-1·mol-1 ): 定容热容 CV (适用条件 :封闭体系、无相变、无化学变化、 W ′=0 定容过程 适用对象 : 任意的气体、液体、固体物质 ) 定压热容 Cp ?=?2 1 ,T T m p dT nC H (适用条件 :封闭体系、无相变、无化学变化、 W ′=0 的定压过程 适用对象 : 任意的气体、液体、固体物质 ) 单原子理想气体: Cv,m = 1.5R , Cp,m = 2.5R 双原子理想气体: Cv,m = 2.5R , Cp,m = 3.5R 多原子理想气体: Cv,m = 3R , Cp,m = 4R 1 221ln ln P P nRT V V nRT =n C C m = ?=?2 1 ,T T m V dT nC U
Cp,m = Cv,m + R 6.理想气体热力学过程ΔU 、ΔH 、Q 、W 和ΔS 的总结 7.定义:△fHm θ(kJ·mol-1)-- 标准摩尔生成焓 △H —焓变; △rHm —反应的摩尔焓变 △rHm θ—298K 时反应的标准摩尔焓变; △fHm θ(B)—298K 时物质B 的标准摩尔生成焓; △cHm θ(B) —298K 时物质B 的标准摩尔燃烧焓。 8.热效应的计算 由物质的标准摩尔生成焓计算反应的标准摩尔焓变 △rH θm = ∑νB △fH θm ,B 由物质的标准摩尔燃烧焓计算反应的标准摩尔焓变 △rH θm = -∑νB △cH θm ,B 9.Kirchhoff (基尔霍夫) 方程 △rHm (T2) = △rHm (T1) + 如果 ΔCp 为常数,则 △rHm (T2) = △rHm (T1) + △Cp ( T2 - T1) 10.热机的效率为 对于卡诺热机 12 11Q Q Q Q W R +=- =η dT C p T T ? ?2 1 1 2 1211Q Q Q Q Q Q W +=+=-=η121T T T -=
化工热力学习题集(附标准答案)
————————————————————————————————作者:————————————————————————————————日期:
模拟题一 一.单项选择题(每题1分,共20分) 本大题解答(用A 或B 或C 或D )请填入下表: 1. T 温度下的纯物质,当压力低于该温度下的饱和蒸汽压时,则气体的状态为(C ) A. 饱和蒸汽 B. 超临界流体 C. 过热蒸汽 2. T 温度下的过冷纯液体的压力P ( A ) A. >()T P s B. <()T P s C. =()T P s 3. T 温度下的过热纯蒸汽的压力P ( B ) A. >()T P s B. <()T P s C. =()T P s 4. 纯物质的第二virial 系数B ( A ) A 仅是T 的函数 B 是T 和P 的函数 C 是T 和V 的函数 D 是任何两强度性质的函数 5. 能表达流体在临界点的P-V 等温线的正确趋势的virial 方程,必须至少用到( ) A. 第三virial 系数 B. 第二virial 系数 C. 无穷项 D. 只需要理想气体方程 6. 液化石油气的主要成分是( A ) A. 丙烷、丁烷和少量的戊烷 B. 甲烷、乙烷 C. 正己烷 7. 立方型状态方程计算V 时如果出现三个根,则最大的根表示( B ) A. 饱和液摩尔体积 B. 饱和汽摩尔体积 C. 无物理意义 8. 偏心因子的定义式( A ) A. 0.7lg()1 s r Tr P ω==-- B. 0.8lg()1 s r Tr P ω==-- C. 1.0lg()s r Tr P ω==- 9. 设Z 为x ,y 的连续函数,,根据欧拉连锁式,有( B ) A. 1x y z Z Z x x y y ???? ?????=- ? ? ?????????? B. 1y x Z Z x y x y Z ????????? =- ? ? ?????????? C. 1y x Z Z x y x y Z ????????? = ? ? ?????????? D. 1y Z x Z y y x x Z ????????? =- ? ? ?????????? 10. 关于偏离函数M R ,理想性质M *,下列公式正确的是( C ) A. *R M M M =+ B. *2R M M M =- C. *R M M M =- D. *R M M M =+ 11. 下面的说法中不正确的是 ( B ) (A )纯物质无偏摩尔量 。 (B )任何偏摩尔性质都是T ,P 的函数。 (C )偏摩尔性质是强度性质。(D )强度性质无偏摩尔量 。 12. 关于逸度的下列说法中不正确的是 ( D ) (A )逸度可称为“校正压力” 。 (B )逸度可称为“有效压力” 。 (C )逸度表达了真实气体对理想气体的偏差 。 (D )逸度可代替压力,使真实气体 的状态方程变为fv=nRT 。 (E )逸度就是物质从系统中逃逸趋势的量度。 题号 1 2 3 4 5 6 7 8 9 10 答案 题号 11 12 13 14 15 16 17 18 19 20 答案
《工程热力学》期末总结 一、闭口系能量方程的表达式有以下几种形式: 1kg 工质经过有限过程:w u q +?= (2-1) 1kg 工质经过微元过程:w du q δδ+= (2-2) mkg 工质经过有限过程:W U Q +?= (2-3) mkg 工质经过微元过程:W dU Q δδ+= (2-4) 以上各式,对闭口系各种过程(可逆过程或不可逆过程)及各种工质都适用。 在应用以上各式时,如果是可逆过程的话,体积功可以表达为: pdv w =δ (2-5) ? = 2 1 pdv w (2-6) pdV W =δ (2-7) ? = 2 1 pdV W (2-8) 闭口系经历一个循环时,由于U 是状态参数,?=0dU ,所以 W Q ??= δδ (2-9) 式(2-9)是闭口系统经历循环时的能量方程,即任意一循环的净吸热量与净功量相等。 二、稳定流动能量方程 t s w h w z g c h q +?=+?+?+?=2 21 (2-10) (适用于稳定流动系的任何工质、任何过程) ? - ?=2 1 vdp h q (2-11) (适用于稳定流动系的任何工质、可逆过程) 三、几种功及相互之间的关系(见表一) 表一 几种功及相互之间的关系
四、比热容 1、比热容的种类(见表二) 。 )/3 kg m 2、平均比热容:1 21 1221 20 t t t t c t t c t t c - -= (2-12) 3、利用平均比热容计算热量:11220 t t c t t c q -= (2-13) 4、理想气体的定值比热容(见表三)
其中:M M R R g 83140= = [J/(kg ·K)] M —气体的摩尔质量,如空气的摩尔质量为28.96kg/kmol 。 空气的kmol /kg 96.28K)kmol /(J 83140?= = M R R g =287[J/(kg ·K)],最好记住空气的气体常数。 引入比热容比k 后,结合梅耶公式,又可得: g p R k k c 1 -= (2-14) g V R k c 1 1-= (2-15) 五、理想气体的热力学能、焓、熵(见表四) (焓的定义:pv u h += kJ/kg , 焓是状态参数) 六、气体主要热力过程的基本计算公式(见表五)
化工热力学(第三版)公式知识总结 vdW 方程 p =RT V?b ?a V 2 RK 方程 p = RT V?b ? a √T ?V(V+b) P R方程 P = RT V?b ? a V (V+b )+b(V?b) 对应态原理 P r = 3 8T r V r ?13??3 V r 2 偏心因子 ω=?1?lgP r s ︱ T r =0.7 普遍化vir ial 方程BP c RT c = B (0)+ωB (1) d U=Td S-p dV dH =Td S+Vdp dA=-Sd T-pdV dG=-Sd T+V dp dZ=MdX+Nd Y (?N ?X )Y =?(?M ?Y )X (?T ?V ) S =?(?P ?S ) V (?S ?P ) T =?(?V ?T ) p 偏离函数定义 M ?M 0ig =M (T,p )?M 0ig (T,p 0) 随状态变化 M (T 2,p 2)?M (T 1,p 1)=[M (T 2,p 2)?M ig (T 2,p 0)]?[M (T 1,p 1)?M ig (T 1,p 0)]+ [M ig (T 2,p 0) ? M ig (T 1,p 0)] G?G 0ig RT ?ln P P 0 = 1RT ∫(V ?RT P )P 0dp 逸度定义 G (T,P )?G 0ig (T,P 0)=RTln f P 0 φ=f P lnφ=ln f p =1RT ∫(V ? RT P )P 0 dp (?lnf ?p )=V RT 饱和蒸汽和液体性质关系M =M sl (1?x )+M sv x 偏摩尔性质 M i ???=(?M t ?n i ) T,p,{n } ≠i 偏摩尔性质表示摩尔性质 M =∑n i n M i ???N i =∑x i M i ???N i 摩尔性质与摩尔性质关系M i ???=M +(1?x)dM dx i M 2????=M ?x 1dM dx i Gi bbs -Duhem 方程在T,p 恒定(∑x i dM i ???N i=1) T,p =0 Leiwis-randa ll 规则 f ?i is =f i X i f ?i is ? =H i,Solvent X i 活度系数 γi =f i ?f i X i lnγi ?=lnγi ?lnγi ∞ 超额性质 G E RT =∑X i lnγi N i ?H =H E =?RT 2∑X i ( ?lnγi ?T ) p,{x }N i
第1章 绪言 一、是否题 1. 孤立体系的热力学能和熵都是一定值。(错。G S H U ??=?=?,,0,0但和 0不一定等于A ?,如一体积等于2V 的绝热刚性容器,被一理想的隔板一分为二,左侧状 态是T ,P 的理想气体,右侧是T 温度的真空。当隔板抽去后,由于Q =W =0, 0=U ?,0=T ?,0=H ?,故体系将在T ,2V ,0.5P 状态下达到平衡,()2ln 5.0ln R P P R S =-=?,2ln RT S T H G -=-=???,2ln RT S T U A -=-=???) 2. 封闭体系的体积为一常数。(错) 3. 理想气体的焓和热容仅是温度的函数。(对) 4. 理想气体的熵和吉氏函数仅是温度的函数。(错。还与压力或摩尔体积有关。) 5. 封闭体系的1mol 气体进行了某一过程,其体积总是变化着的,但是初态和终态的体积相等, 初态和终态的温度分别为T 1和T 2,则该过程的? =2 1 T T V dT C U ?;同样,对于初、终态压力相 等的过程有? =2 1 T T P dT C H ?。(对。状态函数的变化仅决定于初、终态与途径无关。) 6. 自变量与独立变量是一致的,从属变量与函数是一致的。(错。有时可能不一致) 三、填空题 1. 状态函数的特点是:状态函数的变化与途径无关,仅决定于初、终态 。 2. 单相区的纯物质和定组成混合物的自由度数目分别是 2 和 2 。 3. 1MPa=106Pa=10bar=9.8692atm=7500.62mmHg 。 4. 1kJ=1000J=238.10cal=9869.2atm cm 3=10000bar cm 3=1000Pa m 3。 5. 普适气体常数R =8.314MPa cm 3 mol -1 K -1=83.14bar cm 3 mol -1 K -1=8.314 J mol -1 K -1 =1.980cal mol -1 K -1。 第2章P-V-T关系和状态方程 一、是否题 1. 纯物质由蒸汽变成液体,必须经过冷凝的相变化过程。(错。可以通过超临界流体区。) 2. 当压力大于临界压力时,纯物质就以液态存在。(错。若温度也大于临界温度时,则是超临 界流体。) 3. 纯物质的饱和液体的摩尔体积随着温度升高而增大,饱和蒸汽的摩尔体积随着温度的升高而减小。(对。则纯物质的P -V 相图上的饱和汽体系和饱和液体系曲线可知。) 4. 纯物质的三相点随着所处的压力或温度的不同而改变。(错。纯物质的三相平衡时,体系自 由度是零,体系的状态已经确定。)
工程热力学大总结 '
… 第一章基本概念 1.基本概念 热力系统:用界面将所要研究的对象与周围环境分隔开来,这种人为分隔的研究对象,称为热力系统,简称系统。 边界:分隔系统与外界的分界面,称为边界。 外界:边界以外与系统相互作用的物体,称为外界或环境。 闭口系统:没有物质穿过边界的系统称为闭口系统,也称控制质量。 ) 开口系统:有物质流穿过边界的系统称为开口系统,又称控制体积,简称控制体,其界面称为控制界面。 绝热系统:系统与外界之间没有热量传递,称为绝热系统。 孤立系统:系统与外界之间不发生任何能量传递和物质交换,称为孤立系统。 单相系:系统中工质的物理、化学性质都均匀一致的系统称为单相系。 复相系:由两个相以上组成的系统称为复相系,如固、液、气组成的三相系统。 单元系:由一种化学成分组成的系统称为单元系。 多元系:由两种以上不同化学成分组成的系统称为多元系。 } 均匀系:成分和相在整个系统空间呈均匀分布的为均匀系。 非均匀系:成分和相在整个系统空间呈非均匀分布,称非均匀系。 热力状态:系统中某瞬间表现的工质热力性质的总状况,称为工质的热力状态,简称为状态。 平衡状态:系统在不受外界影响的条件下,如果宏观热力性质不随时间而变化,系统内外同时建立了热的和力的平衡,这时系统的状态称为热力平衡状态,简称为平衡状态。 状态参数:描述工质状态特性的各种物理量称为工质的状态参数。如温度(T)、压力(P)、比容(υ)或密度(ρ)、内能(u)、焓(h)、熵(s)、自由能(f)、自由焓(g)等。 基本状态参数:在工质的状态参数中,其中温度、压力、比容或密度可以直接或间接地用仪表测量出来,称为基本状态参数。
第一章绪论 热力学是以热力学第一、第二定律及其他一些基本概 念理论为基础,研究能量、能量转换以及与转换有关的物 质性质相互之间关系的科学。有工程热力学、化学热力学、 化工热力学等重要分支。 化工热力学是将热力学原理应用于化学工程技术领 域。化工热力学主要任务是以热力学第一、第二定律为基 础,研究化工过程中各种能量的相互转化及其有效利用, 研究各种物理和化学变化过程达到平衡的理论极限、条件 和状态。 热力学的研究方法,原则上可采用宏观研究方法和微 观研究方法。以宏观方法研究平衡态体系的热力学称为经 典热力学。 体系与环境:隔离体系,封闭体系,敞开体系 第二章流体的P-V-T关系 在临界点C : 临界点是汽液两相共存的最高温度和最高压力,即临 界温度Tc,临界压力Pc。 纯流体的状态方程(EOS) 是描述流体P-V-T性质的 关系式。由相律可知,对纯流体有: f( P, T, V ) = 0 混合物的状态方程中还包括混合物的组成(通常是摩 尔分数)。 状态方程的应用 (1)用一个状态方程即可精确地代表相当广泛范围内的 P、V、T实验数据,借此可精确地计算所需的P、V、T数 据。 (2)用状态方程可计算不能直接从实验测定的其它热力 学性质。 (3)用状态方程可进行相平衡和化学反应平衡计算。 压缩因子(Z)即:在一定P,T下真实气体的比容与相 同P,T下理想气体的比容的比值. 理想气体方程的应用(1 )在较低压力和较高温度下可用 理想气体方程进行计算。(2 )为真实气体状态方程计算 提供初始值。(3 )判断真实气体状态方程的极限情况的 正确程度,当或者时,任何的状态方程都还原为理想气体 方程。 维里方程式 Virial系数的获取 ( 1 ) 由统计力学进行理论计算目前应用很少 ( 2 ) 由实验测定或者由文献查得精度较高 ( 3 ) 用普遍化关联式计算方便,但精度不如实验测定的 数据 两项维里方程维里方程式Z=PV/RT=1+ B/P (1)用于气相PVT性质计算,对液相不能使用; (2)T
第4章 非均相封闭体系热力学 一、是否题 1. 偏摩尔体积的定义可表示为{}{}i i x P T i n P T i i x V n nV V ≠≠? ??? ????=???? ???=,,,,?。 2. 在一定温度和压力下的理想溶液的组分逸度与其摩尔分数成正比。 3. 理想气体混合物就是一种理想溶液。 4. 对于理想溶液,所有的混合过程性质变化均为零。 5. 对于理想溶液所有的超额性质均为零。 6. 理想溶液中所有组分的活度系数为零。 7. 体系混合过程的性质变化与该体系相应的超额性质是相同的。 8. 对于理想溶液的某一容量性质M ,则__ i i M M =。 9. 理想气体有f=P ,而理想溶液有i i ?? =?。 10. 温度和压力相同的两种理想气体混合后,则温度和压力不变,总体积为原来两气体体积 之和,总热力学能为原两气体热力学能之和,总熵为原来两气体熵之和。 11. 温度和压力相同的两种纯物质混合成理想溶液,则混合过程的温度、压力、焓、热力学 能、吉氏函数的值不变。 12. 因为G E (或活度系数)模型是温度和组成的函数,故理论上i γ与压力无关。 13. 在常温、常压下,将10cm 3的液体水与20 cm 3的液体甲醇混合后,其总体积为 30 cm 3。 14. 纯流体的汽液平衡准则为f v =f l 。
15. 混合物体系达到汽液平衡时,总是有l i v i l v l i v i f f f f f f ===,,??。 16. 均相混合物的总性质与纯组分性质之间的关系总是有 ∑= i i t M n M 。 17. 对于二元混合物体系,当在某浓度范围内组分2符合Henry 规则,则在相同的浓度范围内 组分1符合Lewis-Randall 规则。 18. 二元混合物,当01→x 时,1*1→γ,∞→11γγ,12→γ,∞=2*2/1γγ。 19. 理想溶液一定符合Lewis-Randall 规则和Henry 规则。 20. 符合Lewis-Randall 规则或Henry 规则的溶液一定是理想溶液。 21. 等温、等压下的N 元混合物的Gibbs-Duhem 方程的形式之一是 0ln 0 =??? ? ??∑ =i i N i i dx d x γ。(错。0ln 0 =??? ? ??∑ =j i N i i dx d x γ,N j ~1∈) 等温、等压下的二元混合物的Gibbs-Duhem 方程也可表示成0ln ln * 2 211=+γγd x d x 。 22. 二元溶液的Gibbs-Duhem 方程可以表示成 () () ?? ???????=-==? ? ? ======)1() 0()1()0(210 121111111ln x P x P E x T x T E x x T dP RT V P dT RT H dx 常数常数γγ 23. 下列方程式是成立的:(a )111 1ln ?ln f f RT G G -=-;(b) 1111ln ln γ+=-x RT G G l l ;(c)v l v l f f RT G G 1111?ln ?ln -=-;(d)???? ??=→1111?lim 1x f f x ;(e)??? ? ??=→110,1?lim 1x f H x Solvent 。 24. 因为E H H =?,所以E G G =?。 25. 二元溶液的Henry 常数只与T 、P 有关,而与组成无关,而多元溶液的Henry 常数则与T 、 P 、组成都有关。
2015 至 2016 学年第 1 学期 化工热力学 考试试卷B (答案与评分标准) 考试方式: 闭卷笔试 本试卷考试分数占学生总评成绩的 70 % 一、选择题(本题20分,每题2分) 二、判断题(本题10分,每题1分) 三、填空题(本题10分,每空1分) 1. 8.314,83.14,8.314,1.980 2. 0.243 3. Henry 定律, Lewis-Randall 规则 4. 0.587,0.717 5. 0.334 评分标准:每空1分,除了数字必须完全和以上参考答案相同以外,只要和以上参考答案相近的叙述都可以视为正确答案。 四、计算题(本题50分,每题10分) 1. 一钢瓶的安全工作压力10MPa ,容积为7810cm 3,若装入1000g 的丙烷,且在253.2℃(526.35K )下工作,若钢瓶问是否有危险? (注:以PR 方程计算,PR 方程为:) ()(b V b b V V a b V RT p -++--= ,方程的参数a = 793906.842 6 mol cm MPa ??-;b = 56.293 1 cm mol -?。) 解:1000g 丙烷的物质的量为:100044/g n g mol = (2分) 22.73mol = (1分) 3 781022.73cm V mol -= (2分) 31343.60cm mol --=? (1分)
根据PR 方程,253.2℃(526.35K )下,7810cm 3的钢瓶中装入1000g 的丙烷,其压力应该为: ()()8.314526.35793906.84 343.6056.29343.60(343.6056.29)56.29(343.6056.29)4376.07793906.84793906.8415.23287.31343.60399.8956.29287.31137402.2016172.68RT a p V b V V b b V b = - -++-?=- -?++?-=-=-?+?+ (2分) 10.0610=> (1分) 所以不能安全工作。 (1分) 评分标准:公式和计算方法对但数值略有差错的不扣分;直接代入数据,不写公式且计算正确也得分;仅仅写出公式并罗列数据,但没有计算结果或结果不准确的酌情给分。 2. 三元混合物的各组分摩尔分数分别为0.25,0.3和0.45,在6.585MPa 和348K 下的各组分的逸度系数分别是0.72,0.65和0.91,求混合物的逸度。 解: ?ln ln i i y φφ= ∑ (2分) 0.25ln 0.720.3ln 0.650.45ln 0.910.254=++=- (2分) ()ln ln f P φ= (2分) ln 6.585(0.254) 1.631=+-= (2分) )MPa (109.5=f (2分) 评分标准:公式和计算方法对但数值略有差错的不扣分;直接代入数据,不写公式且计算正确也得分;仅仅写出公式并罗列数据,但没有计算结果或结果不准确的酌情给分。 3. 设已知乙醇(1)-甲苯(2)二元系统在某一气液平衡状态下的实测数据为t = 45℃,p =24.4 kPa ,x 1=0.300,y 1=0.634,并已知组分1和组分2在45℃下的饱和蒸气压为kPa p s 06.231=, kPa p s 05.102=。试采用低压下气液平衡所常用的假设,求: (1) 液相活度系数1γ和2γ; (2) 液相的G E /RT ; 与理想溶液想比,该溶液具有正偏差还是负偏差? 解:(1)由1111γx p py s =,得 (2分)
第三章 催化反应的热力学和动力学 一、催化反应的热力学热力学 化学和酶催化反应和普通化学反应一样,都是受反应物转化为产物过程中的能量变化控制的。因此要涉及到化学热力学、统计学的概念。下面对催化反应热力学作简要介绍。 1.热力学第一定律(又称为能量守恒与转化定律) 实际上是能量守恒和转化定律的说明。能量有各种形式,能够从一种形式转化为另一种形式,从一个物体传递给另一个物体,但在转化和传递中,能量的总量保持不变。如果反应开始时体系的总能量是U 1,终了时增加到U 2,那么,体系的能量变化U ?为: U ?=U 2-U 1 (3-1) 如果体系从环境接受的能量是热,那么,体系还可以膨胀作功,所以体系的能量变化U ?必须同时反映出体系吸收的热`和膨胀所作的功。体系能量的这种变化还可以表示为: U ?=Q -W (3-2) Q 是体系吸收的热能,体系吸热Q 为正值,体系放热(或体系的热量受到损失)Q 为负值;W 是体系所作的功,当体系对环境作功时,W 值是正的,当环境对体系作功时,W 值是负的。体系能量变化U ?仅和始态及终态有关,和转换过程中所取得途径无关,是状态函数。 大多数化学和酶催化反应都在常压下进行,在这一条件下操作的体系,从环境吸收热量时将伴随体积的增加,换言之,体系将完成功。在常压p ,体积增加所作的功为: ??==V p pdV W (3-3) 这里,△V 是体系体积的变化值(即终态和始态时体积的差值)。因此,这时在常压下,体系只作体积功时,热力学一律的表达式为: U ?=V p Q p ?- (3-4) 对在常压下操作的封闭体系,H Q p ?=,△H 是体系热函的变化。因此,对常压下操作的体系:热力学一律的表达式为:V p U H ?+?=? (3-5) △U 和p △V 对描述许多化学反应十分重要。但对发生在水溶液中的反应有其特殊性,因为水溶液中的反应没有明显的体积变化,p △H 接近于零。△H ≈△U ,所以对在水溶液中进行的任何反应,可以用热函的变化△H 来描述总能量的变化,而这个量△H 是可以测定的。
第一章化学反应的方向和限度 第二节化学反应的程度和化学平衡 一可逆反应和化学平衡 1、可逆反应 在同一条件下,既能向一个方向进行,又能向相反方向进行的反应,称为可逆反应。插入视频文件:可逆反应与化学平衡 .swf 严格地说, 可以认为所有的化学反应都具有一定的可逆性, 从微观的角度来看, 反应物分子可以发生有效碰撞, 结合成产物分子;同时, 产物分子也可以发生碰撞,再结合成反应物分子:反应物?产物。 当反应进行到某一程度,恰好逆正υυ=,反应物和产物的浓度都不再随时间而改变。那么,可逆反应的这种状态,就称为化学平衡。 2、化学平衡 正逆反应速率相等时,反应体系所处的状态,称为化学平衡状态。 特点:(1 逆正υυ= (2动态平衡; (3有条件的、相对的平衡(——条件改变,平衡改变。 大量的实验表明:在一定条件下, 处于化学平衡状态的体系, 各物质浓度之间遵守一定的定量关系。这就是平衡常数关系式。 二平衡常数 1、平衡常数
可逆反应在一定温度下达到平衡时,产物浓度的系数次方的乘积与反应物浓度的系数次方的乘积之比是一个常数,这个常数就叫做平衡常数。 :平衡浓度 浓度平衡常数— c K c c c c K b a d g c ( (B(A(D(GdD gG bB aA c ??=+=+ 如果是气体反应,可以用平衡时各组分气体的分压来代替浓度,这时,平衡常数叫做压力平衡常数: (B (A(D(Gp b a d g p p p p K ??= (p :平衡分压★注意:K c 、 K p 一般都有单位,但习惯上不写; K c 一般不等于 K p 。 为了统一和计算方便,规定在平衡常数的表达式中,凡是溶液中的浓度都除以标准态浓度:3θdm mol 1-?=c , θc ——相对浓度 ;若是气体分压,都除以标准态压力:Pa 101325θ=p , θp p ——相对分压 ,这样用相对浓度或相对分压表示的平衡常数,叫 标准平衡常数。 2、标准平衡常数一般如果不作说明,我们提到的平衡常数都是指标准平衡常数。 ★注意 :(1平衡浓度、平衡分压 (2 对有纯固体或纯液体参加的反应, 纯固体或纯液体的浓度视为常数 1, 不 出现在平衡常数的表达式中 (3 溶液中的组分一定用相对浓度θc 表示; 气相一定要用相对分压θp p 表